Principles of Fourier transform ion cyclotron resonance mass spectrometry and its application in structural biology
Mark P. Barrow*, William I. Burkitt and Peter J. Derrick
Institute of Mass Spectrometry and Department of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: M.P.Barrow@warwick.ac.uk; Fax: +44 (0)24 765 24112
First published on 24th August 2004
Abstract
Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry has become increasingly significant within recent years. The inherently ultra-high resolution and mass accuracy allow unequivocal assignments of chemical formulae to be made and further structural elucidation can be conducted through the utilization of tandem mass spectrometry techniques. With the advent of electrospray ionization (ESI), FT-ICR mass spectrometry has become a powerful tool for the investigation of biological macromolecules, such as the study of non-covalent interactions of proteins. In this article, the basic principles are highlighted, some of the techniques employed are described and examples of applications are provided, with particular respect being paid to the field of characterization of biomolecules.
 Mark P. Barrow Mark P. Barrow | Mark Barrow completed his PhD on the study of fullerenes and fullerene derivatives by mass spectrometry at the end of the year 2000. He then began a postdoctoral position which was sponsored by ATOFINA (France), using a 9.4 T FT-ICR at the University of Warwick to investigate polymer, protein, and crude oil samples; the majority of the work entailed the study of naphthenic acids in crude oils, which cause corrosion of refinery apparatus. More recently, he has begun a collaboration with the National Water Research Institute (Canada). |
 William I. Burkitt William I. Burkitt | Will Burkitt graduated from the University of Warwick where he obtained a degree in Chemistry. He stayed at Warwick where he is presently studying for a PhD in physical chemistry, with funding from the EPSRC. His main research interest is the study of non-covalent complexes of proteins such as S100 and anti-TRAP using FT-ICR mass spectrometry. |
 Peter J. Derrick Peter J. Derrick | Peter Derrick is Professor of Chemistry and Chair of the Department of Chemistry at the University of Warwick. He heads a mass spectrometry group which is interested in the design and construction of novel mass spectrometers and ion optical devices and in methodology for the application of mass spectrometry in structural biology and in proteomics. His group has twenty years of experience in FT-ICR, and was one of the first to apply FT-ICR to the studies of macromolecules and clusters. |
Introduction
Mass spectrometry1–3 has been playing an increasingly important role in science,4 particularly during the past decade, and is used within fields such as forensics, environmental analysis, the military, fullerene research, drug development, and space exploration. Practical examples of recent uses of mass spectrometry include the sequencing of a 55,000 year old protein from a bison bone in Siberia5 and the use of accelerator mass spectrometry for analyzing 30,000 year old cave art in France.6 Mass spectrometry is particularly used for the study of proteins and the study of complex mixtures. Quantitative analysis of multicomponent mixtures is a current challenge, and is frequently associated with metabolomics,7 proteomics,8 and the petroleum industry. The term “petroleomics”9 has been coined to describe multicomponent analysis in the context of crude oils and other petroleum based products, where the analysis of the corrosive components within crude oils (such as naphthenic acids) is one significant example,10 as these components increase the cost of oil by several dollars per barrel.11 High resolution and high mass accuracy prove to be invaluable when trying to establish the identities of hundreds or thousands of components within a complex mixture, as demonstrated by Marshall and co-workers during the resolution of 10,000 distinct components in a complex mixture arising from a coal extract.12Principles of Fourier transform ion cyclotron resonance mass spectrometry
Of the different varieties of mass spectrometer available, it is the Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer13–19 which holds the greatest potential at the forefront of research, due to the inherently ultra-high resolution and mass accuracy which can be achieved. Such performance allows unequivocal mass assignment and resolution of species which could not otherwise be distinguished if using some other types of mass spectrometer. All varieties of mass spectrometer must involve the analysis of gas-phase ions to obtain a mass-to-charge ratio (m/z) and ion sources must be used to generate the gas-phase ions from a neutral sample. The analyte can be introduced as a solid, liquid, or a gas, depending on the type of ion source used. The ions are extracted into the mass analyzer region of the mass spectrometer and are ultimately detected. In the case of FT-ICR mass spectrometry, ions are usually generated externally in a separate ion source and then injected into a container known as the “cell,” which is typically cubical or cylindrical in geometry. The FT-ICR analyzer cell is located within a strong magnetic field, typically generated using a superconducting magnet. Fig. 1 shows a schematic representation of a cylindrical FT-ICR analyzer cell, such as the Infinity Cell used in FT-ICR mass spectrometers manufactured by Bruker Daltonics.20 Charged particles moving in the presence of a magnetic field, with a component of their velocity that is perpendicular to the magnetic field axis, will experience a force known as the “Lorentz Force.” The axes of the force, motion of the (positively) charged particle, and magnetic field are mutually perpendicular, as dictated by Fleming's Left Hand Rule. The Lorentz Force acting on a charged particle in the presence of a magnetic field alone is given by eqn. (1):| F = qνBsinθ | (1) |
| F = qνB | (1a) |
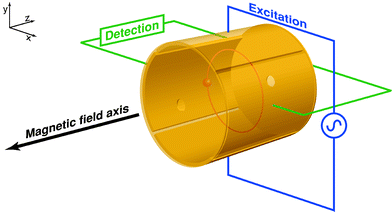 | ||
| Fig. 1 Schematic representation of a cylindrical FT-ICR analyzer cell. The cell is aligned with the bore of the magnet so that the magnetic field axis is coaxial with the trapping axis (z-axis). In this particular example, the excitation plates are located along the y-axis and the detection plates along the x-axis. The trapping electrodes are located at each end of the cell, and the orbiting ions are shown in red. | ||
The total charge on an ion can be described by eqn. (2):
| q = ze | (2) |
A charged particle, such as an ion, will begin to precess about the center of the magnetic field axis, resulting in an orbit. This motion is known as the “cyclotron motion.” The cyclotron frequency is related to the mass-to-charge ratio of the ion and is shown in eqns. (3a) and (3b):
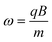 | (3a) |
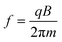 | (3b) |
Detection of the ions occurs as the ion packets pass two detector plates. As the ion packets move past these plates, charge moves within the detection circuit to counteract the proximity of the ions. The potential (voltage) change between the detection plates can be measured as a function of time and it is from here that the raw data (known as a “transient,” “time-domain” data, or sometimes as an “FID,” which is the acronym for “free induction decay”) is obtained. It should be noted that the ions repeatedly pass the detector plates for the duration of the acquisition time, as non-destructive detection is employed. The magnitude of the signal is proportional to the total charge and the proximity of the ions to the detection plates (orbital radius), and is independent of magnetic field strength. The raw data will represent the detection at the same time of all the ions, with their different cyclotron frequencies. It is therefore necessary to extract data about the different ion packets. This is done through usage of the mathematical procedure known as a “Fourier transform” (FT)21,22 where frequency information is obtained from time-domain data. A spectrum is produced where signal magnitude is plotted as a function of the cyclotron frequencies of the ions present. From eqns. (3a) and (3b), it can be seen that the cyclotron frequency is related to m/z, and it is therefore possible to convert the plot, perform a calibration,23 and create a mass spectrum in which signal magnitude is plotted as a function of m/z. Fig. 2 illustrates the process of obtaining time-domain data, utilizing a Fourier transform to convert to the frequency domain, and then calibration of the data in terms of m/z. For the purposes of calibration, an unknown sample can be doped with a standard and the mass spectrum can be “internally” calibrated, or a mass spectrum of a standard can be calibrated and the calibration information (in the form of constants) can be computationally exported to spectra of unknown samples (“external calibration”). Furthermore, Fig. 2 highlights the fact that the relationship between m/z and frequency is non-linear, as first demonstrated by eqns. (3a) and (3b).
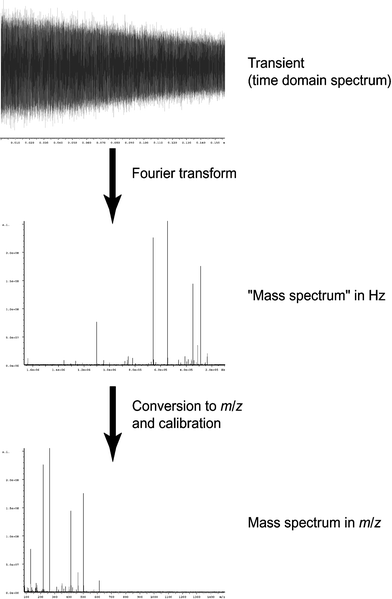 | ||
| Fig. 2 Illustration of the processing of raw data. A Fourier transform is performed on the time-domain data to convert it to the frequency domain, and this resulting spectrum is then calibrated in terms of m/z. | ||
When the ions enter the FT-ICR analyzer cell, however, the radii of the cyclotron orbits are too small to be detectable. The ions must be excited to detectable radii and this occurs through usage of a radio frequency (RF) potential applied to two excitation plates at the resonant frequency (i.e. resonant with the cyclotron frequency) of the ions, and the excitation additionally results in coherence of the ion packets. Fig. 3 shows a schematic representation of a cross section of an FT-ICR analyzer cell, where ions are being excited by the RF potential applied to the excitation electrodes. This is effected by a linear increase or sweep through the resonant frequency range of interest at constant amplitude, also known as an “RF chirp.” Ions of different m/z values are excited to orbits of the same radius, though their cyclotron frequencies differ.
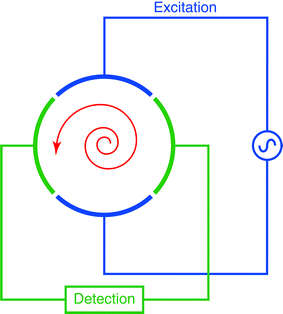 | ||
| Fig. 3 A cross section of a cylindrical FT-ICR analyzer cell, depicting excitation of ions (in red) through the application of an RF potential to the excitation electrodes. Once the ions have been excited to a cyclotron motion of suitable radius, the image current of the orbiting ions can be detected on the detection plates. | ||
The FT-ICR analyzer cell (see Fig. 1) consists of the two excitation and two detection plates, but, in order to restrain the ions' motion along the axis of the magnetic field, it is necessary to also include two trapping plates. A low potential (typically of an order of 1 V) is applied to the trapping plates to restrain the ions within the FT-ICR analyzer cell, so that they may be excited and detected. An undesirable side-effect of having such a trapping potential is that, though the excitation and detection plates may not have potentials applied, the presence of the electrostatic field from the trapping plates results in a non-zero potential at the center of the FT-ICR analyzer cell, which can offset the center of the cyclotron motion. The trapping potentials give rise to the “magnetron motion,”18,19 and the cyclotron motion is offset from the center of the cell as a result of this additional orbiting motion. Cyclotron frequencies are typically within the kHz to MHz range, whilst magnetron frequencies are typically in the region of a few hundred Hz. The relationship describing the magnetron frequency is given by eqn. (4), and it can be seen that the magnetron frequency depends upon the cell dimensions, the cell design, the trapping potentials applied, and the magnetic field strength. The net effect is that the motion of the ions will include three components: the cyclotron motion, the magnetron motion, and the oscillation between the trapping plates along the magnetic field axis. The magnetron motion17,24–29 (and the overall, complex ion motion18,19) will not be covered in further depth in this tutorial.
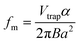 | (4) |
Electrospray ionization
Electrospray ionization (ESI)32–36 is the ionization technique nowadays most frequently coupled with FT-ICR mass spectrometry. ESI has the advantage of being a “soft” ionization technique, resulting in the vaporization and ionization of a sample whilst minimizing fragmentation. The minimization of fragmentation is particularly important for labile molecules such as biological molecules, and for the study of complex mixtures where fragments must not interfere with the mass spectrum when trying to determine the original constituents. For example, Zhan and Fenn have detailed the use of ESI to study fossil fuels,37 as fragmentation would be undesirable due to further complication of the mass spectra. ESI frequently leads to the formation of multiply charged ions, particularly in the case of large biomolecules. As mass spectrometry is based upon the determination of m/z, not mass alone, it is frequently the case that a mass spectrum will contain the same molecules but in a variety of charge states, and therefore at several different m/z values. As ions become more highly charged, the m/z becomes lower and the spacing between the “isotopomers” (peaks due to the presence of other isotopes) becomes narrower. As a result, it becomes more difficult to resolve the signals and the resolution of the mass analyzer becomes more important. Note that the charge state of an ion can be determined by examining the spacing between the isotopomer signals. The growth of electrospray ionization can, therefore, be seen to be promoting the growth in FT-ICR mass spectrometry, for FT-ICR mass spectrometers offer the ultra-high resolution which is particularly well suited to a coupling with ESI. For these reasons, ESI FT-ICR mass spectrometry has become increasingly important for the study of biological molecules in recent years.Mass accuracy and resolution
FT-ICR mass spectrometers are best known for their excellent mass accuracy and resolution. Mass accuracy (measured in ppm) is a measurement of how well the observed m/z correlates with the “true value,” and is particularly important in situations in which many possible elemental compositions could be assigned to a nominal, integer m/z value. Many other types of modern commercial mass spectrometers cite performance specifications for mass accuracies of the order of tens of ppm over a specific mass range, whereas specifications for FT-ICR mass spectrometers typically cite mass accuracies of 1 ppm or better (lower). Eqn (5) is the relationship for determining the mass accuracy of a result when comparing the observed m/z with the value believed to be “true,” called here the theoretical m/z: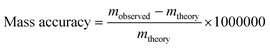 | (5) |
Resolution is extremely important for resolving closely spaced signals, such as in the cases of complex mixtures or multiply-charged ions (as generated by electrospray ionization, for instance). The ability to use instruments with high resolution can sometimes make the difference between identifying and not identifying a species of interest. An example of this is a case where the high resolution of a 9.4 T FT-ICR mass spectrometer allowed a fullerene derivative which contained nitrogen to be identified as it was possible to resolve 12C5914N+ (theoretical m/z 722.002525) and 12C5813C2+ (theoretical m/z 722.006161).38 Without the usage of such instrumentation, the presence of 12C5914N+ could not have been confirmed, as it may have been assumed only one signal existed and was associated with the 12C5813C2+ ion. Eqn. (6) defines resolution:
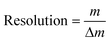 | (6) |
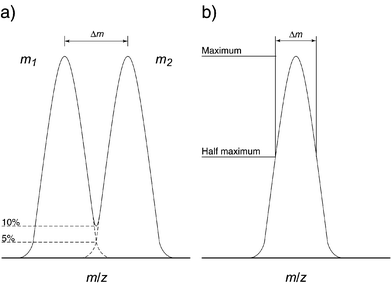 | ||
| Fig. 4 Definitions of resolution: (a) shows the “10% valley” definition, most commonly used in conjunction with magnetic-sector instruments, where two separate peaks of similar intensities overlap at a peak height of 5% of their maxima, therefore summing to be 10% at the overlap; (b) shows the “full width at half maximum” definition, which is most typically used in conjunction with time-of-flight and FT-ICR mass spectrometers. | ||
Whilst the resolution of many commercial time-of-flight mass spectrometers may approach 10,000 and many commercial magnetic-sector mass spectrometers may approach a resolution of 100,000, FT-ICR mass spectrometers can routinely reach resolutions of hundreds of thousands in “broadband mode” (“normal” experimental conditions) or even reach a resolution of a few million in “heterodyne mode” (where a very narrow m/z range is studied but under very high resolution conditions). With FT-ICR mass spectrometry, mass spectra are recorded as a finite number of data points, determined by the user, on the workstation prior to an acquisition. This determines the number of data points used in the time-domain spectrum. This time-domain dataset will be “filled up” at a rate determined by the sampling frequency used. The sampling frequency is determined by the highest cyclotron frequency (i.e. lowest m/z) set in the mass spectrum, and the sampling frequency must be equal to at least twice that of the highest frequency being recorded in accordance with the Nyquist theorem.22 The “acquisition time,” the duration of the detection phase, is therefore determined by the dataset size and the sampling frequency which must be used (determined from the low m/z cut-off point), according to eqn. (7):
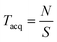 | (7) |
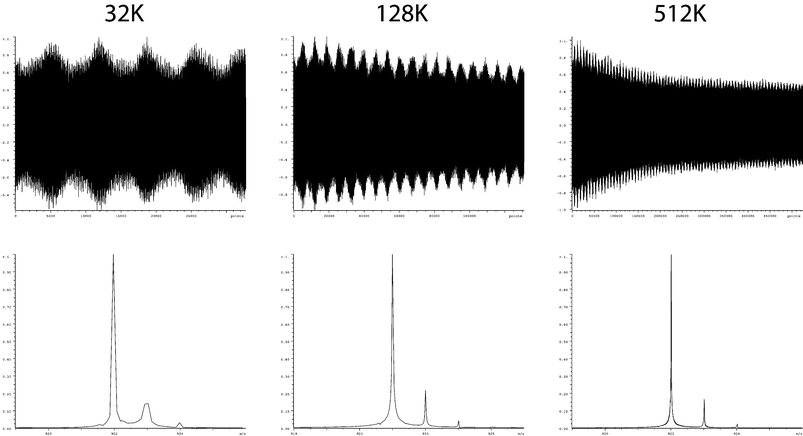 | ||
| Fig. 5 Demonstration of the effect of dataset size (and therefore acquisition time) upon resolution. The three spectra shown all result from the same raw data, where the only differences are the proportion of the raw data (512K in total) used during the Fourier transform. | ||
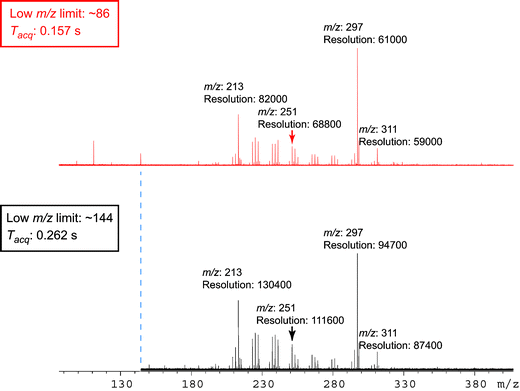 | ||
| Fig. 6 A multicomponent mixture is shown, highlighting the dependence of resolution on both m/z and the low m/z cut-off, where the change in the low m/z cut-off is associated with a change in acquisition time. For the purpose of simplicity, the resolution for each signal is listed to the nearest hundred and m/z is quoted to the nearest integer. It can be seen that increasing the low m/z cut-off enhances the overall resolution throughout a mass spectrum, but that the resolution decreases throughout a given spectrum with increasing m/z. | ||
Traditionally in mass spectrometry, users “tune” for the strongest signal they can obtain. A series of ion optical lenses (essentially electrodes to which potentials are applied) are used to focus and steer the ion beams in mass spectrometers to a desired location, much as more traditional lenses can be used to focus and steer beams of light. Typically, ions are guided from an ion source and through the flight tube towards a detector or, in the case of FT-ICR mass spectrometry, from the ion source and into the FT-ICR analyzer cell. The process of changing the potentials applied to the lenses and monitoring signal strength changes is known as “tuning.” In contrast to the traditional desire to maximize signal strength, having a large number of different ion packets in an FT-ICR can cause problems, as the ion clouds will interact through Coulombic repulsion. These interactions, of which there are different forms, are collectively known as space-charge effects.39–44 Space-charge effects typically conspire to decrease mass accuracy and resolution, and a user must be aware of such effects in order to minimize them. Some of the methods a user can employ (for a given instrument) include: minimizing the trapping potentials in order to allow the ion packets to be spread along the z-axis (trapping axis), exciting to large cyclotron radii to increase spacing between ion packets, and carefully regulating the number of ions present in the FT-ICR analyzer cell, for example through control of accumulation times in an external ion trap such as a hexapole.
Tandem mass spectrometry
For structural elucidation, “tandem mass spectrometry” experiments are performed, where particular ions can be selected and dissociated, and the resulting dissociation pattern provides structural information. One of the most common forms of tandem mass spectrometry experiment performed on FT-ICR mass spectrometers is sustained off-resonance irradiation collision-induced dissociation (SORI-CID).18,45,46 The term “collisionally activated dissociation” (CAD) can also be used instead of CID. An ion packet of a given m/z is “selected” by ejecting all other ions through excitation. Only the desired ions are not excited, leaving them alone in the FT-ICR analyzer cell. A collision gas is introduced into the FT-ICR analyzer cell, usually through a solenoid valve which can be pulsed open for a specified time. While in the presence of the gas, the selected ions are excited off-resonance. Off-resonance excitation results in the ions' orbit expanding and contracting as a function of time, rather than continuing to expand until hitting the cell electrodes (as occurs during the on-resonance excitation of unwanted ions during selection). As the ions' orbit expands and contracts, multiple, low-energy collisions with the gas particles occur. This causes a “slow heating effect” and the ions' internal energies are increased, leading to dissociation. The user can obtain structural information about the “parent ions” (the selected species) through the determination of the elemental composition of the fragments.Other varieties of tandem mass spectrometry experiment exist in FT-ICR mass spectrometry. Such experimental methods include infra-red multiphoton dissociation (IRMPD) and electron capture dissociation (ECD). IRMPD47,48 entails the usage of a laser, most commonly a CO2 laser, to irradiate ions whilst trapped in the FT-ICR analyzer cell. The absorption of the photons slowly heats the ions, increasing the internal energy and leading to dissociation. This can be compared with the “slow heating effect” of SORI-CID, though IRMPD has the advantage of avoiding the requirement to put a collision gas into the FT-ICR analyzer cell, as a higher pressure leads to a more rapid damping of the transient (time-domain data) and therefore decreases the resolution. SORI-CID and IRMPD are usually considered to be ergodic processes,49 where the weakest bonds, such as backbone amide bonds and post-translational modifications when studying peptides and proteins, tend to be cleaved. Both techniques tend to lead to the formation of b-fragments and y-fragments from peptides and proteins.50 ECD51–53 involves a beam of low-energy electrons (0.2 eV or less, for instance) being emitted into the cell. The ions capture these low-energy electrons and the mechanism of dissociation is thought to be a non-ergodic process, in which N–Cα and S–S bonds are preferentially cleaved. Utilization of ECD leads primarily to the formation of c and z˙ fragment ions, when studying peptides and proteins, and one advantage is that labile bonds, such as non-covalent bonds and phosphorylation sites, are left intact. ECD mass spectra can therefore be relatively less complex due to avoidance of unwanted fragmentation.
Experimental
All mass spectra shown were acquired using a Bruker (Bruker Daltonics, Billerica, MA, USA) BioAPEX II 9.4 T FT-ICR mass spectrometer,46 coupled with an Analytica (Analytica of Branford, Branford, CT, USA) electrospray ion source or its nanospray variant. Typical experimental parameters for the instrument at the University of Warwick are outlined in the following to relate aforementioned theory with experimental methodology. The duration of time for which ions would be allowed to accumulate in the hexapole, “D1,” would typically be maintained within the range of 1–4 s. Accumulating ions for long periods in the hexapole results in a strong signal, but users should also be aware that doing so may also result in unwanted side-effects, including fragmentation.54 One important parameter, which is sometimes overlooked, is known as “P2.” P2, which follows after D1, sets the time during which ions are extracted from the hexapole and allowed to traverse the ion optics and be accumulated in the FT-ICR analyzer cell. A side-effect of this parameter is a “time-of-flight effect” from the hexapole to the FT-ICR analyzer cell, whereby changing this parameter induces a bias towards higher or lower m/z.10,55,56 A P2 of approximately 3000 µs may be used for studying small molecules, whereas a P2 of approximately 5000 µs may be used for studying high m/z species such as large proteins; it should be noted that the precise value of P2 will depend heavily upon the user's particular ion source parameters. Cell parameters would include: the trapping potentials (“PV1” and “PV2”) being maintained at 1.5 V (for positive-ion mode, and −1.5 V for negative-ion mode), the “dwell time” for the excitation (“P3”) set to 12 µs, the RF attenuation for excitation (“PL3”) set to approximately “11.2” (where the peak-to-peak output potential can be calculated to approximately equal 76 V;45 users should note that smaller values of PL3 result in higher excitation, and that the relationship between PL3 and the output potential is logarithmic, not linear), and “XBB,” a constant which adjusts the step size within the frequency list for the RF chirp, is maintained at 200, although this should be adjusted for instruments using different magnetic field strengths (100–200 is typically advisable for 9.4 T instruments). Mass spectra are then typically collected as “512K” datasets (524288 data points; 512 multiplied by 1024), and the acquisition time will vary, as the m/z range selected will depend on the particular sample being studied. When choosing the excitation range (in terms of m/z), users can set the excitation range to be wider than the detection range to avoid some of the fluctuations at the edges of the RF chirp; users of Bruker instruments may wish to try running the “Wave Detect” pulse program to see appearance of the RF chirps that they use. The number of scans accumulated during an acquisition (rather than running in tune mode), set by the command “ns,” will be varied greatly according to the signal strength for a particular sample, but would typically be between 32 and 256 scans, for instance.Application to biological macromolecules
Non-covalent/covalent interactions
Mass spectrometry has in recent years become a well-established technique for the sequencing of biomolecules. However, combining the “soft” ionization afforded by electrospray ionization32,33 with mass spectrometry has allowed some solution-phase properties to be probed. Electrospray ionization makes possible the dispersion and ionization of non-covalently bound complexes from solution to the gas-phase,57 and as a result it is possible to study the intrinsic interactions of biomolecules with their ligands by mass spectrometry.58 The high resolution and mass accuracy afforded by FT-ICR mass spectrometry offers decisive advantages when studying such interactions, as will be illustrated below.Mass spectrometry measures the mass-to-charge ratio of the ions, consequently if the charge is not known the mass remains uncertain. The high resolution afforded by FT-ICR removes the uncertainty about the charge state of the ions as the isotopomers of each species can be resolved, for species up to about 100 kDa in mass when using an instrument with a 9.4 T magnet.59 This makes it possible to count the number of peaks from that species that appear in one m/z unit to reveal the ion's charge, and hence the mass of the molecule. Mass analysis techniques where the resolution obtained is not great enough for the charge state of high-mass molecules to be measured directly cannot definitively identify the mass of the analyte.
Proteins are often found to form dimers or other higher-order assemblies, and such assemblies can be both covalently and non-covalently bound. Distinguishing between a monomer and a dimer with twice as many charges is not possible if the isotopes are not resolved. Overlapping monomers and dimers are easily distinguishable using FT-ICR as isotopic peaks of dimers have half the spacing of monomers, and hence appear in synchrony with the isotopomers as well as half way between the isotopomers of the monomers. Fig. 7 shows an ESI spectrum of the protein S100A4 in an aqueous buffered solution at pH 5.8.60 It should also be noted that the isotope distribution of the dimer is offset slightly to that of the monomer, which is due to the dimer being covalently bound with a cysteine–cysteine bond, reducing the mass of the complex by 2 Da. Also shown is a close up of the tetramer peak, overlapping with a dimer peak. The tetramer peak is offset from the dimer peak, indicating the presence of disulfide bonds. Such details are only observed when using the high-resolution currently available routinely for biomacromolecules only with FT-ICR mass spectrometry.
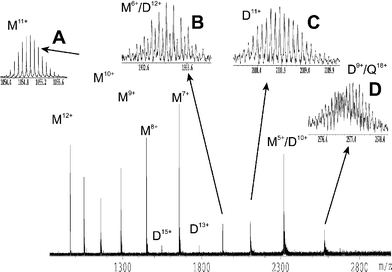 | ||
| Fig. 7 ESI FT-ICR mass spectrum of 60 µM S100A4 in 5 mM ammonium acetate buffer. M = S100A4 monomers, D = S100A4 dimers and Q = S100A4 tetramers. Insets, A: S100A4 monomer in the 11+ charge state. B: overlap of the 6+ charge state of S100A4 monomer and the 12+ charge states of covalently bound S100A4 dimer. C: 11+ charge state of non-covalently bound S100A4 dimers. D: overlapping isotope distributions of the 9+ charge state of non-covalently bound S100A4 dimers and the 18+ charge state of covalently bound S100A4 tetramers. | ||
Metal ion binding
The binding of ligands and of metal ions to protein receptors are involved in the regulation of most biological processes. The interactions of metal ions with proteins have been studied using ESI FT-ICR61–65 where the high resolution achieved permits the analysis of complex mixtures of proteins and metal ions. Fig. 8 shows a spectrum of the protein S100A12,66 in the presence of zinc ions. From this single spectrum it is possible to determine that S100A12 forms non-covalent dimers and that the binding of zinc is cooperative, in the sense that the binding of the first zinc ion increases the complexes affinity for a second zinc ion. This is evidenced from the spectrum, as the peaks arising from the protein dimer with one zinc ion bound have lower intensities than those from either the two-zinc bound or no-zinc bound dimer. The high resolution was necessary as the protein sample contained two monomers that differed by one methionine residue on the N terminus, and consequently three dimers were formed each differing in mass from one of the others by the mass of a methionine residue. The combined nominal mass of two zinc ions is identical to that of a methionine residue (131 Da in each). In the spectra, a methionine residue contributed 131 Da, whereas two zinc ions contributed 127 Da as a result of the loss of four protons due to the two charges on each of the zinc ions. The average masses of certain species differed at most by only a few Da, which resulted in the isotope distributions of the two species overlapping. The high resolution achieved however allowed the overlapping distributions to be resolved. The difference in mass between the isotopic peaks of these distributions was 0.2 Da, caused mostly by the nuclear packing mass deficiency of the zinc atoms. The mass difference of the protein dimers was about 21 kDa.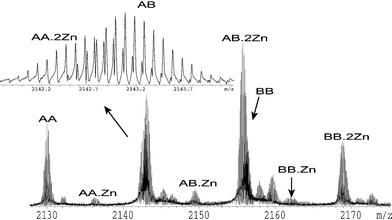 | ||
| Fig. 8 10+ charge state of an ESI FT-ICR mass spectrum of S100A12 in 10 mM ammonium acetate buffer with 1 µM zinc chloride. AA indicates the non-covalent dimer of S100A12 with two methionine-deficient monomers. AB indicates the non-covalent dimer of S100A12 with one methionine-deficient monomer and one monomer containing an N-terminal methionine residue. BB indicates the non-covalent dimer of S100A12 with two monomers containing an N-terminal methionine residues. The number of zincs bound to each dimer is indicated. | ||
Multiple conformations
The ions produced by electrospray ionization are often generated in a range of charge states. The charge states populated by a particular species and the degree to which each charge state is populated in a mass spectrum are referred to as the species' charge state distribution. The charge state distribution of a particular protein sample depends upon a number of factors including the protein's conformation in the solution-phase.67 Unfolded conformations are believed to result in the formation of higher charge states than folded conformations. This has led to the suggestion that a protein sample containing multiple conformations may be detected by examining the species' charge state distribution.68The presence of multiple conformations of a protein in solution may, however, be detected more directly by using H/D exchange. To perform a H/D exchange experiment, a protein sample is placed into an excess of deuterated solvent (for example D2O), which results in the exposed residues of the protein exchanging H atoms with the solvent. Each exchanged H atom increases the mass of the protein by 1 Da. Each protein conformation exchanges hydrogen atoms at a different rate, as each conformation has different residues accessible to the solvent. The result of this is that, in the case of a protein with multiple conformations, multiple isotope distributions are observed in the mass spectra of that protein.69 The high mass-accuracy and resolution afforded by FT-ICR allows the resolution of any overlapping isotope distributions that rather than resulting from deuterium incorporation result from the attachment of ligands such as metal ions.
An analogous experiment may be performed in the gas-phase.70 In this case, ions are trapped in the FT-ICR analyzer cell and a deuterated gas, such as ND3 or D2O, is leaked into the cell. These gases can then exchange deuterium atoms for hydrogen atoms within the ions of interest. This produces multiple isotope distributions of one species in the mass spectra if multiple conformations are present.71 This is feasible because the ions of proteins produced by electrospray ionization can retain a “memory” of their solution-phase conformations in the gas-phase.71 Performing such experiments in the gas-phase allows individual species to be isolated before the gas-phase H/D occurs. To determine the rates of exchange of conformations the ions are trapped for differing lengths of time before the mass analysis occurs. The ions may be trapped for hours if necessary.
Low-resolution mapping of protein structure is possible using solution H/D exchange and mass spectrometry.72 This is achieved by quenching the H/D exchange after a certain time by lowering the pH and cooling the sample to near-freezing. Proteolytic enzymes are then used to digest the protein into smaller peptides and the resulting peptides are mass analyzed.72 Further fragmentation of the peptides may be performed within the mass spectrometer to give residue-specific information.73 The extent of deuteration measured for specific fragments reveals which residues have been exposed to solvent and which were buried within the protein.74 Here the mass accuracy and resolving power of FT-ICR have the distinct advantage of allowing more fragments to be identified from what are potentially very complex samples.73
A less direct method was demonstrated by Nousiainen et al.75 who determined the presence of two protein conformations in a sample of troponin C by examining its calcium-ion binding pattern. ESI mass spectra of troponin C in buffered solution and a range of calcium concentrations were recorded. It was found that the pattern of calcium binding that was observed could only be rationalised if it was considered that troponin C was present in two conformations, each with its own individual affinity for calcium. It was found that one conformation bound calcium in a manner characteristic of properly folded troponin C, while the calcium binding of the second conformation implied structural changes in the calcium-binding pockets of the C-terminus.
Electron capture dissociation
Electron capture dissociation51 is one of the most promising new tandem mass spectrometry techniques to emerge in recent years.53 ECD is performed on multiply charged ions [M+nH]n+ trapped in the FT-ICR analyzer cell of the mass spectrometer. Trapping the ions in the FT-ICR analyzer cell allows the parent ion to be pre-selected before fragmentation. Low-energy electrons (<1 eV) are generated from electron sources behind the FT-ICR analyzer cell, and are allowed to interact with the trapped ions. The neutralization of an electron at a protonated site releases ∼6 eV which is sufficient for dissociation to occur on non-ergodic time scales (<10−12 s).76,77 The dissociation is selective and does not dissociate non-covalent bonds, which may be present within the analyte.The dissociation induced by ECD cleaves a larger proportion of inter-residue bonds than slow heating techniques and yields a greater number of fragments. Hence with peptides and proteins greater sequence coverage is achieved.78 With peptides and proteins, there appears to be little preference shown as to which residues are cleaved by ECD, other than an affinity towards cleavage on the C-terminal side of tryptophan.52 Proline due to its cyclic structure does not yield fragments for cleavage on its N-terminal side. Disulfide bonds are preferably cleaved by ECD,52 unlike other dissociative techniques. This is advantageous, as a fragment that is cleaved but still linked to the parent ion by a disulfide bond cannot be identified. Unlike other dissociation techniques, the fragments produced by ECD tend to retain their post-translational modifications. Sites of phosphorylation,79 sulfated side chains,80N-glycosylation81 and O-glycosylation82 may be determined. Further improvement to the sequence coverage obtained using ECD has been achieved by first activating the ions.83 This can be done either by heating the ions using techniques such as SORI-CID, black-body infra-red dissociation (BIRD)84 or IRMPD, or by colliding the ions with a gas pulse as they enter the FT-ICR analyzer cell. The improvement is attained because activating the ions has the effect of breaking non-covalent bonds that would otherwise hold the fragment to the parent ion.
When sequencing proteins or peptides, high mass-accuracy is needed to distinguish between amino acid residues, for example the masses of a Gln and Lys residues differ by only 0.037 Da. With the highest-field magnets, FT-ICR can routinely provide the mass accuracy necessary for this particular distinction with fragments up to the size of a few kDa. Distinguishing between the isomers Leu and Ile on the basis of mass alone though is not possible. However, capture of 10 eV electrons, so called hot-electron capture dissociation (HECD),85 results in secondary fragmentation of the z˙ ions, characteristic fragmentation of Leu and Ile residues, and yields different w ions.
Using ECD, McLafferty and co-workers have made the de novo assignment of the complete sequence of the 8.6 kDa protein ubiquitin and the 2.8 kDa protein mellitin.86 Inter-residue cleavage of 250 of the 258 inter-residue locations was achieved for the 29 kDa protein carbonic anhydrase.87 These results demonstrated the potential for the top-down approach to protein sequencing by FT-ICR.
SORI-CID of protein complexes
It has been demonstrated that having been ionized and dispersed into the gas-phase using ESI, protein ions retain a “memory” of their solution-phase structure in the gas-phase.71 Examples of the evidence for this includes the investigation by Douglas et al. of the gas-phase stability of heme binding to apomyoglobin and apocytochrome b5 mutants.88 The number of hydrogen bonds between the heme propionate groups and surface residues was systematically reduced in the protein mutants used. The solution-phase stability of heme binding to these mutants was measured, and the gas-phase stability of the complexes was determined by dissociating the complexes in the orifice-skimmer region. There was a correlation between the stability of heme binding in the solution-phase and the voltage in the orifice-skimmer region of the ion source required to dissociate the ion of the heme-protein complexes.Nousiainen et al. studied the stability in the gas-phase of the complex between calmodulin, calcium and a set of point-mutated RS20 peptide analogs by dissociating these complexes in between the capillary and the skimmer of the ESI source. Plotting the ratio of the relative intensities of the calmodulin, calcium and peptide complex against the calmodulin and calcium complex that was the product of the gas-phase dissociation of the full complex revealed the gas-phase stability of the complexes. It was concluded that the basic arginine 16 residue played a more important role in the gas-phase than the nonpolar valine 11 and alanine 13 residues.89
Robinson and co-workers used mass spectrometry to study the protein released from intact E. coli. ribosomes in the gas-phase.90 The release of proteins from the complex was studied using a range of solution conditions. The proteins released were found to vary as a result of changes in pH, replacement of Mg2+ by Li+ and the use of the inhibitor thiostrepton, while use of the inhibitor fusidic acid resulted only in limited changes.
Because proteins retain a “memory” of their solution-phase structure, FT-ICR mass spectrometry is beginning to be used to probe the quaternary structure of proteins. In one approach, protein complexes are isolated in the FT-ICR analyzer cell of an FT-ICR mass spectrometer and slowly heated using SORI-CID. The complex is heated though multiple collisions with gas molecules until the complex dissociates. The hope is that the resulting fragments can be used to probe the quaternary structure of protein complexes. This is an active area of research. Jurchen and Williams have recently demonstrated that the SORI-CID dissociation of protein homodimers is dependent upon the solution conditions from which the proteins are electrospayed.91
Dissociation of the complex between calmodulin, the synthetic peptide RS20 and four calcium ions by SORI-CID was performed by Nousiainen et al.92 Two reaction pathways were observed for the 8+ charge state of the complex, either calmodulin with four calcium ions in the 6+ charge state and RS20 in the 2+ charge state or calmodulin with four calcium ions in the 5+ charge state and RS20 in the 3+ charge state were formed. It was concluded that the binding of RS20 to calmodulin was specific and that gas-phase dissociation of calmodulin-RS20-Ca4 complex reflected the existence of in vacuo salt bridges.
Conclusions
Fourier transform ion cyclotron resonance mass spectrometry represents an increasingly powerful tool for the study of biological molecules, especially within “proteomics.”93–95 Improvements in FT-ICR analyzer cell design, increases in magnetic field strength, addition of new features such as types of tandem mass spectrometry experiment and the coupling with electrospray have led to improvements in mass accuracy, resolution, structural elucidation and the range of samples that can be analyzed. The ultra-high resolution and mass accuracy associated with FT-ICR mass spectrometers make them unique, and such strengths make them ideally suited for unequivocal mass assignment. It has, however, proved difficult from a users point of view, for instrument manufacturers to represent the instruments as “black boxes” and it remains the case that users of FT-ICR mass spectrometers get the most out of their instruments through understanding the physical principles involved. The future looks set for a continuation of the trend of increased investment in FT-ICR mass spectrometry and the wider ownership of such instruments, with particular interest in biological samples and petroleum-related products.Acknowledgements
The Engineering and Physical Sciences Research Council (UK) and the National Water Research Institute (Saskatoon, Canada) are gratefully acknowledged for providing funding. The authors would also like to thank I. Jonathan Amster for valuable discussion and John V. Headley for providing permission to publish the data on the multicomponent mixture.References
- E. de Hoffmann, J. Charette and V. Stroobant, in Mass Spectrometry: Principles and Applications, John Wiley and Sons Ltd., Chichester, 1996 Search PubMed.
- J. R. Chapman, in Practical Organic Mass Spectrometry: A Guide for Chemical and Biochemical Analysis, John Wiley and Sons Ltd., Chichester, 1993 Search PubMed.
- K. R. Jennings and G. G. Dolnikowski, Methods Enzymol., 1990, 193, 37–61 CrossRef CAS.
- G. L. Glish and R. W. Vachet, Nat. Rev. Drug Discovery, 2003, 2, 140–150 CrossRef CAS.
- C. M. Nielsen-Marsh, P. H. Ostrom, H. Gandhi, B. Shapiro, A. Cooper, P. V. Hauschka and M. J. Collins, Geology, 1998, 30, 1099–1102 CrossRef.
- H. Valladas, J. Clottes, J.-M. Geneste, M. A. Garcia, M. Arnold, H. Cachier and N. Tisnérat-Laborde, Nature, 2001, 413, 479 CrossRef CAS.
- W. Weckwerth, Annu. Rev. Plant Biol., 2003, 54, 669–689 Search PubMed.
- P. L. Ferguson and R. D. Smith, Annu. Rev. Biophys. Biomol., 2003, 32, 399–424 CrossRef CAS.
- C. M. Henry, Chem. Eng. News, 2003, 81, 39.
- M. P. Barrow, L. A. McDonnell, X. Feng, J. Walker and P. J. Derrick, Anal. Chem., 2003, 75, 860–866 CrossRef CAS.
- A. Turnbull, E. Slavcheva and B. Shone, Corrosion, 1998, 54, 922–930 Search PubMed.
- Z. G. Wu, S. Jernstrom, C. A. Hughey, R. P. Rodgers and A. G. Marshall, Energy Fuels, 2003, 17, 946–953 CrossRef CAS.
- M. B. Comisarow and A. G. Marshall, Chem. Phys. Lett., 1974, 25, 282–283 CrossRef CAS.
- M. B. Comisarow and A. G. Marshall, Can. J. Chem., 1974, 52, 1997–1999 CAS.
- M. B. Comisarow and A. G. Marshall, Chem. Phys. Lett., 1974, 26, 489–490 CrossRef CAS.
- C. B. Jacoby, C. L. Holliman and M. L. Gross, Fourier Transform Mass Spectrometry: Features, Principles, Capabilities, and Limitations, ed. M. L. Gross, Kluwer Academic Publishers, Netherlands, 1992, ch. 5 Search PubMed.
- A. G. Marshall and L. Schweikhard, Int. J. Mass Spectrom. Ion Processes, 1992, 118/119, 37–70 CrossRef.
- I. J. Amster, J. Mass Spectrom., 1996, 31, 1325–1337 CrossRef.
- A. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1–35 CrossRef CAS.
- P. Caravatti and M. Allemann, Org. Mass Spectrom., 1991, 26, 514–518 CAS.
- A. G. Marshall and M. B. Comisarow, Anal. Chem., 1975, 47, 491A–504A CAS.
- A. G. Marshall and F. R. Verdun, in Fourier transforms in NMR, optical, and mass spectrometry: a user's handbook, Elsevier, Amsterdam, Oxford, 1990 Search PubMed.
- S. D. H. Shi, J. J. Drader, M. A. Freitas, C. L. Hendrickson and A. G. Marshall, Int. J. Mass Spectrom, 2000, 196, 591–598 CrossRef.
- K. Hübner, H. Klein, C. Lichtenberg, G. Marx and G. Werth, Europhys. Lett., 1997, 37, 459–463 CrossRef.
- R. C. Dunbar, J. H. Chen and J. D. Hays, Int. J. Mass Spectrom. Ion Processes, 1984, 57, 39–56 CrossRef CAS.
- R. C. Dunbar, Int. J. Mass Spectrom. Ion Processes, 1984, 56, 1–9 CrossRef CAS.
- T. Dienes, S. J. Pastor, S. Schürch, J. R. Scott, J. Yao, S. Cui and C. L. Wilkins, Mass Spectrom. Rev., 1996, 15, 163–211 CrossRef CAS.
- L. Schweikhard, S. Guan and A. G. Marshall, Int. J. Mass Spectrom. Ion Processes, 1992, 120, 71–83 CrossRef CAS.
- V. H. Vartanian and D. A. Laude, Int. J. Mass Spectrom., 1998, 178, 173–186 CrossRef CAS.
- G. S. Jackson, J. D. Canterbury, S. Guan and A. G. Marshall, J. Am. Soc. Mass Spectrom., 1997, 8, 283–293 CrossRef CAS.
- T. E. Sharp, J. R. Eyler and E. Li, Int. J. Mass Spectrom. Ion Phys., 1972, 9, 421–439 CrossRef CAS.
- M. Dole, L. L. Mack, R. L. Hines, R. C. Mobley, L. D. Ferguson and M. B. Alice, J. Chem. Phys., 1968, 49, 2240–2249 CrossRef CAS.
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4451–4459 CrossRef CAS.
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4671–4675 CrossRef CAS.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS.
- M. Mann, Electrospray Mass Spectrometry, ed. M. L. Gross, Kluwer Academic Publishers, Netherlands, 1992, ch. 8 Search PubMed.
- D. Zhan and J. B. Fenn, Int. J. Mass Spectrom., 2000, 194, 197–208 CrossRef CAS.
- X. Feng, N. Clipston, T. Brown, H. Cooper, U. Reuther, A. Hirsch, P. J. Derrick and T. Drewello, Rapid Commun. Mass Spectrom., 2000, 14, 368–370 CrossRef CAS.
- S.-P. Chen and M. B. Comisarow, Rapid Commun. Mass Spectrom., 1991, 5, 450–455 CAS.
- S.-P. Chen and M. B. Comisarow, Rapid Commun. Mass Spectrom., 1992, 6, 1–3 CAS.
- M. L. Easterling, T. H. Mize and I. J. Amster, Anal. Chem., 1999, 71, 624–632 CrossRef CAS.
- S. Guan, M. C. Wahl and A. G. Marshall, Anal. Chem., 1993, 65, 3647–3653 CrossRef CAS.
- A. J. Perrung and R. T. Kouzes, Int. J. Mass Spectrom. Ion Processes, 1995, 145, 139–153 CrossRef.
- J. T. Stults, Anal. Chem., 1997, 69, 1815–1819 CrossRef CAS.
- J. Laskin, M. Byrd and J. Futrell, Int. J. Mass Spectrom., 2000, 195/196, 285–302 CrossRef.
- M. Palmblad, K. Hakansson, P. Hakansson, X. Feng, H. J. Cooper, A. E. Giannakopulos, P. S. Green and P. J. Derrick, Eur. J. Mass Spectrom., 2000, 6, 267–275 CrossRef CAS.
- Y. O. Tsybin, M. Witt, G. Baykut, F. Kjeldsen and P. Hakansson, Rapid Commun. Mass Spectrom., 2003, 17, 1759–1768 CrossRef CAS.
- A. J. Stace, J. Chem. Phys., 1998, 109, 7214–7223 CrossRef CAS.
- P. J. Derrick, P. M. Lloyd and J. R. Christie, Physical Chemistry of Ion Reactions, 13th International Mass Spectrometry Conference, ed. I. Cornides, G. Horváth and K. Vékey, John Wiley & Sons, 1995, vol. 13, pp. 23–52 Search PubMed.
- M. M. Sheil, M. Guilhaus and P. J. Derrick, Org. Mass Spectrom., 1990, 25, 671–680 CAS.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS.
- R. A. Zubarev, N. A. Kruger, E. K. Fridriksson, M. A. Lewis, D. M. Horn, B. K. Carpenter and F. W. McLafferty, J. Am. Chem. Soc., 1999, 121, 2857–2862 CrossRef CAS.
- R. A. Zubarev, Mass Spectrom. Rev., 2003, 22, 57–77 CrossRef CAS.
- L. A. McDonnell, A. E. Giannakopulos, P. J. Derrick, Y. O. Tsybin and P. Hakansson, Eur. J. Mass Spectrom., 2002, 8, 181–189 CrossRef CAS.
- T.-P. E. Sze and T.-W. D. Chan, Rapid Commun. Mass Spectrom., 1999, 13, 398–406 CrossRef CAS.
- K. Miyabayashi, N. Yasuhide, M. Miyake and K. Tsujimoto, Eur. J. Mass Spectrom., 2000, 6, 251–258 CAS.
- B. Ganem, Y.-T. Li and J. D. Henion, J. Am. Chem. Soc., 1991, 113, 6294–6296 CrossRef CAS.
- J. A. Loo, Mass Spectrom. Rev., 1997, 16, 1–23 CrossRef CAS.
- N. L. Kelleher, M. W. Senko, M. M. Siegel and F. W. McLafferty, J. Am. Soc. Mass Spectrom., 1997, 8, 380–383 CrossRef CAS.
- W. I. Burkitt, P. J. Derrick, D. Lafitte and I. Bronstein, Biochem. Soc. Trans., 2003, 31, 985–989 CrossRef CAS.
- P. K. Taylor, B. A. Parks, D. M. K. Jr. and I. J. Amster, J. Biol. Inorg. Chem., 2001, 6, 201–206 Search PubMed.
- K. A. Johnson and I. J. Amster, J. Am. Soc. Mass Spectrom., 2001, 12, 819–825 CrossRef CAS.
- P. K. Taylor, D. M. K. Jr and I. J. Amster, Int. J. Mass Spectrom, 2001, 210/211, 651–663 CrossRef.
- S. Tarabykina, M. Kriajevska, D. J. Scott, T. J. Hill, D. Lafitte, P. J. Derrick, G. G. Dodson, E. Lukanidin and I. Bronstein, FEBS Lett., 2000, 475, 187–191 CrossRef CAS.
- S. Tarabykina, D. J. Scott, P. Herzyk, T. J. Hill, J. R. H. Tame, M. Kriajevska, D. Lafitte, P. J. Derrick, G. G. Dodson, N. J. Maitland, E. M. Lukanidin and I. B. Bronstein, J. Biol. Chem., 2001, 276, 24212–24222 CrossRef CAS.
- O. V. Moroz, G. G. Dodson, K. S. Wilson, E. Lukanidin and I. B. Bronstein, Microsc. Res. Tech., 2003, 60, 581–592 CrossRef CAS.
- J. B. Fenn, J. Am. Soc. Mass Spectrom., 1993, 4, 524–535 CrossRef CAS.
- A. Dobo and I. Kaltashov, Anal. Chem., 2001, 73, 4763–4773 CrossRef CAS.
- A. Miranker, C. V. Robinson, S. E. Radford, R. T. Aplin and C. M. Dobson, Science, 1993, 262, 896–900 CrossRef CAS.
- B. E. Winger, K. J. Light-Wahl, A. L. Rockwood and R. D. Smith, J. Am. Chem. Soc., 1992, 114, 5898–5900 CrossRef CAS.
- F. Wang, M. A. Freitas, A. G. Marshall and B. D. Sykes, Int. J. Mass Spectrom., 1999, 192, 319–325 CrossRef CAS.
- Z. Zhang and D. L. Smith, Protein Sci., 1993, 2, 522–531 CAS.
- S. J. Eyles, J. P. Speir, G. H. Kruppa, L. M. Gierasch and I. A. Kaltashov, J. Am. Chem. Soc., 2000, 122, 495–500 CrossRef CAS.
- F. Wang, J. S. Blanchard and X.-J. Tang, Biochemistry, 1997, 36, 3755–3759 CrossRef CAS.
- M. Nousiainen, P. Vainiotalo, H. J. Cooper, A. Hoxha, P. J. Derrick, D. Fati, H. R. Trayer, D. G. Ward and I. P. Trayer, Eur. J. Mass. Spectrom, 2002, 8, 471–481 CrossRef CAS.
- P. J. Derrick, Ion Lifetimes, 5, ed. A. Maccoll, Butterworths, London, 1975 Search PubMed.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1997, 120, 3265–3266 CrossRef CAS.
- D. M. Horn, R. A. Zubarev and F. W. McLafferty, Proc. Natl. Acad. Sci. USA, 2000, 97, 10313–10317 CrossRef CAS.
- S. D.-H. Shi, M. E. Hemling, S. A. Carr, D. M. Horn, I. Lindh and F. W. McLafferty, Anal. Chem., 2000, 73, 19–22 CrossRef CAS.
- N. L. Kelleher, R. A. Zubarev, K. Bush, B. Furie, B. C. Furie, F. W. McLafferty and C. T. Walsh, Anal. Chem., 1999, 71, 4250–4253 CrossRef CAS.
- K. Håkansson, H. J. Cooper, M. R. Emmett, C. E. Costello, A. G. Marshall and C. L. Nilsson, Anal. Chem., 2001, 73, 4530 CrossRef.
- E. Mirgorodskaya, P. Roepstorff and R. A. Zubarev, Anal. Chem., 1999, 71, 4431–4436 CrossRef CAS.
- D. M. Horn, Y. Ge and F. W. McLafferty, Anal. Chem., 2000, 72, 4778–4784 CrossRef CAS.
- R. C. Dunbar, Mass Spectrom. Rev., 2004, 23, 127–158 CrossRef CAS.
- F. Kjeldsen, K. F. Haselmann, B. A. Budnik, F. Jensen and R. A. Zubarev, Chem. Phys. Lett., 2002, 356, 201–206 CAS.
- R. A. Zubarev, D. M. Horn, E. K. Fridriksson, N. L. Kelleher, N. A. Kruger, M. A. Lewis, B. K. Carpenter and F. W. McLafferty, Anal. Chem., 2000, 72, 563–573 CrossRef CAS.
- S. K. Sze, Y. Ge, H. Oh and F. W. McLafferty, Proc. Natl. Acad. Sci. USA, 2002, 99, 1774–1779 CrossRef CAS.
- C. L. Hunter, A. G. Mauk and D. J. Douglas, Biochemistry, 1997, 36, 1018–1025 CrossRef CAS.
- M. Nousiainen, P. J. Derrick, D. Lafitte and P. Vainiotalo, Biophys. J., 2003, 85, 491–500 CrossRef CAS.
- C. L. Hanson, P. Fucini, L. L. Ilag, K. H. Nierhaus and C. V. Robinson, J. Biol. Chem., 2003, 278, 1259–1267 CrossRef CAS.
- J. C. Jurchen and E. R. Williams, J. Am. Chem. Soc., 2003, 125, 2817–2826 CrossRef CAS.
- M. Nousiainen, P. Vainiotalo, X. Feng and P. J. Derrick, Eur. J. Mass. Spectrom, 2001, 7, 293–298.
- M. Tyers and M. Mann, Nature, 2003, 422, 193–197 CrossRef CAS.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS.
- A. Sali, R. Glaeser, T. Earnest and W. Baumeisters, Nature, 2003, 422, 216–225 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |