Synthesis and characterization of covalently immobilized bis-crown ether based potassium ionophore
Róbert
Bereczki
a,
Róbert E.
Gyurcsányi
*b,
Béla
Ágai
c and
Klára
Tóth
*ab
aResearch Group for Technical Analytical Chemistry of the Hungarian Academy of Sciences, Budapest University of Technology and Economics, Szt. Gellért tér 4, Budapest, 1111-Hungary. E-mail: robertgy@mail.bme.hu; ktoth@mail.bme.hu
bInstitute for General and Analytical Chemistry, Budapest University of Technology and Economics, Szt. Gellért tér 4, Budapest, 1111-Hungary
cDepartment of Organic Chemical Technology, Budapest University of Technology and Economics, Műegyetem rkp. 3, Budapest, 1111- Hungary
First published on 26th November 2004
Abstract
The synthesis of a novel covalently immobilized crown ether based potassium ionophore is presented. Apart from previously proposed methods for the preparation of PVC linked ionophores based on the chemical modification of functionalized PVC polymers, the hereby proposed procedure involves the direct copolymerization of a suitable derivative of the bis-crown ether type potassium ionophore (BME 44) and vinyl chloride monomer. The analytical performance of the potentiometric ion selective electrodes incorporating the PVC bound ionophore were optimized and determined. Compared with electrodes based on other bis-crown ether type immobilized potassium selective ionophores a slightly improved logKpotK,Na and a longer lifetime was found. Spectral imaging and chronoamperometry were used to study the mobility of different bis-crown ether derivatives in plasticized PVC membranes.
1. Introduction
Solvent polymeric membranes containing neutral ionophores1 are widely used for the selective determination of various ions, especially in clinical, on-line process and environmental analysis. The active components (ionophores and different additives) of such membranes need to be highly lipophilic and are implicitly hosted in a hydrophobic polymeric matrix.2 Despite the hydrophobic character of solvent polymeric membranes, during extended exposure to the aqueous sample solution, a gradual loss of membrane components (ionophores, plasticizer, additives) cannot be avoided.3 Clearly, the loss of the active components (leaching) affects the selectivity and response function of the ion selective electrodes (ISEs), ultimately leading to the disappearance of their ion measuring capability.3 The leaching process is especially severe in case of optodes (since the optical signal is directly dependent on the concentration of the membrane components),4 miniaturized electrodes, flow through systems and samples with lipophilic character, such as biological fluids, that favor more enhanced extraction of the membrane components.5 The leaching process besides limiting the lifetime of the electrodes might also cause toxic problems in case of in-vivo measurements.6 Therefore the mechanism of the leaching process was thoroughly studied and mathematical formulations have been derived on the basis of mass transport laws in the aqueous and organic (membrane) phase, the partition coefficient of the membrane components at the membrane/solution interface, and the geometry of the system.3 The models developed were successfully used to predict the lifetime of ISEs as well as to formulate minimal requirements for an ISE system to achieve the desired operation time. On the experimental side, solutions have been proposed to eliminate or at least to slow down the leaching process. Due to the essential importance of ionophores in ISEs, many of these studies were focused to avoid their loss from the membrane and as a consequence various solutions such as increasing the lipophilicity of the ionophores7 or immobilizing them to the polymeric matrix have been proposed. The covalent immobilization provides the most effective mean to prevent the leaching and methods for practically all the membrane components (plasticizer,8 lipophilic additive9–11 and ionophores4,12) have been reported. Furthermore, attempts have also been made to substitute the plasticized polymers with the so-called self-plasticized matrices.13Recently with the advent of mass transport controlled electrodes14–16 the immobilization of ionophores was recognized as a viable strategy to design rugged ISEs with improved detection limits. It has been demonstrated that by using immobile ionophores the zero current ion fluxes are significantly reduced and therefore even with conventional inner filling solutions nanomolar detection limit could be obtained for lead selective electrodes.17 Although various polymeric matrices were used for the immobilization of ionophores, such as different functionalized PVCs, polyurethanes, silicon rubbers, polyacrylates, the most common formulations of an ion selective membrane are based on plasticized high molecular weight poly(vinyl chloride) (PVC). Beyond its compliance with the essential requirements for the generation of a potentiometric response and thorough characterization, the popularity of PVC based membranes can also be explained with their good mechanical properties as well as their easy handling and compatibility with different electrode designs. However, until now, the covalent attachment of ionophores to PVC backbones could only be accomplished by using functionalized PVCs such as PVC–COOH.12 This approach is rather convenient and simple, but the reaction product often contains unreacted functional groups that have been reported to affect the selectivity and response time of the relevant ISEs.18 Similar results were obtained with a lead ionophore attached to polyurethane since the functional groups of polyurethane provide coordinating sites for alkali and alkaline earth metal cations.17 Therefore, we have been interested to develop a novel method for the synthesis of PVC linked ionophores by direct copolymerization of the ionophore and the vinyl chloride monomer avoiding the drawbacks of previously described methods. A suitable bis-crown ether based ionophore (BR 0297) having a chemical structure similar to the commercially available BME 44 potassium ionophore (introduced earlier by our group),19 bearing a terminal alkenyl-chain was synthesized20 and copolymerized with vinyl chloride. Although macrocyclic crown ether based ionophores are widely used in ion selective electrodes,1 only a few attempts have been done for covalent immobilization of their suitably tailored derivatives to polymeric backbones. According to our knowledge the first experiments in this field involved different crown or benzocrown ethers bearing a pendant amino group. Thus Daunert and co-workers synthesized 4′-aminobenzo-15-crown-5 and then covalently attached it to carbodiimide activated carboxylic-PVC to prepare K+-sensor.12 A value of −2.58 was reported for the potassium vs. sodium selectivity (logKpotK,Na), however, the lifetime of the sensor was limited and a gradual deterioration of the detection limit and slope was observed. As an explanation the authors pointed out that a slow hydrolysis of the amide bond linking the ionophore to the polymeric backbone might occur. Cross and co-workers synthesized different 18-crown-6 derivatives grafted to carboxy-PVC or poly(acrylic acid), and blended this “active” polymer with high molecular weight PVC.21 However, in the case of polymer blends, phase separation of the active and support polymer can take place ultimately leading to poor potentiometric response (logKpotK,Na = −0.6). Further attempts to fabricate durable K+ ion sensors have been made by Hall and co-workers. During the polymerization process they attached acryloylamido-benzocrown-ethers to a methyl-methacrylate/n-butyl-acrylate copolymer matrix (logKpotK,Na = −1.9).13
It is clear that the selectivity values lag well behind of those obtained with commercial potassium ionophores. This is most likely explained either with the poor selectivity of the grafted crown ethers or with the adverse effect of the polymeric matrix, and better analytical characteristics are expected when ionophores with proper selectivity are covalently attached to an “inert” polymeric matrix. This can be considered a challenging task since to our knowledge the direct copolymerization of an ionophore with the vinyl chloride monomer has never been attempted. Therefore, here we report the synthesis and potentiometric characterization of a novel PVC linked ionophore (PVC-BR0297) obtained by copolymerization of BR 0297 ionophore and vinyl chloride monomer. The mobility of the different bis-crown ether derivatives in plasticized PVC membranes were investigated by spectral imaging technique and chronoamperometry.
2. Experimental
2.1. Chemicals and reagents
Selectophore grade BME 44 ([2-dodecyl-2-methyl-1,3-propanediyl-bis[N-(5′-nitro(benzo-15-crown-5)-4′-yl)carbamate]]), potassium tetrakis(4-chlorophenyl)borate (KTpClPB), sodium tetrakis[3,5-bis(trifluromethyl)phenyl]borate (NaTFPB), high molecular weight poly(vinyl chloride) (PVC HMW), bis(2-ethylhexyl)sebacate (DOS), ortho-nitrophenyl-octylether (o-NPOE) and tetrahydrofuran (THF, puriss p.a.) were purchased from Fluka (Fluka, Buchs, Switzerland). The potassium ionophore BR 0297 was synthesized in the laboratory of the Department of Organic Chemical Technology at Budapest University of Technology and Economics.18 All aqueous solutions used were prepared from chemicals of analytical grade and double quartz distilled water.2.2. Synthesis of covalently bound potassium ionophore
PVC-BR0297 was synthesized via thermally initiated free radical suspension polymerization in 5 dm3 jacketed stainless steel reactor (Büchi AG, Switzerland) in a pilot laboratory of BorsodChem Inc. (Kazincbarcika, Hungary). Ten g ionophore (BR 0297) dissolved in 100 g ethyl acetate, 1000 g vinyl chloride monomer (VCM), 2200 g distilled water together with suspending agents (0.9 g poly(vinyl alcohol) and 0.1 g (hydroxypropyl)methyl cellulose ether) were introduced in the reactor and strongly agitated (650 min−1) at 58 °C and 10 bar. Using 0.8 g bis(2-ethylhexyl)peroxydicarbonate and 1.2 g 2,2′-azo-bis(2,4-dimetil valeronitrile) initiators dissolved in the VCM, after 12 h 30 wt.% of the monomer was transformed. The product, PVC-BR0297 copolymer, was separated from the unreacted BR 0297 ionophore by dissolving the crude product of the polymerization reaction in THF and precipitating it with MeOH. The process was repeated at least four times and the purity of the PVC-BR0297 was tested by analyzing the polymeric fraction with gel permeation chromatography.2.3. Polymer characterization
The molecular weight distribution of PVC-BR0297 copolymer was determined by gel permeation chromatography (GPC) in THF, using a setup consisting of Waters 510 pump, Waters Styragel HR 5E column and a Waters 410 refractometer detector (Waters Corporation, Milford, MA, USA). The instrument was calibrated with PVC standards (Sp2 Scientific Polymer Products Inc., Ontario, NY, USA). The ionophore content of the purified PVC-BR0297 polymer was determined with a Unicam UV-2 UV-Visible spectrometer at 391 nm using the standard addition method.2.4. Preparation of the potassium selective membranes
The membranes containing the free ionophore (BR 0297) typically consisted of 0.25–2 wt.% ligand, 34–39 wt.% PVC powder, 61–64 wt.% plasticizer (o-NPOE or DOS) and lipophilic anionic sites (KTpClPB, 0–100 mol% with respect to the ligand). The membrane components were dissolved in 1 ml THF and cast in 30 mm diameter glass rings. Membranes from the covalently immobilized ionophore (PVC-BR0297) with 0.65 wt.% ionophore content were prepared according to the same procedure as described for the free ionophore. Some of the membrane compositions used in this study are summarized in Table 1.| Membranea | PVC/mg | Plasticizer/mg | Ionophore/mg; mmol kg−1 | Additive/mg; mol% | ||||
|---|---|---|---|---|---|---|---|---|
| HMW PVC | PVC-BR0297 | DOS | o-NPOE | BR 0297 “free” | BR 0297 “bound” | BME 44 | ||
| a BR (BR 0297), PBR (PVC-BR0297), BME (BME 44), D (DOS), N (o-NPOE). b NaTFPB. | ||||||||
| BR-D-1 | 66.36 | 124.45 | 2.35; 13.75 | 0.40; 30 | ||||
| BR-D-2 | 74.40 | 117.60 | 1.14; 6.68 | |||||
| BR-D-3 | 57.75 | 134.46 | 1.16; 6.79 | |||||
| BR-D-4 | 86.86 | 106.46 | 1.18; 6.87 | |||||
| BR-D-5 | 64.26 | 128.05 | 3.50; 20.19 | 0.49; 25 | ||||
| BR-D-6 | 64.67 | 128.01 | 0.43; 2.52 | 0.066; 25 | ||||
| BR-N-1 | 74.40 | 116.30 | 0.50; 2.96 | 0.08; 30 | ||||
| BR-N-2 | 64.48 | 121.64 | 4.18; 24.64 | 1.77; 70 | ||||
| PBR-D-1 | 86.67 | 101.75 | 0.56; 3.36 | 0.16; 50 | ||||
| PBR-D-2 | 74.41 | 117.65 | 0.48; 2.83 | |||||
| PBR-D-3 | 64.57 | 128.04 | 0.42; 2.47 | 0.059; 25 | ||||
| PBR-D-4 | 64.02 | 128.00 | 0.42; 2.47 | 0.105; 25b | ||||
| PBR-N-1 | 74.50 | 116.43 | 0.48; 2.84 | 0.08; 30 | ||||
| BME-D-1 | 65.40 | 122.43 | 2.40; 13.02 | 0.37; 30 | ||||
| BME-D-2 | 75.06 | 117.10 | 1.20; 6.42 | |||||
| BME-D-3 | 64.25 | 127.08 | 3.81; 20.13 | 0.49; 25 | ||||
| BME-N-1 | 74.42 | 116.58 | 0.49; 2.64 | 0.08; 30 | ||||
2.5. Chronoamperometric measurements
An Autolab Pgstat10 potentiostat-galvanostat (Ecochemie, Utrecht, The Netherlands) controlled by a Pentium II personal computer (Dell Dimension V333c) was used to perform the chronoamperometric studies. A transport cell made of Perspex® consisting of two 30 mL compartments was used. Each compartment accommodated a disk shaped Ag/AgCl electrode (A = 0.785 cm2) connected to the potentiostat. The membranes were conditioned overnight in 10−2 M KCl solution prior to the measurement. After placing them between the two compartments the leakage free operation was secured by means of three adjusting screws. Approximately 25 mL of 10−2 M potassium chloride solution was injected in each compartment and stirred throughout the experiment. Voltage steps typically ranging between 0.75 and 2.5 V were applied across the membrane and the resulting current transients were recorded. In the case of repeated polarizations applied to the same membrane relaxation periods with continuous monitoring of the potential were included. This ensured that the original equilibrium value of 0 mV in the electrochemical cell has been reached before applying the next voltage step.2.6. Spectroscopic imaging of the diffusion process
The bis-crown ether derivatives bearing nitro groups incorporated in PVC membranes absorb light in the visible range, which provided the basis for the optical determination of their diffusion coefficients.22 For this purpose two membranes one with the ionophore and the other one without (blank) but both having the same PVC/DOS ratios were used. Approximately 2 mm wide and 15 mm long slices were cut from these membranes and glued together along their shorter side with THF and placed on a microscope slide. A PARISS® Spectral Imaging System (http://www.lightforminc.com, Lightform Inc., Hillsborough, NJ, USA) mounted on an Olympus IX71 Inverted Research Microscope (Olympus Hungary Kft., Budapest, Hungary) was used to follow the diffusion of ionophores. The light from a selected line of the microscope's field of view is wavelength dispersed by the spectrometer and focused on a Peltier cooled charge coupled device (CCD). The recorded image (240 × 750 pixels) consists of intensity values with their spatial and wavelength coordinates. In other words the spectral imaging system provides simultaneously 240 full spectra uniformly distributed along a conveniently selected spatial direction. In these investigations the observation line (projection of the entrance slit) was chosen to be perpendicular on the joint interface of the two membranes and aligned in the direction of the diffusion. Further adjustments were made to position the separation line between the two membranes in the middle of the spectral image. The absorbance profile of the ionophore across the joint interface was obtained by recording the spectral image and plotting the absorbance values at 420 nm for each pixel as a function of their spatial coordinates. The evolution of the diffusion could be monitored by repeating the exposure according to a preprogrammed time scheme.2.7. Potentiometric measurements
Solvent polymeric membranes (diameter 5 mm) were incorporated into conventional ISE electrode bodies (Type IS 561; Philips, Eindhoven, The Netherlands). The inner electrolyte was 10−3 M KCl. Simultaneous electromotive force (EMF) measurements with 6 electrodes were carried out at room temperature with Lawson Labs 16-channel pH-meter (Lawson Labs Inc., Malvern, PA, USA) versus a double junction Ag/AgCl reference electrode (Metrohm 6.0726.100) having 0.1 M lithium acetate as salt bridge electrolyte. The potentiometric selectivity coefficients were determined with the separate solution method (SSM) from the EMF data measured in 0.1 M solutions of the chloride (Ba2+, Ni2+, Rb+, Cs+, NH4+, Na+, Sr2+, Zn2+, Ca2+, Li+, Mg2+) and nitrate (Cd2+, Cu2+, Pb2+) salts of the interfering ions and the primary ion, K+. The activity coefficients were calculated using the extended Debye–Hückel equation, while the liquid junction potential was estimated using the Henderson formalism.For the determination of complex stability constants the procedure described by Bakker et al. was used.23 Briefly, the ionophore containing membranes under study were matched with membranes having exactly the same composition, but without ionophore (blank). The membranes were conditioned in 0.01 M KCl or NaCl for at least one day and after placing them in Philips electrode bodies their potential was determined in the solution used for conditioning. The sandwich membrane was prepared by mechanically pressing together the ionophore loaded and its correspondent blank membrane immediately after their individual membrane potentials were determined. Then the sandwich membrane was incorporated in the Philips electrode body and the potential was recorded.
3. Results and discussion
The synthesis of PVC bound potassium selective ionophore involved first the preparation of a suitable molecule (BR 0297) having the same bis(15-crown-5) nitrourethane structure like BME 44 but bearing an allyl-type chain instead of the lipophilic tail (Fig. 1). This provides the possibility for copolymerization with different monomers to obtain polymers with covalently bound ionophores. The difficulty in copolymerizing BR 0297 and vinyl chloride relies in the strong radical-capturing properties of the NO2 groups of the ionophore, which has an inhibiting effect on the polymerization. Therefore the copolymerization of BR 0297 with vinyl-chloride monomer was carefully optimized and required longer reaction time and higher amount of initiator than conventional vinyl-chloride polymerization. A further consequence of the radical-capturing effect was the fragmentation of the polymeric chains resulting in a relatively low molecular weight PVC-BR0297 product (MWPVC-BR0297 = 73 × 103) when compared with Fluka HMW PVC (MWPVC HMW = 136 × 103). Due to this fragmentation, the PVC-BR0297 needed less plasticizer to obtain membranes with mechanical characteristics similar to those prepared from HMW-PVC. The weight percent of the ionophore in the PVC-BR0297 polymer was 0.65% that resulted in IS membranes typically with 0.25–0.29 wt.% (∼2.8 mmol kg−1) ionophore. This is lower than the most often used membrane formulations, which usually contain 1 wt.% ionophore (∼5–10 mmol kg−1). With this relatively low ionophore loading it is important to evaluate the mol equivalent of intrinsic anionic sites in the polymeric compound. Using a spectroscopic method developed earlier by our group,24 based on the determination of the protonation degree of pH selective chromoionophore (ETH 5294) integrated in the polymeric matrix, the concentration of ionic sites in PVCs prepared by BorsodChem's technology was determined to be 0.14 ± 0.02 mmol kg−1 PVC. For easier interpretation, this value lies between the Selectophore grade (0.06 mmol kg−1) and purum (0.36 mmol kg−1) PVCs, both manufactured by Fluka and represents only ∼5.5 mol% of the ionophore amount in PVC-BR0297 based membranes. | ||
| Fig. 1 Structures of BME 44, the mobile (BR 0297) and the immobilized (PVC-BR0297) bis-crown ether based potassium selective ionophores. | ||
3.1. Potentiometric measurements
The potentiometric potassium response of all membranes involved in this study was determined and close to Nernstian responses were obtained. The slopes of the membranes with the covalently linked ionophore (PVC-BR0297) (∼54 mV decade−1) were slightly smaller than those with the mobile BR 0297 ionophore, which were theoretical. The selectivity of the PVC-BR0297 ionophore was determined for a large number of cations, potential interferents in biological or environmental samples. However, the most important criterion is certainly the selectivity for potassium versus sodium ions and therefore most of the discussion will refer to the logKpotK,Na value. As known the selectivity coefficient depends on the concentration of active constituents (ionophores, anionic additive) in the membrane, stoichiometry and stability constants of the formed complexes. While many of these factors are intrinsic properties of the compounds used, the anionic site-ionophore ratios can be experimentally adjusted to obtain optimal selectivity.25 The optimum ratios are generally indicative of the stoichiometry of ion : ionophore complex. First PVC-BR0297 membranes with the same ionophore content, but with different mol% of KTpClPB (0, 30, 50, 70, 100) were prepared and the potassium selectivity was determined for the ions listed in the Experimental. The response times of ISEs based on PVC-BR0297 were longer than those based on their mobile analogues when changing between the interfering and primary ion solutions. However, even for such dramatic changes of the primary ion concentration (involving partial reconditioning of the membrane) the t95% has not exceeded five minutes.The selectivity for practically all ions showed (logKpotK,Mvs. mol% of anionic sites with respect of the ionophore content) an optimal selectivity at 50 mol% lipohilic anion content (result are not shown) and membranes having this composition were used for further comparisons with the other bis-crown ether type ionophore (BME 44 and BR 0297) containing membranes. The logKpotK,Na value of −2.61 as determined for PVC-BR0297 membranes was significantly worst than the value of −3.35 obtained for BME 44. When performing the same set of measurements with mobile BR 0297 ionophore containing membranes, the logKpotK,Na value was identical with the one determined for BME 44 (−3.35). Moreover, in contrary with covalently bound ionophore based membranes, for membranes consisting of the mobile BR 0297 ionophore the selectivity was practically not influenced by the ionic additive/ionophore ratio until it was lower than 100% (data not shown). It should be also noted that no selectivity decrease was observed for mobile ionophores containing membranes even when containing lower concentrations of ionophore (ca. 0.25 wt.%). By adding to the PVC-BR0297 containing membranes BR 0297 ionophore the selectivity improved and the values obtained for free ionophore containing membranes (−3.35) were regained. This suggests that the selectivity loss observed for the covalently linked ionophore is not due to an intrinsic selectivity modifying effect of the copolymer matrix.
To further investigate the difference in the selectivity behavior of covalently linked ionophores as starting point the equation  can be used, where KIJ is the equilibrium constant of the ion-exchange between the membrane and aqueous phase, while
can be used, where KIJ is the equilibrium constant of the ion-exchange between the membrane and aqueous phase, while 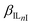 and
and  are the complex formation constants of the ionophore-primary ion and interfering ion complexes, respectively. This is the simplified form of a more general equation2 and it is only valid when the primary and interfering ions have equal charge (zI
=
zJ) and form equal stoichiometry complexes (nI
=
nJ), which is the case here when discussing the potassium vs. sodium selectivity. Since the selectivity coefficient is a function of the ratio of the respective complex formation constants we were interested to determine their values. The complex formation constants can be assessed with the sandwich membrane method23 and calculated according to the equation:
are the complex formation constants of the ionophore-primary ion and interfering ion complexes, respectively. This is the simplified form of a more general equation2 and it is only valid when the primary and interfering ions have equal charge (zI
=
zJ) and form equal stoichiometry complexes (nI
=
nJ), which is the case here when discussing the potassium vs. sodium selectivity. Since the selectivity coefficient is a function of the ratio of the respective complex formation constants we were interested to determine their values. The complex formation constants can be assessed with the sandwich membrane method23 and calculated according to the equation:
 | (1) |

The data for BME 44–potassium complex log = 7.75 ± 0.03 is in good agreement with previously published values (7.84).23 At 95% confidence level the complex formation constant for BR 0297–potassium ion complex is not significantly different than the value determined for BME 44 and also a decrease of almost an order of magnitude in the ionophore content does not significantly alter the logβKL value. In the case of covalently linked ionophore the formation constant for the potassium–ionophore complex was significantly smaller (about an order of magnitude) than for the mobile BR 0297 ionophore. However, the selectivity behavior is governed by the ratio of complex formation constants for potassium and sodium. In the case of BME 44 we have used the complex formation constants determined by Bakker's group (logβNaL
= 6.00),23 which gave a ratio of  , while for PVC-BR0297 the same ratio was 1.35 × 10−2. These results show that while the complex formation constants are decreasing if the ionophore is linked to the PVC matrix, the difference in the corresponding
, while for PVC-BR0297 the same ratio was 1.35 × 10−2. These results show that while the complex formation constants are decreasing if the ionophore is linked to the PVC matrix, the difference in the corresponding  ratios are within the experimental error. Therefore it seems that the complex formation constants are not responsible for the poorer potassium selectivity of the PVC-BR0297 membranes. On the other hand, both the selectivity data and the sub-Nernstian slopes can suggest slow phase transfer kinetics at the membrane/solution interface.26 It has been also reported that in plasticized PVC membrane an exudation of the plasticizer occurs at the membrane/solution interface.27,28 In this layer generally an enrichment of mobile ionophores and ionic sites has been observed. However, since in our case the selective complexing sites are linked to PVC, the phase segregation at the surface of the ion selective membrane obviously should result in a depletion of the ionophore at the interface with respect to the membrane bulk. In this case the potassium uptake/exchange at the membrane interface instead of the direct one step reaction with the ionophore should involve an extensive mediation by the anionic sites.29 Most likely the absence of the ionophore in the outmost layer of the membrane causes the very poor selectivity coefficients of membranes with no added extrinsic lipophilic ionic sites. The ionophore depletion at the membrane surface could also significantly bias the ionic additive/ionophore ratio at the membrane surface from the nominal value established in the membrane bulk. This could explain in some extent the selectivity loss experienced with membranes having ionic additive concentrations higher than 50 mol% with respect to the ionophore. Most likely the phase segregation effect diminishes at higher ionophore contents, but since we could not increase the ionophore content of the PVC-BR0297 the validity of this assumption could not be demonstrated. However, by decreasing the content of covalently linked ionophore in the membrane (dilution with Selectophore® grade HMW PVC) as expected the selectivity further deteriorated. In conclusion it seems that the selectivity decrease in the case of PVC-BR0297 is caused by the corroborated effect of both the low concentration of the PVC-BR0297 ionophore in the membrane and the phase segregation at the membrane surface, but direct evidence cannot be supplied at the present stage.
ratios are within the experimental error. Therefore it seems that the complex formation constants are not responsible for the poorer potassium selectivity of the PVC-BR0297 membranes. On the other hand, both the selectivity data and the sub-Nernstian slopes can suggest slow phase transfer kinetics at the membrane/solution interface.26 It has been also reported that in plasticized PVC membrane an exudation of the plasticizer occurs at the membrane/solution interface.27,28 In this layer generally an enrichment of mobile ionophores and ionic sites has been observed. However, since in our case the selective complexing sites are linked to PVC, the phase segregation at the surface of the ion selective membrane obviously should result in a depletion of the ionophore at the interface with respect to the membrane bulk. In this case the potassium uptake/exchange at the membrane interface instead of the direct one step reaction with the ionophore should involve an extensive mediation by the anionic sites.29 Most likely the absence of the ionophore in the outmost layer of the membrane causes the very poor selectivity coefficients of membranes with no added extrinsic lipophilic ionic sites. The ionophore depletion at the membrane surface could also significantly bias the ionic additive/ionophore ratio at the membrane surface from the nominal value established in the membrane bulk. This could explain in some extent the selectivity loss experienced with membranes having ionic additive concentrations higher than 50 mol% with respect to the ionophore. Most likely the phase segregation effect diminishes at higher ionophore contents, but since we could not increase the ionophore content of the PVC-BR0297 the validity of this assumption could not be demonstrated. However, by decreasing the content of covalently linked ionophore in the membrane (dilution with Selectophore® grade HMW PVC) as expected the selectivity further deteriorated. In conclusion it seems that the selectivity decrease in the case of PVC-BR0297 is caused by the corroborated effect of both the low concentration of the PVC-BR0297 ionophore in the membrane and the phase segregation at the membrane surface, but direct evidence cannot be supplied at the present stage.
On the positive side, however, a remarkable stability in time of the analytical performances of PVC-BR0297 based membranes was observed. The long term stability of the electrodes was determined, by storing the ISEs in aqueous solution (10−3 M KCl) and following their performance for a period of 120 days. No selectivity decrease has been observed at the end of the study (Table 3).
| IS Membranes | PBR-D-1 | PBR-D-1a | BR-N-2 | BME-D-1 | BR-D-1 |
|---|---|---|---|---|---|
| a Values were determined after 120 days of storage in 10−3 M KCl solution. | |||||
| Slope/mV decade−1 | 53.8 | 54.1 | 60.3 | 58.9 | 58.4 |
| Linear measuring range/M | 10−1–10−6 | 10−1–10−6 | 10−1–10−6 | 10−1–10−6 | 10−1–10−6 |
| Interfering ion, J | logKpotK,J | ||||
| Na+ | −2.61 | −2.44 | −3.38 | −3.35 | −3.27 |
| Li+ | −3.32 | −3.24 | −4.11 | −3.93 | −3.85 |
| NH4+ | −1.77 | −1.59 | −2.16 | −2.14 | −2.09 |
| Mg2+ | −2.53 | −2.44 | −2.70 | −2.69 | −2.68 |
| Cs+ | −1.98 | −1.73 | −2.54 | −2.47 | −2.44 |
| Ca2+ | −3.93 | −4.51 | −5.72 | −4.64 | −4.54 |
| Sr2+ | −3.80 | −4.07 | −5.08 | −4.33 | −4.29 |
| Pb2+ | −2.55 | −2.15 | −4.01 | −4.33 | −4.28 |
| Zn2+ | −3.75 | −4.02 | −4.90 | −4.36 | −4.35 |
| Ba2+ | −3.76 | −3.75 | −4.35 | −4.39 | −4.40 |
| Ni2+ | −3.77 | −3.96 | −4.65 | −4.38 | −4.41 |
| Cd2+ | −3.18 | −3.64 | −4.45 | −4.18 | −4.19 |
| Rb+ | −0.68 | −0.59 | −0.86 | −0.80 | −0.84 |
| Cu2+ | −3.41 | −3.40 | −4.61 | −4.37 | −4.36 |
3.2. Mobility of the bis-crown ether type ionophores
We have used optical and chronoamperometric techniques to study the mobility of bis-crown ether type ionophores in the PVC based membranes. For optical measurements a method previously described by the group of Pretsch22,30 has been adapted and improved by using a hyperspectral imaging system to follow the absorbance changes in the membranes. The diffusion of the ionophore from an ionophore loaded membrane into a blank membrane (without ionophore but otherwise having exactly the same composition) can be described by Fick's first law,31 which combined with Lambert–Beer's law results in the following expression: | (2) |
 | ||
| Fig. 2 Fitted absorbance gradients across the conjoined membranes at different time periods for mobile BR 0297 (A) and covalently immobilized PVC-BR0297 (B) ionophores. | ||
| Membrane | D/cm2 s−1 | Standard deviation | R/kΩ |
|---|---|---|---|
| a The diffusion coefficients were determined by chronoamperometry (ca) or spectral imaging (si). | |||
| BR-N-1 (ca) | 1.44 × 10−8 | 8 × 10−10 | 199 |
| BR-D-2 (si) | 0.61 × 10−8 | 3 × 10−10 | — |
| BR-D-3 (si) | 1.39 × 10−8 | 6 × 10−10 | — |
| BR-D-4 (si) | 0.25 × 10−8 | 2 × 10−10 | — |
| PBR-N-1 (ca) | No breakpoint was observed | 663 | |
| PBR-D-2 (si) | No diffusion was observed | — | |
| BME-N-1 (ca) | 1.47 × 10−8 | 4 × 10−10 | 223 |
| BME-D-2 (si) | 0.54 × 10−8 | 9 × 10−10 | — |
For o-NPOE based membranes, where the use of spectral imaging method could not be performed with enough reliability due to spectral overlapping, the mobility of the ionophores were investigated by chronoamperometry.32,33 The chronoamperometric technique is based on the electrical field induced transport of ions from one side of the IS membrane to the other, while recording the resulting current. The selectivity and permselectivity of the transport is given by the potassium ionophores and anionic sites incorporated in the IS membrane, respectively, providing transference numbers very close to 1 (1.02 ± 0.04) for the potassium.34 The mechanism of the ion transport process in solvent polymeric membranes induced by applied electrical field has been previously discussed in detail for mobile, neutral carrier membranes.33,35,36 The electrical field induced transport across the membrane results in the concentration polarization of all active constituents of the IS membrane: free ionophore, ion-ionophore complex and lipophilic additive.37 The free ionophore concentration decreases at the positively polarized side of the membrane due to the uptake of potassium ions, with concomitant formation of the ionophore–potassium complex, while increasing at the negative side (release of potassium ions). If the voltage is sufficiently high,35 after a certain time the back diffusion of the free ionophores cannot compensate for their depletion at the negatively polarized side of the membrane and at this point the resistance of the membrane quickly increases and implicitly the current decreases.37 Therefore the recorded current time curves exhibit a characteristic breakpoint, and the time (τ) needed to achieve this point depends on the diffusion coefficient of the free ionophore (Dionophore), the applied voltage (Vappl), the membrane resistance (Rohm) and on the concentration of the free ionophore (Cfreeionophore, calculated by subtracting the concentration of the anionic site from the total ionophore concentration).33,35
 | (3) |
Representative current–voltage curves are shown in Fig. 3 for solvent polymeric membranes based on BR 0297 and PVC-BR0297 ionophores. The theory (eqn. (3)) was found applicable for the mobile ionophores, demonstrated also by the linearity of τ1/2vs. the reciprocal of the applied voltage (results not shown). The initial current was used to calculate the bulk resistance of the different membranes. While similar composition BME 44 and BR 0297 based membranes had practically the same bulk resistance (∼200 kΩ) the PVC-BR0297 ionophore showed higher resistance (∼660 kΩ). The chronoamperometric transients obtained for PVC-BR0297 as a result of applied voltages in the range 0.75 and 10 V exhibited no breakpoint in the time scale of the measurements (15 min) providing further proof for the immobility of the PVC-BR0297 ionophore.
 | ||
| Fig. 3 Chronoamperometric transients of mobile BR 0297 (A) and immobile PVC-BR0297 (B) as result of an applied voltage of 1 V. Note the different current scale of the chronoamperometric transients. The insert shows the voltage–current curves used to calculate the bulk membrane resistances. | ||
The diffusion coefficient of BME 44 (1.47 × 10−8 cm2 s−1, BME-N-1) and BR 0297 (1.44 × 10−8 cm2 s−1, BR-N-1) were practically the same in o-NPOE plasticized membranes. There was also no significant difference between the diffusion coefficient of the two compounds in DOS plasticized membranes (0.54 × 10−8 cm2 s−1, BME-D-2 and 0.61 × 10−8 cm2 s−1, BR-D-2). This means that the hydrodynamic radius of the two ionophores is very similar and despite BME-44's long lipohilic chain, it is most likely determined by the bis-crown ether moiety. There is however, a noticeable difference between the diffusion coefficients of the ionophores recorded in o-NPOE and DOS plasticized PVC membranes.38 This difference is most likely due to the difference in the physical properties of the plasticizers since the diffusion coefficient, according to Einstein–Stokes equation, is a function of the dynamic viscosity (η) of the media in which the diffusion takes place ( , k—Boltzmann's constant, r—radius of the diffusing species). Although the viscosities of the o-NPOE and DOS plasticized PVC membranes are not known, the dynamic viscosities of the pure o-NPOE and DOS plasticizers are significantly different 13.8 × 10−2 and 20.2 × 10−2 g cm−1 s−1, respectively.3 Due to their high plasticizer content the viscosity of the PVC membranes should be highly dependent on the viscosity of the plasticizer, and therefore lower diffusion coefficients are to be expected in the PVC-DOS compared with the PVC–o-NPOE membranes, which is in agreement with the experimental data.
, k—Boltzmann's constant, r—radius of the diffusing species). Although the viscosities of the o-NPOE and DOS plasticized PVC membranes are not known, the dynamic viscosities of the pure o-NPOE and DOS plasticizers are significantly different 13.8 × 10−2 and 20.2 × 10−2 g cm−1 s−1, respectively.3 Due to their high plasticizer content the viscosity of the PVC membranes should be highly dependent on the viscosity of the plasticizer, and therefore lower diffusion coefficients are to be expected in the PVC-DOS compared with the PVC–o-NPOE membranes, which is in agreement with the experimental data.
4. Conclusion
The synthesis of PVC bound ionophore by direct copolymerization of an ionophore derivative and vinyl chloride monomer is reported for the first time. The method was found to be a viable way for the preparation of immobile ionophores. The strong radical capturing effect of the nitro groups on the BR 0297 monomer limited to 0.65 the weight percent of the ionophore with respect to the resulting polymer. However, it is reasonable to assume that the method will result in higher yields when ionophore structures without radical capturing functionalities are used. The logKpotK,Na values of immobilized ionophore containing PVC membranes were found inferior to similar structure but mobile potassium ionophores. Both the stability constants of covalently immobilized ionophore–potassium and sodium complexes are lower than that of the corresponding mobile ionophore complexes, however their ratio is the same. Thus the decreased logKpotK,Na cannot be ascribed to the decrease of the ratio of the stability constants of ionophore-relevant ion complexes, but rather to the change of the ionophore/mobile site concentration ratios in the membrane surface layer as compared to membrane bulk. The diffusion coefficients of the bis-crown type mobile ionophores have been determined with spectral imaging techniques and chronoamperometry. The spectral imaging studies demonstrated the immobility of the covalently linked ionophores in the solvent polymeric membrane.Acknowledgements
We are grateful to József Kupai (BorsodChem Inc., Kazincbarcika, Hungary) for his indispensable assistance in the preparation of the PVC-BR0297 polymer and to Andrásné Szesztay for the GPC measurements. This work has received financial support from the Hungarian Scientific Foundation (OTKA: F037977, F034431, M041969) and OTKA-NSF 46146 grants. R. E. G. gratefully acknowledges the Bolyai János and Varga József (Jorge Balla) fellowships. R. B. thanks the Varga József Foundation for financial support.References
- P. Bühlmann, E. Pretsch and E. Bakker, Chem. Rev., 1998, 98, 1593–1687 CrossRef.
- E. Bakker, P. Bühlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083–3132 CrossRef CAS.
- U. Oesch and W. Simon, Anal. Chem., 1980, 52, 692–700 CrossRef.
- T. Rosatzin, P. Holy, K. Seiler, B. Rusterholz and W. Simon, Anal. Chem., 1992, 64, 2029–2035 CrossRef CAS.
- O. Dinten, U. E. Spichiger, N. Chaniotakis, P. Gehrig, B. Rusterholz, W. E. Morf and W. Simon, Anal. Chem., 1991, 63, 596–603 CrossRef CAS.
- E. Lindner, V. V. Cosofret, S. Ufer, R. P. Buck, W. J. Kao, M. R. Neuman and J. M. Anderson, J. Biomed. Mater. Res., 1994, 28, 591–601 CrossRef.
- P. Gehrig, B. Rusterholz and W. Simon, Anal. Chim. Acta, 1990, 233, 295–298 CrossRef CAS.
- D. J. Harrison, A. Teclemariam and L. L. Cunningham, Anal. Chem., 1989, 61, 246–251 CrossRef CAS.
- D. N. Reinhoudt, J. F. J. Engbersen, Z. Brzozka, H. H. Vandenvlekkert, G. W. N. Honig, H. A. J. Holterman and U. H. Verkerk, Anal. Chem., 1994, 66, 3618–3623 CrossRef CAS.
- J. A. J. Brunink, R. J. W. Lugtenberg, Z. Brzozka, J. F. J. Engbersen and D. N. Reinhoudt, J. Electroanal. Chem., 1994, 378, 185–200 CrossRef CAS.
- Y. Qin and E. Bakker, Anal. Chem., 2003, 75, 6002–6010 CrossRef CAS.
- S. Daunert and L. G. Bachas, Anal. Chem., 1990, 62, 1428–1431 CrossRef CAS.
- L. Y. Heng and E. A. H. Hall, Anal. Chem., 2000, 72, 42–51 CrossRef CAS.
- T. Sokalski, A. Ceresa, T. Zwickl and E. Pretsh, J. Am. Chem. Soc., 1997, 119, 11347–11348 CrossRef CAS.
- E. Lindner, R. E. Gyurcsányi and R. P. Buck, Electroanalysis, 1999, 11, 695–702 CrossRef CAS.
- E. Bakker and E. Pretsch, Anal. Chem., 2002, 74, 420A–426A CrossRef CAS.
- M. Püntener, T. Vigassy, E. Baier, A. Ceresa and E. Pretsch, Anal. Chim. Acta, 2004, 503, 187–194 CrossRef CAS.
- E. Lindner, V. V. Cosofret, R. P. Kusy, R. P. Buck, T. Rosatzin, U. Schaller, W. Simon, J. Jeney, K. Tóth and E. Pungor, Talanta, 1993, 40, 957–967 CrossRef CAS.
- K. Tóth, E. Lindner, M. Horváth, J. Jeney, I. Bitter, B. Ágai, T. Meisel and L. Tõke, Anal. Lett., 1989, 22, 1185–1207 CAS.
- R. Bereczki, B. Ágai and I. Bitter, J. Incl. Phenom. Macrocycl. Chem., 2003, 47, 53–58 CrossRef CAS.
- G. G. Cross, T. M. Fyles and V. V. Suresh, Talanta, 1994, 41, 1589–1595 CrossRef CAS.
- M. Püntener, M. Fibbioli, E. Bakker and E. Pretsch, Electroanalysis, 2002, 14, 1329–1338 CrossRef.
- Y. M. Mi and E. Bakker, Anal. Chem., 1999, 71, 5279–5287 CrossRef CAS.
- R. E. Gyurcsányi and E. Lindner, Anal. Chem., 2002, 74, 4060–4068 CrossRef CAS.
- R. Eugster, P. M. Gehrig, W. E. Morf, U. E. Spichiger and W. Simon, Anal. Chem., 1991, 63, 2285–2289 CrossRef CAS.
- E. Lindner, K. Tóth and E. Pungor, Dynamic characteristics of ion-selective electrodes, CRC Press, Boca Raton, FL, 1988 Search PubMed.
- R. Kellner, E. Zippel, E. Pungor, K. Tóth and E. Lindner, Fresenius’ Z. Anal. Chem., 1987, 328, 464–468 CrossRef CAS.
- G. Horvai, E. Graf, K. Tóth, E. Pungor and R. P. Buck, Anal. Chem., 1986, 58, 2735–2740 CrossRef CAS.
- R. D. Armstrong, Electrochim. Acta, 1987, 32, 1549–1552 CrossRef CAS.
- B. Schneider, T. Zwickl, B. Federer, E. Pretsch and E. Lindner, Anal. Chem., 1996, 68, 4342–4350 CrossRef CAS.
- J. Crank, Clarendon Press, Oxford, UK, 2nd edn., 1975, p. viii, 414 pp.
- B. D. Pendley and E. Lindner, Anal. Chem., 1999, 71, 3673–3676 CrossRef CAS.
- B. D. Pendley, R. E. Gyurcsányi, R. P. Buck and E. Lindner, Anal. Chem., 2001, 73, 4599–4606 CrossRef CAS.
- A. P. Thoma, A. Vivianinauer, S. Arvanitis, W. E. Morf and W. Simon, Anal. Chem., 1977, 49, 1567–1572 CrossRef CAS.
- M. L. Iglehart, R. P. Buck, G. Horvai and E. Pungor, Anal. Chem., 1988, 60, 1018–1022 CrossRef CAS.
- T. M. Nahir and R. P. Buck, J. Phys. Chem., 1993, 97, 12363–12372 CrossRef CAS.
- E. Lindner, R. E. Gyurcsányi and B. D. Pendley, Pure Appl. Chem., 2001, 73, 17–22 CAS.
- R. Long and E. Bakker, Anal. Chim. Acta, 2004, 511, 91–95 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |