Thermogravimetric calibration of permeation tubes used for the preparation of gas standards for air pollution analysis
Simonetta
Tumbiolo
a,
Luc
Vincent
b,
Jean-François
Gal
*a and
Pierre-Charles
Maria
a
aLaboratoire de Radiochimie, Sciences Analytiques et Environnement, Faculté des Sciences, Université de Nice-Sophia Antipolis, Parc Valrose, 06108 Nice Cedex 2, France. E-mail: gal@unice.fr; Fax: +33 (0)492 07 61 11; Tel: +33 (0)492 07 61 10
bChimie des Matériaux Organiques et Métalliques, Faculté des Sciences, Université de Nice-Sophia Antipolis, Parc Valrose, 06108 Nice Cedex 2, France
First published on 30th August 2005
Abstract
Sources of VOC (Volatile Organic Compounds) reference-materials at ppm and ppb levels are needed for calibration of air monitoring instruments. The permeation-tube technique is considered effective for the preparation of low concentration standards of high accuracy and stability. In this work, purpose-built PTFE permeation tubes, containing benzene, toluene, ethylbenzene, o-xylene or m-xylene (BTEX) were accurately and rapidly calibrated. Using the sensitive thermo-balance of a thermogravimetric apparatus, very low permeation rates were determined by the continuous monitoring of the tube weight loss as a function of time. Permeation rates in the range from 25 to 350 ng min−1 were determined with precision. Thermogravimetry appears to be a rapid method for the measurement of weight loss at constant temperature, allowing rapid characterization and recalibration of permeation tubes. A detailed study on toluene, chosen as a typical case, showed that there are variations of the permeation rate in the long term. The temperature dependence of the permeation coefficient was also explored and permeation rates were shown to display an Arrhenius behavior in the temperature range 304–324 K. Thermodynamic parameters influencing the permeation were discussed.
Introduction
Volatile organic compounds (VOCs) are considered as one of the major sources of indoor and outdoor air pollution. In particular, BTEX (benzene, toluene, ethylbenzene, xylenes) compounds can cause acute and long-term toxic effects on human health, especially benzene that is recognized as a carcinogenic compound. Regulations limit the amount of VOCs in the environment and, in several countries, the continuous measurement of benzene in ambient air is now mandatory.1,2 Development of analytical methods for sampling and analyzing BTEX in air requires the use of multi-component gas standards, which allow appropriate simulation of the environment of interest. Gaseous standard mixtures can be prepared by either static or dynamic methods. Static sample preparation is easily realized by injecting into a container a known weight of organic liquid, sufficiently volatile to be completely vaporized. The principal pitfall of this method is that the sources of errors are not well defined. As the concentration of an individual component decreases, the stability of the standard mixture often decreases: polar or reactive components can interact or adsorb on container walls; high molecular weight or high boiling point compounds can condense in the container; different components may react in the gas phase or on the walls. Moreover, several dilution steps may be necessary.3 For these reasons, the use of dynamic methods is generally preferred for generating standard mixtures. Several advantages over batch-type systems may be cited: minimization of the effects of adsorption on the surfaces of the system; continuous dilution range; ease of flow generation and analyte mixing; possibility of air sample conditioning for temperature, relative humidity, and control of air velocity.4 There are several methods for continuous generation of VOC gas standards, including:– Mixing of gas stream, also called “dilution of streams”, that are based on the mixing of two or more gas streams, coming from certified cylinders, at known flow rates and regulated by means of appropriate flow-meters.3–5
– Syringe continuous injection, where a volatile organic liquid is injected continuously at a constant rate into an air stream, by means of a mechanically driven syringe or pump.6
– Capillary injection, where liquid VOCs are added to the dilution flow by a capillary line.7,8
– Diffusion methods, where the gaseous pollutant is incorporated into the stream by means of a calibrated diffusion cell containing the pollutant in the liquid state, its vapor diffusing through a capillary tube.9–11
– Evaporation methods that use diluent gas bubbled through, or passed over, a liquid.5
– Electrolytic methods, that may be set to generate gases such as NO, NO2, CO2.5
– Thermal decomposition of silica based surface compounds, that may be used to generate vapours or gases such as thiols, isothiocyanates, CO, CO2, ethene and acetaldehyde.12–15
– Permeation methods,17–21 that use a sealed container, that can be made of a piece of polymer tube sealed at each end, filled with an analyte which permeates at controlled temperature through the polymer. The permeation process involves the dissolution of the analyte into the polymer, its diffusion through the polymer wall, and its release out of the polymer. Permeation tubes provide a simple method for preparing gas mixtures containing one or several gases at low concentration, at the part per million to part per billion levels. Usually, the permeable membrane is made of polytetrafluoroethylene or a fluorinated ethylene–propylene copolymer, although other materials, such as polyethylene, polyvinyl acetate, polyamide, polyester, polyethylene terephthalate or silicone polymers, could be used.16 Most permeation devices are two-phase (vapor + liquid) systems, although single-phase high-pressure gaseous tubes have been prepared occasionally.5,21 Permeation works well for analytes capable of maintaining gas–liquid equilibrium inside a container. The permeation device is then “conditioned” at a constant suitable temperature to saturate the polymer with the fluid, leading to a constant permeation rate. After a temperature change, some delay is required to re-establish a constant permeation rate. Control of permeation rate may be achieved through changes in geometry or temperature. Increasing the operating temperature or the surface area of the polymer membrane increases the rate. Conversely, increasing the thickness of the membrane decreases the pollutant permeation rate. Further adjustments of the compound concentrations may be obtained by adjustments of the diluent flow-rate.22,23 Combining permeation tubes in parallel allows the generation of multi-component mixtures.
A key advantage of permeation tubes resides in the range of compounds for which the method can be used, including condensable gases, permanent gases, liquids, sublimating solids, and many semi-volatile compounds. A recent report describes the application of permeation tubes for generating standard gas mixtures for malodorous substances such as volatile fatty acids.24 The two major constraints for analytes used in permeation devices are their availability in pure form and their long-term stability at a temperature allowing generation of a vapor pressure of at least 102 Pa.3
Calibration of permeation tubes or other permeation devices is an important step in the analytical process. Several techniques have been employed to quantify the emission rate of trace level gases from permeation devices. NH3 and HNO3 emission rates from permeation tubes have been determined using the absorption of ultraviolet light.25 Ion chromatography (IC) has also been employed for the purpose of determining permeation-tube emission rates in the case of water-soluble species.26 The output stream of the permeation tube is allowed to bubble through water and the solution is then analyzed by IC. However, the long period required for collecting a sufficiently large sample and the analysis time make this technique unsuitable for assessing permeation tube stability. Permeation tubes are most often calibrated using gravimetry, which consists of determining the tube weight loss as a function of time.3,5,18,27 The fact that calibration is referred directly to easily traceable weight measurements is the principal advantage over other calibration methods.
This paper presents the use of a thermobalance for monitoring the weight loss of permeation tubes. The aim of this work was to generate continuous and stable BTEX gas flows, which could be used for generating standard gas mixtures at trace levels. Tests were conducted on permeation tubes fabricated in the laboratory, and designed to be compatible with the thermobalance. The procedures evolved during the past two years, with the extension of our previous studies on benzene28 to the other members of the BTEX family (toluene, ethylbenzene, xylenes). One aim of the present study was to replace, by permeation tubes, cylinders containing certified gas standards, used for calibration of SPME (Solid Phase Micro-Extraction) fibers.29 The ruggedness of the performance of the permeation tubes was evaluated in connection with their practical use, and the temperature dependence of the weight-loss rate was investigated in the case of toluene. In the literature, potential sources of errors other than temperature changes have been evaluated in special cases. For example, a carbonyl sulfide permeation device was shown to simultaneously produce carbon dioxide, due to the decomposition of the main constituent.30 In the case of BTEX, decomposition can be neglected in the absence of light and oxidant. Initial impurities are in trace amounts, and even if they permeate faster than the principal component, their contribution to the amount of emitted vapors is negligible.
Experimental
Permeation tubes fabrication
All permeation tubes were made from lengths of Teflon® tubing (PTFE) (Fisher–Bioblock Scientific™) of different diameters. This polymer is inert, sufficiently elastic to be sealed with plugs and commercially available in a wide variety of diameters and wall thicknesses. Each tube was stopped by two stainless-steel (AISI 420) balls, a material chosen for its durability and low reactivity.22 Steel balls are commercially available in many sizes that can be easily introduced into the tubes, resulting in a leak-proof seal. This design of the permeation tubes was chosen for reasons of simplicity, but also for minimizing weight and volume, considering the characteristics of the thermobalance. BTEX permeation tubes used in our preliminary work were stopped with two machined brass plugs.28 After one year, the liquid exhibited a blue coloration and a blue solid was present in the liquid. This was attributed to oxidation of the brass plugs.Therefore, we manufactured a new series of BTEX permeation tubes stopped with stainless steel balls. Before sealing, each tube was partially filled, approximately to 80 percent of the tube volume, with one of the analytical-grade BTEX compounds. For each tube, an extra length was left above the plug. In this part of the PTFE tube above the ball, two small holes were drilled for the suspension of the permeation tube in the thermobalance oven. One year after fabrication, the balls and tubes appeared to be intact, without any type of visible alterations. Table 1 reports the dimensions and the initial weights for each tube.
SAFETY NOTE: benzene is classified as a carcinogen; BTEX liquid compounds must be handled under a well-vented hood.
Weight-loss monitoring of permeation tubes
Continuous weight loss was monitored using a thermobalance (SETARAM, France, model 92-12) with a digital resolution of 0.2 µg, a 1 µg detection limit and a maximum load of 20 g. The chamber housing the balance and the connected oven constitute a gastight system, allowing weight-loss monitoring under vacuum or under a controlled circulating atmosphere (Fig. 1). The oven temperature was regulated to ±0.2 °C. Each permeation tube was suspended to the balance beam using a platinum wire in such a way as to position the tube close to the oven temperature probe. Measurements were carried out under isothermal conditions, under a ∼0.5 L h−1 flow of zero-air. Precise measurement of the gas flow was not necessary, because the exact composition of the evolved gas was not of concern for this study. The zero-air gas exhaust from the oven, containing traces of the analytes, was sent to a vented hood. For each tube, weight loss was monitored during a week. For each experiment, the permeation tube was left in position under the constant conditions given above. The experimental points were recorded after a two-day stabilization period. Permeation tubes necessitate a conditioning period after their fabrication, or when operating conditions are modified.16,28 This period is easily visualized on the weight vs. time curves.28 | ||
| Fig. 1 Scheme of the thermobalance used for the weight-loss monitoring of permeation tubes. | ||
Results and discussion
The permeation tubes weight loss vs. elapsed time, at 31.0 °C, is shown in Fig. 2 for each of the BTEX compounds. The internal clock of the instrument computer gives elapsed time, and uncertainties on time measurements are considered negligible. The mass scale of the instrument can be calibrated to better than 0.1%, and the resulting absolute accuracy of our results should be in this range. In fact, the largest source of random and systematic error is the temperature, which is considered later. Rather constant weight-loss rates, or permeation rates, were observed. They are reported in Table 2, in units of nanograms per minute, as is common practice for permeation tubes. For each compound, the slope of the least-squares regression line (weight loss vs. elapsed time) is given (all determination coefficients, R2 > 0.999), along with the 95% confidence interval. Permeation rates varied between 26 ng min−1 and 350 ng min−1, depending on the compound and tube size. Such low permeation rates enable generation of standard gaseous mixtures at low concentrations of analytes. The very small standard deviations, and the associated small 95% confidence interval, are attributed to the large number of acquired data points (about 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500). This means also that the temperature of the oven is constant; see below. The accuracy of the permeation rate is determined by the accuracy of the mass calibration, which is easily traceable to primary standards. Comparisons were made between data obtained for this series of experiments with data previously reported.28 In our previous work, as a result of software limitations, data were acquired during five days, but acquisition had to be restarted manually every 24 hours. Small daily fluctuations were observed corresponding to the restart operation. In the present work, acquisitions were performed continuously during five days. The small daily fluctuations were drastically reduced, as compared with previous data obtained with restarts every 24 hours.
500). This means also that the temperature of the oven is constant; see below. The accuracy of the permeation rate is determined by the accuracy of the mass calibration, which is easily traceable to primary standards. Comparisons were made between data obtained for this series of experiments with data previously reported.28 In our previous work, as a result of software limitations, data were acquired during five days, but acquisition had to be restarted manually every 24 hours. Small daily fluctuations were observed corresponding to the restart operation. In the present work, acquisitions were performed continuously during five days. The small daily fluctuations were drastically reduced, as compared with previous data obtained with restarts every 24 hours.
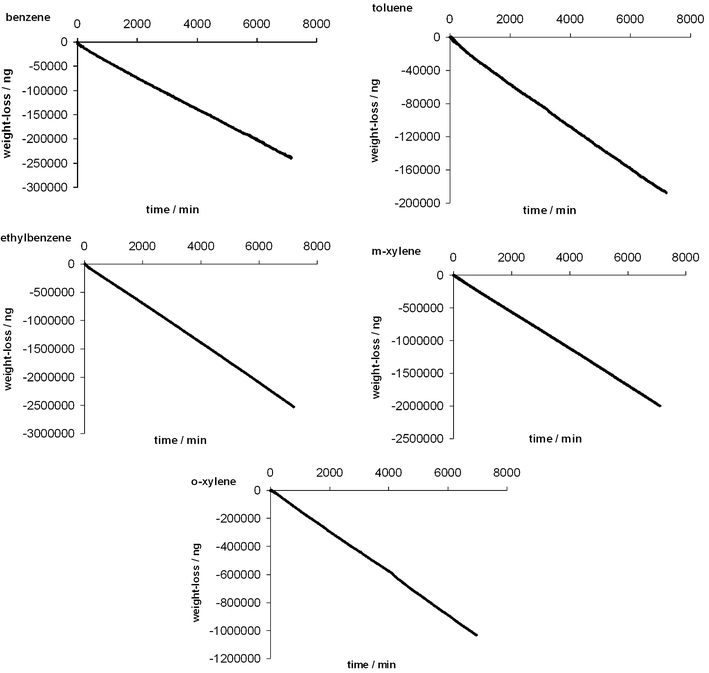 | ||
| Fig. 2 Thermogravimetric monitoring of benzene, toluene, ethylbenzene, o-xylene and m-xylene permeation tubes: plots of the weight loss vs. time, corresponding to a non-interrupted 120 h acquisition period. | ||
Temperature effects on permeation rate
Small temperature fluctuations alter the bath gas density (buoyant force) and affect the apparent weight. This effect can be neglected in our case. The most important temperature effect is on permeation rates. A series of experiments were realized to evaluate the effect of temperature changes on a tube filled with toluene. Weight-loss rates at three different temperatures (31 °C, 41 °C and 51 °C) are given in Table 3 with their corresponding 95% confidence intervals.| Temperature | Permeation rate/ng min−1 |
|---|---|
| 31 °C | 71.50 ± 0.03 |
| 41 °C | 130.43 ± 0.01 |
| 51 °C | 276.22 ± 0.03 |
The experimental conditions were the same as above. Again, rather constant weight-loss rates were observed after a stabilization period of two days. It should be noted that the stabilization period could be visualized by recording the weight vs. time curve. It is worth mentioning that, after a one-year ageing the weight-loss rate of the toluene tube at 31 °C increased from 25.76 ng min−1 to 71.50 ng min−1. This variation may be attributed to a swelling phenomenon, to which polymers are subjected when in contact with an organic solvent for a long period.31–36 The monitoring of the evolution of the permeation rate could be done by calibrating the permeation tubes at regular intervals, e.g. once a month. A referee pointed out the interest of carrying out such a complementary study, which could allow a better estimation of the tube lifetime and the validity delay of a given calibration. In a technical note,28 we have previously suggested that on-line calibration of permeation tubes using thermogravimetry would be the most accurate way for generation of trace concentration gas standards.
Permeation tubes in which the liquid is not in direct contact with the polymer, as the refillable design of Teckentrup and Klockow,37 may give more stable emissions in the long term. The principal aim of our work was the validation of the thermobalance method for calibration of permeation tubes. Consequently, such designs, larger and heavier than our tubes, were not considered for this work, because of the size of the oven and the maximum balance load.
Permeation theory relies on Fick's diffusion law. Accordingly, the amount of sample material (fluid) q (in unit of mass × (area × time)−1) that passes through the permeation membrane can be expressed by eqn (1):5,21
 | (1) |
 | (2) |
The quantity measured is in fact the lost mass per unit of time (permeation rate) corresponding to  . The dependence of B on temperature is expected to follow an Arrhenius-type equation:
. The dependence of B on temperature is expected to follow an Arrhenius-type equation:
 | (3) |
 | (4) |
 | (5) |
Therefore, an Arrhenius-like dependence of permeation rate against temperature is expected. In our work, for the toluene tube, permeation rate variations were observed by exploring a temperature range of 31–51 °C, as reported in Table 3. A least square fit for the three experimental data sets leads to eqn (6):
 | (6) |
 of the toluene tube is expressed in ng min−1 and the temperature in K. From the exponential term of eqn (6), we can infer a value of the apparent activation energy equal to 55.3 kJ mol−1. As reported in the literature, the value of the permeation rate varies by about 10% with a change in temperature of 1 °C.5,17 From eqn (6), we obtain a change of 6–7% per degree in the whole experimental temperature range. In the complete measurement process involving permeation tubes, there are two kinds of errors due to temperature: temperature fluctuations (precision) during tube calibration and gas standard generation, and the possible temperature difference (accuracy) between the thermogravimetric oven and the oven used for gas generation, if they are different. From the toluene data, it is estimated that overall uncertainties of less than 2 to 3% can be obtained if the temperatures of calibration and gas generation systems are controlled and regulated to ±0.1 °C.
of the toluene tube is expressed in ng min−1 and the temperature in K. From the exponential term of eqn (6), we can infer a value of the apparent activation energy equal to 55.3 kJ mol−1. As reported in the literature, the value of the permeation rate varies by about 10% with a change in temperature of 1 °C.5,17 From eqn (6), we obtain a change of 6–7% per degree in the whole experimental temperature range. In the complete measurement process involving permeation tubes, there are two kinds of errors due to temperature: temperature fluctuations (precision) during tube calibration and gas standard generation, and the possible temperature difference (accuracy) between the thermogravimetric oven and the oven used for gas generation, if they are different. From the toluene data, it is estimated that overall uncertainties of less than 2 to 3% can be obtained if the temperatures of calibration and gas generation systems are controlled and regulated to ±0.1 °C.
Conclusions
The analyte source is a critical component of an apparatus for generating standard gaseous mixtures containing pollutants at trace levels. Permeation tubes permit the production of a continuous flow of calibration gas mixture. Directions are suggested for the preparation, calibration and method of use of easily made permeation devices. Thermogravimetry is proposed as an attractive alternative method to the classical discontinuous gravimetric calibration, and for rapid screening of tube quality. The advantages of continuous weight-loss monitoring of permeation tubes, put forward in a previous technical note,28 are confirmed, and the calibration method was developed and improved in this work. After a stabilization period that is easily visualized, the permeation rates can be measured, within relatively short measurement periods. Rather constant weight-loss rates were observed and small daily fluctuations, due to minimal perturbations of the measurement conditions, were reduced. The method allows an easy visualization of the stabilization period after any change in the working conditions.The combination of a thermobalance with other instruments such as a mass spectrometer, a Fourier transform infrared spectrometer, or a gas chromatograph, entail crucial calibration steps. A system, equipped with accurate flow controls of carrier gases, may be used for the calibration of spectrometers or GCs. The use of a dedicated instrument with greater sensitivity and better oven temperature regulation may help to reduce the short- and long-term mass measurement noise.
The temperature dependence of the weight-loss rate was shown to follow an Arrhenius-type law. This relationship may be useful for the prediction of permeation rate at other temperatures than those of the measurements, as well as the evaluation of errors related to temperature changes.
Acknowledgements
Financial support by ADEME (Agence de l'Environnement et de la Maîtrise de l'Energie) is gratefully acknowledged. We thank Dr Hugues Frey (Calibrage Company, St. Chamas, France) for useful discussions.References
- US Environmental Protection Agency; Technology Transfer Network; National Air Toxics Assessment, http://www.epa.gov/ttn/atw/nata/.
- Directive 2000/69/EC of the European Parliament and of the Council of 16 November 2000 relating to limit values for benzene and carbon monoxide in ambient air, Official Journal of the European Communities , 2000, L313, 12.
- J. J. McKinley and R. E. Majors, LC–GC, 2000, 10, 1024–1033.
- J. A. Koziel, P. A. Martos and J. Pawliszyn, J. Chromatogr., A, 2004, 1025, 3–9 CrossRef CAS.
- J. Namiesnick, J. Chromatogr., 1984, 300, 79–108 CrossRef CAS.
- H. Rothweiler, P. A. Wager and C. Schlatter, Atmos. Environ., 1991, 25B, 231–235 CAS.
- E. Goelen, M. Lambrechts, F. Geyskens and T. Rymen, Int. J. Environ. Anal. Chem., 1992, 47, 217–225 CrossRef CAS.
- H. Hori and Y. Yanagisawa, Environ. Sci. Technol., 1993, 27, 2023–2030 CrossRef CAS.
- M. Gautrois and R. Koppmann, J. Chromatogr., A, 1999, 848, 239–249 CrossRef CAS.
- P. R. Fielden and G. M. Greenway, Anal. Chem., 1989, 61, 1993–1996 CrossRef CAS.
- J. M. Wong, N. Y. Kado, P. A. Kuzmicky, H.-S. Ning, J. E. Woodrow, D. P. H. Hsieh and J. N. Seiber, Anal. Chem., 1991, 63, 1644–1650 CrossRef CAS.
- M. Prokopowicz, P. Konieczka and J. Namiesnik, Environ. Technol., 1999, 20, 1065–1073 CrossRef CAS.
- P. Konieczka, M. Prokopowicz, B. Zygmunt, J. F. Biernat and J. Namiesnik, Chromatographia, 2000, 51, S249–S260 CrossRef CAS.
- E. Przyk, P. Konieczka, J. Szczygelska-Tao, R. Teschner, J. F. Biernat and J. Namiesnik, J. Chromatogr., A, 2001, 928, 99–108 CrossRef CAS.
- E. Przyk, A. Switaj-Zawadka, J. Szczygelska-Tao, A. Przyjazny, J. F. Biernat and J. Namiesnik, Crit. Rev. Anal. Chem., 2003, 33, 249–267 CrossRef CAS.
- J. Namiesnik, D. Gorlo, L. Wolska and B. Zygmunt, Chem. Anal. (Warsaw), 1999, 44, 201–213 CAS.
- W. Janicki, L. Wolska, T. Gorecki and J. Namiesnik, Chem. Anal. (Warsaw), 1993, 38, 423–428.
- J. J. McKinley, Annu. ISA Anal. Div. Symp.—Proc., 1998, 31, 115–128 Search PubMed.
- J. R. Hopkins, A. C. Lewis and K. A. Read, J. Environ. Monit., 2003, 5, 8–13 RSC.
- P. H. Huang and R. Kacker, J. Res. Natl. Inst. Stand. Technol., 2003, 108, 235–240 Search PubMed.
- T. J. Bruno, J. Chromatogr., A, 1995, 704, 157–162 CrossRef CAS.
- A. E. O'Keeffe and G. C. Ortman, Anal. Chem., 1966, 38, 760–763 CrossRef CAS.
- F. P. Scaringelli, A. E. O'Keeffe, E. Rosenberg and J. P. Bell, Anal. Chem., 1970, 42, 871–876 CrossRef CAS.
- J. P. Spinhirne and J. A. Koziel, Trans. ASAE, 2003, 46, 1639–1646 Search PubMed.
- J. A. Neuman, T. B. Ryerson, L. G. Huey, R. Jakoubek, J. B. Nowak, C. Simons and F. C. Fehsenfeld, Environ. Sci. Technol., 2003, 37, 2975–2981 CrossRef CAS.
- R. A. Washenfelder, C. M. Roehl, K. A. McKinney, R. R. Julian and P. O. Wennberg, Rev. Sci. Instrum., 2003, 74, 3151–3154 CrossRef CAS.
- D. Knopf, Accredit. Qual. Assur., 2001, 6, 113–119 Search PubMed.
- P.-C. Maria, J.-F. Gal, M. Balza, E. Peré-Trepat, S. Tumbiolo and J.-M. Couret, Anal. Chem., 2002, 74, 305–307 CrossRef CAS.
- S. Tumbiolo, J.-F. Gal, P.-C. Maria and O. Zerbinati, Anal. Bioanal. Chem., 2004, 380, 824–830 CrossRef CAS.
- A. Fried, B. Henry and S. Sewell, J. Geophys. Res., 1998, 103, 18895–18906 CrossRef CAS.
- R. Makitra, Y. Pyrih, E. Sagladko, A. Turovskiy and G. Zaikov, J. Appl. Polym. Sci., 2001, 81, 3133–3140 CrossRef CAS.
- F. Bénière, Diffus. Defect Data, Pt. A, 2001, 194–199, 897–908 Search PubMed.
- S. R. Lustig and N. A. Peppas, J. Appl. Polym. Sci., 1987, 33, 533–549 CrossRef CAS.
- J. Brandrup and E. H. Immergut, Polymer Handbook, Wiley, New York, 3rd edn, 1989 Search PubMed.
- L. H. Sperling, Introduction to Physical Polymer Science, Wiley, New York, 2nd edn, 1992 Search PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, New York, 1953 Search PubMed.
- A. Teckentrup and D. Klockow, Anal. Chem., 1978, 50, 1728 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |