Hyphenating liquid phase separation techniques with mass spectrometry: on-line or off-line
Emma
Edwards
and
Jane
Thomas-Oates
Department of Chemistry, University of York, Heslington, York, UK YO10 5DD
First published on 19th August 2004
Abstract
The advantages of hyphenating analytical separation techniques to mass spectrometers have been recognised since the 1970s. The first attempts to couple liquid phase separations to mass spectrometry (MS) were problematic, but the development of electrospray ionisation (ES) in 1984 facilitated the production of routinely used, commercial interfaces. This review considers the most recent major developments in hyphenating liquid chromatographic and electrophoretic separations to mass spectrometry, and considers the role the post genomic sciences have played in driving them. It also considers why, despite all of the advances in hyphenation, the future appears to hold a major role for off-line analysis.
1 Introduction
The completion of the human genome sequence at the turn of this millennium has sparked an explosion in science and discovery that has spawned several new research fields in what is now termed post genomic science. Quantitative and qualitative analysis of the gene expression products especially mRNA, proteins and the metabolites they produce are now the subject of intensive study. The term proteome defines the entire protein complement in a given cell, tissue or organism at a particular time or physiological state. Proteomics is the study of protein expression, modifications and localization, and interactions of proteins in complexes. Metabolomics is the study of generally low-molecular-weight compounds such as lipids, carbohydrates, vitamins, and hormones that carry out much of the cell's business.Advances in these post genomic sciences have been vastly aided by significant developments in separations science (liquid chromatographic and electrophoretic separations) and mass spectrometry. However, the need in these areas for fully automated systems that enable rapid high throughput analyses, with ever more demanding limits of detection is now providing the driving force for rapid advances in the coupling of these two critical technologies in hyphenated systems, which are the subject of this review.
2 On-line hyphenation
2.1 Liquid chromatographic separations
In the early 1970s the first attempts to couple liquid phase separations such as liquid chromatography (LC) to mass spectrometry (MS) were made. However, coupling LC to MS proved more problematic than had the coupling of gas chromatography (GC) to MS because the high liquid flow rates made it difficult to maintain the mass spectrometer's vacuum. Therefore, to couple the two techniques together a suitable interface was required.Early methods used to interface LC to MS included thermospray, plasma spray, the moving belt, the pneumatic nebuliser interface, direct liquid introduction, the particle beam interface and dynamic FAB. These interfaces were not user friendly and the development of improved liquid based ionisation techniques was required. The introduction of atmospheric pressure liquid ionisation techniques such as atmospheric pressure chemical ionisation (APCI) and electrospray (ES) has led to the development of easy to use interfaces that have improved ionisation efficiencies, and rapidly resulted in the demise of the other earlier LC-MS interfaces. APCI and ES techniques are now the method of choice for many LC-MS applications.
The APCI source is a robust ion source, most suitable for the analysis of thermally stable, non-polar, small molecules. APCI tends to almost exclusively produce [M + H]+ ions. APCI is much more compatible with traditional higher flow LC than is ES as the flow rates are similar. An extensive literature search of LC-APCI-MS revealed no new significant developments in interface design in recent years. This is indicative of how well the available instrumentation works, and does not we believe negate the fact of the high volume of its usage. However, a relatively new atmospheric ionisation technique, atmospheric pressure photoionisation (APPI),1 has been found to efficiently ionise non-polar compounds at low flow rates and it has been suggested that this may become an important ionisation technique in the future for interfacing capillary LC and CE to mass spectrometry.
ES ionisation enables large, non-volatile molecules to be analysed directly from the liquid phase, making it ideal for the analysis of involatile, thermally labile biomolecules. In most currently used electrospray interfaces analyte solution is pumped through a capillary (typical flow rates 0.1–10 µL min−1), which is held at a high potential (typically 2–5 kV). Many commercial LC-MS interfaces (such as that shown in Fig. 1A) are available. If the LC eluent flow rate is compatible with the ES flow rate, a transfer capillary is all that is required to directly transfer all of the eluent from the LC column to the ES needle. If the eluent flow rate is too high for the ES interface, then it can be split; only the appropriate proportion of the eluent is sent to the mass spectrometer.
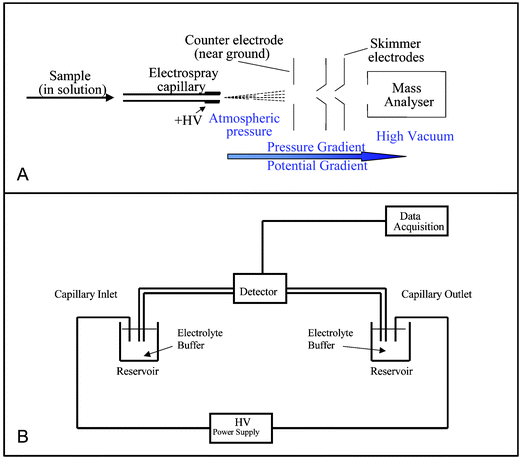 | ||
| Fig. 1 Panel A: Features of an electrospray interface, Panel B: Schematic of capillary electrophoresis instrumentation. | ||
It is largely thanks to subsequent developments of the original electrospray experiment that such huge advances in hyphenating liquid separation systems to mass spectrometers have been made. Probably the most important of those developments is nanospray.2 Wilm and Mann used the term nanospray to describe the spray observed from the use of static needles filled with solution with no external pumping. The flow from these needles is induced by the electrospray process itself, giving flows of tens of nL per minute. However the definition of the term nanospray has recently been refined to refer to ES regimes with flow rates of less than 50 nL min−1.1 Nanospray results in higher ion signal intensities and it offers a better tolerance towards salt contamination, e.g. from buffers, than normal ES. This is because smaller droplets are formed through the nanospray process, so that fewer generations of offspring droplets are required before ions are formed. The larger droplets formed by ES require solvent evaporation to occur before sufficient charge density for droplet fission can build up, concentrating salts to levels higher than those found in the small droplets formed by nanospray. Recent experiments have shown that suppression effects are pronounced even at flow rates of 50 nL min−1 and above. Low flow spray conditions above about 20 nL min−1 were found to offer only the advantage of low sample consumption.3 Consequently, these authors propose that for true benefits of nanospray, flows must be below 20 nL min−1.
A further consideration of LC-MS coupling via ES is that limit of detection is approximately inversely related to the LC flow rate. Therefore better limits of detection are achieved with lower flow rates until ionisation efficiency is limited by the absolute amount of analyte.4,5 Nanospray interfaces enable the use of these very low flow rates. The use of shorter columns with smaller internal diameters enables much lower flow rates to be used for chromatographic separation. Capillary LC flow rates can range from nL min−1 to a few µL min−1; flow rates at the lower end of this range are too low for conventional ES, and so low flow ES must be used. A key advantage of capillary LC-MS is that it only requires very small volumes of sample to be injected.6
Capillary LC columns are conventionally made from random close packing of small diameter spherical particles packed under high pressure into the fused silica capillaries. The use of small sphere packings (1.5–3 µm) increases separation efficiency. However, it results in very high backpressure and so the column length must be kept short or expensive ultra high pressure LC equipment is required. The main problem with restricting the length of these capillary columns is that it is not always possible to separate complex mixtures in such short columns.7
The use of monolithic columns in capillary LC enables longer column lengths to be used as they are more permeable than packed columns, and so result in lower backpressures. Monoliths are formed by in situ polymerisation or consolidation of particulate packings. They consist of a single rod containing a network of highly interconnected pores through which the mobile phase can flow. Monoliths are particularly well suited for the fabrication of capillary columns, because the monolith is immobilised on the capillary wall and so frits are not required to contain the packing.8 As backpressure and length of column present fewer limitations over the use of packed columns, it is expected that the use of monolithic capillary LC columns for analysis of complex sample mixtures will grow rapidly.
ES and MALDI are complementary ionisation techniques very widely used for analysis of a broad range of biomolecules. MALDI has the advantage that it is more tolerant towards sample contaminants such as buffers and salts, and uses very small volumes of sample. It also typically produces singly charged protonated molecules as opposed to the multiply charged ions produced by ES. Interfacing LC directly to a MALDI ion source is fairly complex, as matrix-sample co-crystals must be formed before the sample can be analysed. Recently, several interfaces have been described to enable MALDI MS interfacing. On line methods have included using continuous flow probes, aerosol interfaces, moving wheel and moving ball methods. These methods have had varying degrees of success. However, they are unlikely to be developed into routinely-used interfaces as the limits of detection are much worse than those achieved using LC-ES
2.2 Capillary electrophoretic separations
Capillary electrophoresis (CE) is a liquid phase separation technique in which separation is based on the electrophoretic mobility of the analytes. Typical CE flow rates are in the nano flow range of 1–100 nL min−1. CE has been used with FAB9 and MALDI.10 Few examples of hyphenating CE-MS through an APCI interface exist because of the inherent incompatibility of the low flow from the CE with the high flow required for APCI.11 However all of the commercially available CE-MS interfaces and recently developed novel interfaces make use of ES ionisation; these are summarised below.The basic CE instrumental configuration is simple; the ends of a capillary are placed into buffer reservoirs, each containing an electrode connected to a high voltage power supply (Fig. 1B). A small window is burned into the capillary's polyimide coating enabling UV detection. In on-line CE-ES-MS the cathode end of the capillary cannot be placed in a background electrolyte vial as it must be connected to the mass spectrometer, and so an alternative electrical connection must be used. The background electrolyte (BGE) systems typically used in CE (e.g. phosphate buffers) are non-volatile and cause salts to crystallise around the mass spectrometer's sample cone. This causes a reduction in sensitivity and spray stability and may result in ion suppression.
The first experiments that resulted in successful coupling of CE-MS via ES were carried out by Olivares et al.12 in 1987. The separation capillary was surrounded by a stainless steel capillary, which functioned as the electrospray needle and the CE cathode, and was held at a potential of 3–5 kV relative to the mass spectrometer's counter electrode. To enable compatible flow rates between the CE and the ES interface, large diameter (100 µm internal diameter (id)) capillaries were used.
An improved interface was described in 1988,13 which used a sheath flow surrounding the separation capillary to provide the electrical contact for the ES voltage and act as the CE cathode. This type of interface, now referred to as the coaxial sheath flow interface (Fig. 2A), is generally used in most commercial designs, as it is the most robust design currently available. The high sheath flow (typically 3–5 µL min−1) results in a relatively stable ion current, but the concentration limits of detection are compromised by the resulting dilution. The liquid junction interface (Fig. 2B)14 requires a lower sheath flow rate, which helps to address this limitation. However, it is difficult to obtain good alignment of the CE capillary and the spray tip, which can result in a large dead volume, which reduces separation efficiency.
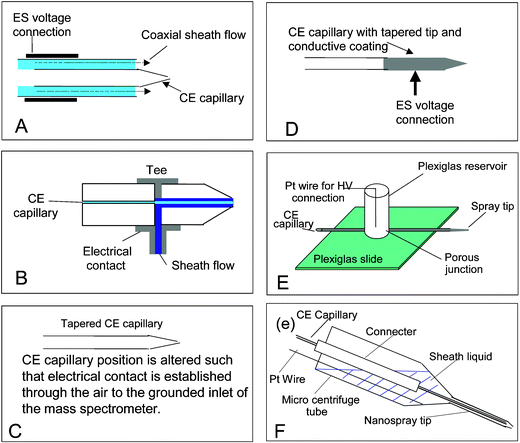 | ||
| Fig. 2 CE-MS Interfaces: Panel A: Co-axial sheathflow, Panel B: Liquid junction, Panel C: Tapered capillary, Panel D: Coated capillary, Panel E: Porous junction, Panel F: Micro-sheathflow. | ||
Theoretically, a sheathless interface that provides similar flow rates to nanospray would result in no dilution of the analyte and high ionisation efficiencies; hence, one would expect optimum limits of detection. The quest for a robust sheathless nanospray interface has led to many different designs. A successful interface must close the CE separation electrical circuit and provide an electrical potential, which enables the ES process.
Mazereeuw et al.15 (Fig. 2C) simply tapered the end of the CE capillary and positioned it close to the inlet capillary of the MS. Electrical contact was made through the air. In this design the background electrolyte must be of a low concentration to reduce the chance of electrical discharge occurring between the capillary and the MS inlet. CE separations are more efficient when a high concentration background electrolyte is used; therefore this method is of limited use.
Many designs (Fig. 2D) establish an electrical contact by coating the capillary outlet with a conductive coating or polymer. The use of the fairy dust16 method and graphite coatings17,18 are well documented. Common problems associated with coated capillaries are sputtering of the coating resulting in a loss of performance, and lack of reproducibility of manufacture, for example variations in the amount, smoothness etc of the coating. Designs in which a wire is inserted into the capillary either through a hole drilled into the capillary wall19 or inserted into the end of the capillary have also been described. It is difficult to drill a hole in the capillary as it is such a small diameter and inserting a wire into the end of the capillary affects the profile the electrospray Taylor cone.20
One of the most recent designs of CE interface describes the use of an electrically porous junction21 (Fig. 2E). HF is used to etch a small section of the capillary such that an electrical current can pass through it. The authors claim that this method resulted in no detectable deterioration in separation or electrospray performance for over two weeks and an injection of 25 fmol of [Glu1]-fibrinopeptide B gave an electropherogram with a signal-to-noise ratio of over 100∶1.
Whitt and Moini22 have also developed an electrically porous interface using HF etching. In their protocol, the porous interface design is achieved by drilling a well in the capillary wall using a dentist's drill, until a capillary wall thickness of 10–20 µM remains and 49% HF is used to etch the wall to achieve electrical porosity. It is reported that the interface can be fabricated in a few minutes, provides reproducible results and last several days.
Chen et al.23 described a low makeup flow interface, which used makeup flow rates of several hundred nL min−1. The interface consists of a borosilicate nanospray tip, with the end removed, and connected to a liquid reservoir, a microcentrifuge tube (Fig. 2F). The sheath liquid is placed into the reservoir and the CE capillary is inserted into the nanospray tip. A platinum wire electrode is placed into the reservoir to complete the ES electrical circuit. The authors claim that this design combines the versatility of liquid sheath design and limit of detection of the sheathless designs.
Commercial CE-MS interfaces make use of coaxial sheathflow because they provide the best compromise of ease of use, robustness, separation efficiency and limits of detection. However, an ideal system would utilise a sheathless nanospray interface to take advantage of the better limits of detection. To date, these sheathless interfaces add complexity to the instrumentation and are generally not robust and so are not commercially available; new ideas are continually being explored and are published in the literature.
3 Off-line hyphenation
3.1 Liquid chromatographic separations
ES is currently the most routinely used ionisation technique for LC-MS interfacing. However, when analysing complex mixtures, the time scale imposed by the on-line nature of this analysis can restrict the data that can be collected. Data dependent scanning involves scanning the mass range and then automatically switching to carry out collision induced dissociation (CID) tandem MS analysis (MS/MS) analysis of the most abundant precursor ions. Problems may arise if:(i) High peak resolution from the separation system results in a short time available for analysis of all components in a peak; by the time the MS/MS is being carried out the peak may have passed.
(ii) While MS/MS is being carried out, subsequent closely eluting peaks may be missed.
(iii) All necessary data must be obtained from each component on this very short timescale—further data can only be obtained by repeating the whole LC-MS analysis.
If the LC eluent could be deposited as MALDI spots such problems would be prevented as, once spotted, MALDI plates can be retained and if necessary the sample can easily be reanalysed, employing different instrument parameters. As mentioned in section 2.1 on-line MALDI LC-MS has had relatively little success. However, manual off-line LC-MALDI can be easily achieved; the LC eluent is fractionated and collected into microtubes. Then each fraction is concentrated and spotted onto a target and analysed by MALDI MS. This process is labour intensive and can result in sample loss.24,25 Though methods that rely on fractionation are easy to implement, a certain amount of chromatographic resolution must be sacrificed26 and this may be problematic. However, the complex nature of samples requiring analysis, and improvements in separation efficiency and peak resolution in separation science, are creating a need for off-line mass spectrometric analysis and this has become a driving force for off-line LC/MALDI interfacing.
The introduction of nanoflow separation techniques such as capillary LC, means that sample volumes from the LC system are directly compatible with spotting onto a MALDI target without pre-concentration; therefore the process can now be automated. In recent years, the introduction of laboratory robots such as the Probot fractionation system (Dionex, Sunnyvale, CA) has enabled full automation. Fractions are deposited by a microfraction collector and the MALDI matrix is added coaxially at the needle tip. Moseley et al27 used such a system for the evaluation of proteome coverage of complex protein mixtures gained by analysing samples using both LC-ES-MS/MS and LC-MALDI-MS/MS. In this comparison a Probot fractionation system was used with a post column splitter, so that 80% of the nano LC eluent was analysed on-line by ES-MS/MS and the other 20% was used for spotting onto the MALDI target for off-line analysis. From a proteolytic digest of the large 39S sub-unit of the bovine mitochondrial ribosome, 51 proteins were detected, eight of which were only identified by LC-ES-MS/MS, 11 were identified only by LC-MALDI-MS/MS and, 32 were identified by both.
An alternative, comparable, commercially-available system (Waters Corp. Milford, Massachusetts) uses atmospheric deposition. The LC eluent passes into a heated nebulizer that produces a fine spray of droplets that are evaporated onto specialised MALDI plates that are pre-coated with matrix. The chromatographic separation is maintained, as the sample is deposited along a continuous track rather than being fractionated. As the sample is concentrated into narrow tracks, this acts as a method of pre-concentration.28 However, the laser must be able to scan the length of the track. A disadvantage of this system is that the precoated plates can only be used once.
A fully automatable system enabling novel simultaneous non-contact liquid deposition has been developed. In this design the eluent from the LC forms an electrically grounded droplet. A negative voltage (−20 kV) pulse is applied to the target causing polarization of the droplet; the electric field generated pulls the droplet onto the target. Bruker Anchor plates were used as targets.26
In spite of the demonstrated power of using on-line LC-MS/MS for proteomics applications, historically proteomic studies have relied on 2D gel electrophoresis followed by excision of the spot from the gel. The protein(s) contained within the spot is subjected to protease digestion and MALDI MS/MS is used to identify the peptides released. Though 2D gels are widely used, they have a number of shortcomings:29
• Proteins with extreme molecular masses and pIs tend to be underrepresented on 2D gels.
• Post translational modifications of proteins, e.g. phosphorylation, glycosylation, introduce variation in the size and charge of proteins (microheterogeneity). Hence the same protein can be focused over a wide pH range.
• Some proteins may not be detected due to the low copy number of the protein i.e. the dynamic range of gels is limited.
• 2D gels can be time consuming to prepare, run, and analyse, suffer from variable reproducibility, and a certain amount of specialised skill is required to run them successfully.
Hence there is a need for an alternative to 2D gel separations. The advances in off-line LC-MALDI interfacing have increased the popularity of separation techniques that combine the ease of use of a 1D gel, with the separation power of LC to resolve the mixture of proteolytic peptides produced on treating the gel band with protease. This combination has become known as Gel-C-MS (/MS).30 As this technique does not suffer the limitations of 2D gels it could become the method of choice in many proteomic applications. However, the main limitation of Gel-C-MS is the inability to characterise the expressed protein abundance.
3.2 Capillary electrophoretic separations
The advances made in nano LC-MALDI have been used to develop automated off-line CE-MALDI applications. A recent example is the analysis of a heterogeneous non-ionic surfactant.31 A fractionating robot was used to spot the CE eluent and matrix onto a MALDI target during the electrophoretic separation. However, for on-line coupling CE, like LC, is far more easily interfaced with electrospray.4 Perspectives for the future
There have been many advances made in hyphenating liquid separation techniques to mass spectrometry. There are commercially available interfaces for LC, nano LC and CE. Whilst they do not always offer the best limits of detection they are the most robust. Literature describing novel interfaces is frequently published. However, only interfaces that are reliable, robust, enable reproducibility and high-throughput analyses will stand any chance of commercial success, no matter how good the individual data they generate. In our opinion it is ironic that despite all of the advances in hyphenation, the problems caused by the time scale imposed by analysing complex mixtures on-line seem to be heading towards developments of ‘off-line interfacing’ techniques such as off-line LC-MALDI. Though the post genomic sciences are no longer in their infancy, they continue to be a major driving force for technological advances in this field.References
- K. A. Hanold, S. M. Fischer, P. H. Cormia, C. E. Miller and J. A. Syage, Anal. Chem., 2004, 76, 2842–2851 CrossRef CAS.
- M. Wilm and M. Mann, Anal. Chem., 1996, 68, 1–8 CrossRef CAS.
- A. Schmidt, M. Karas and T. Dülcks, J. Am. Soc. Mass Spectrom., 2003, 14, 492–500 CrossRef CAS.
- K. J. Oosterkamp, E. Gelpi and J. Abian, J. Am. Soc. Mass Spectrom., 1998, 33, 976–983 CrossRef.
- J. H. Wahl, D. R. Goodlett, H. R. Udseth and R. D. Smith, Electrophoresis, 1993, 14, 448–457 CAS.
- R. Freitag, J. Chromatogr. A, 2004, 1033, 267–273 CrossRef CAS.
- B. Barroso, D. Lubda and R. Bischoff, J. Proteome Res., 2003, 2, 633–642 CrossRef CAS.
- H. Oberacher and C. G. Huber, Trends Anal. Chem., 2002, 21, 166–174 CrossRef CAS.
- M. A. Mosely, L. J. Deterding, B. K. Tomber and J. W. Jorgenson, J. Chromatogr. A, 1989, 480, 197–209 CrossRef CAS.
- J. Priesler, F. Foret and B. L. Karger, Anal. Chem., 1998, 70, 5278–5287 CrossRef CAS.
- Y. Tanaka, K. Otsuka and S. Terabe, J. Pharm. Biomed. Anal., 2003, 30, 1889–1895 CrossRef CAS.
- J. A. Olivares, N. T. Nguyen, C. R. Yonker and R. D. Smith, Anal. Chem., 1987, 59, 1230–1232 CrossRef CAS.
- R. D. Smith, J. A. Olivares, N. T. Nguyen and H. R. Udseth, Anal. Chem., 1988, 60, 436–441 CrossRef CAS.
- E. D. Lee, W. Muck, J. D. Henion and T. D. Covey, Biomed. Environ. Mass Spectrom., 1989, 18, 844–850 CAS.
- M. Mazereeuw, M. Hofte, A. J. P. Tjaden and U. R. van der Greef, Rapid Commun. Mass Spectrom., 1997, 11, 981–992 CrossRef CAS.
- D. R. Barnidge, S. Nilsson and K. E. Markides, Anal. Chem., 1999, 71, 4115–4118 CrossRef CAS.
- S. Nilsson, M. Wetterhall, J. Bergquist, L. Nyholm and K. E. Markides, Rapid Commun. Mass Spectrom., 2001, 15, 1997–2000 CrossRef CAS.
- Y. Z. Chang and G. R. Her, Anal. Chem., 2000, 72, 626–630 CrossRef CAS.
- P. Cao and M. Moini, J. Am. Soc. Mass Spectrom., 1998, 9, 1081–1088 CrossRef CAS.
- L. Fang, R. Zhang, E. R. Williams and R. N. Zare, Anal. Chem., 1994, 66, 3696–3701 CrossRef CAS.
- G. M. Janini, T. P. Conrads, K. L. Wilkens, H. J. Issaq and T. D. Veenstra, Anal. Chem., 2003, 75, 615–1619.
- J. T. Whitt and M. Moini, Anal. Chem., 2003, 75, 2188–2191 CrossRef CAS.
- Y. Chen, M. Tseng, Y. Chang and G. Her, Anal. Chem., 2003, 75, 503–508 CrossRef CAS.
- S. Udiavar, A. Apffel, J. Chakel, S. Swedberg, W. S. Hancock and E. J. Pungor, Anal. Chem., 1998, 70, 3572–3578 CrossRef CAS.
- T. J. Griffin, S. P. Gygi, B. Rist, R. Aebersold, A. Lobada, A. Jilkine, W. Ens and K. G. Standing, Anal. Chem., 2001, 73, 978–986 CrossRef CAS.
- C. Ericson, Q. T. Phung, D. M. Horn, E. C. Peters, J. R. Fitchett, S. B. Ficarro, A. R. Salomon, L. M. Brill and A. Brock, Anal. Chem., 2003, 75, 2309–2315 CrossRef CAS.
- W. M. Bodnar, R. K. Blackburn, J. M. Krise and M. A. Moseley, J. Am. Soc. Mass Spectrom., 2003, 14, 971–979 CrossRef CAS.
- D. B. Wall, S. J. Berger, J. W. Finch, S. A. Cohen, K. Richardson, R. Chapman, D. Drabble, J. Brown and D. Gostick, Electrophoresis, 2002, 23, 3193–3204 CrossRef CAS.
- R. J. Simpson, L. M. Connolly, J. S. Eddes, J. J. Pereira, R. L. Moritz and G. E. Reid, Electrophoresis, 2000, 21, 707–1732.
- J. Froehlich, C. G. Wilkerson, W. K. Ray, R. S. McAndrew, K.W. Oysteryoung, D. A. Gage and B. S. Phinney, J. Proteome Res., 2003, 2, 413–425 CrossRef.
- T. Meyer, D. Waidelich and A. W. Frahm, J. Pharm. Biomed. Anal., 2002, 30, 263–271 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |