
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteratomic Fe–Cu cooperation in nitrogen-doped carbon for enhanced oxygen reduction†
Xiang
Ao
 abc,
Linfeng
Li
c,
Yong
Ding
b,
Gyutae
Nam
b,
Bote
Zhao
*bd,
Chundong
Wang
*c and
Meilin
Liu
*b
abc,
Linfeng
Li
c,
Yong
Ding
b,
Gyutae
Nam
b,
Bote
Zhao
*bd,
Chundong
Wang
*c and
Meilin
Liu
*b
aState Key Laboratory of Electrical Insulation and Power Equipment, Center of Nanomaterials for Renewable Energy (CNRE), School of Electrical Engineering, Xi’an Jiaotong University, Xi’an, 710049, China
bSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: meilin.liu@mse.gatech.edu
cSchool of Integrated Circuits, State Key Laboratory of New Textile Materials and Advanced Processing, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: apcdwang@hust.edu.cn
dSchool of Environment and Energy, South China University of Technology, Guangzhou, 510006, China. E-mail: botezhao@scut.edu.cn
First published on 20th June 2025
Abstract
The development of robust and electrocatalytically active catalysts for the oxygen reduction reaction (ORR) remains a significant challenge in advancing electrochemical energy technologies. Here, we report a Fe–Cu dual-metal catalyst embedded in nitrogen-doped porous carbon (FeCu–NC), synthesized via a controllable host–guest encapsulation strategy to enhance charge and mass transfer in the ORR. The FeCu–NC catalyst exhibits impressive ORR performance, with half-wave potentials of 0.918 V and 0.805 V in alkaline and acidic media, respectively, surpassing that of commercial Pt/C (0.889 V) in alkaline media and approaching its activity (0.835 V) under acidic conditions. Moreover, the catalyst demonstrates remarkable stability with negligible degradation in accelerated degradation testing. Density functional theory calculations reveal strong Fe–Cu interactions that optimize intermediate adsorption energies, enhancing catalytic efficiency. In practical applications, the FeCu–NC catalyst delivers high peak power densities of 250.3 mW cm−2 in zinc–air batteries and 0.58 W cm−2 in proton exchange membrane fuel cells. It also exhibits impressive long-term stability compared to other reported non-precious metal catalysts. These findings provide valuable insights for designing advanced catalysts for a wide range of electrocatalytic processes.
Broader contextThe oxygen reduction reaction (ORR) is a key process in energy technologies such as fuel cells and metal–air batteries, which are essential for sustainable energy systems. Platinum-based catalysts have been the benchmark catalysts for the ORR due to their superior catalytic performance. However, their high cost and limited stability necessitate the development of alternative catalysts based on more abundant and cost-effective materials. Recent advances in metal–nitrogen–carbon catalysts have shown promise, but challenges remain in optimizing their performance. This study introduces a novel Fe–Cu dual-metal catalyst embedded in nitrogen-doped porous carbon (FeCu–NC), which leverages the synergistic interactions between iron and copper atoms to significantly enhance ORR performance. The FeCu–NC catalyst exhibits remarkable activity and stability, outperforming traditional platinum-based catalysts in alkaline environments and demonstrating promising performance under acidic conditions as well. This work offers valuable insights into the design of advanced electrocatalysts, potentially paving the way for their application in next-generation renewable energy devices. |
Introduction
The oxygen reduction reaction (ORR) is a fundamental process in various electrochemical energy conversion and storage systems, such as fuel cells and metal–air batteries.1–3 However, its inherently sluggish kinetics significantly hinder the efficiency, necessitating the development of electrocatalysts to accelerate reaction rates and enhance device performance.4,5 Despite their superior catalytic activity for the ORR, platinum-based materials suffer from high cost and insufficient stability, limiting their widespread application.6,7 This challenge has driven extensive research into cost-effective alternatives based on earth-abundant elements. Despite notable advancements in catalyst design, achieving both high activity and durability, particularly under acidic conditions, remains challenging.8–10 Therefore, the rational development of affordable ORR catalysts with high activity and durability is of critical importance.11,12Catalysts with atomically dispersed metal sites anchored on nitrogen-doped carbon (M–N–C) have emerged as promising candidates to replace platinum-based ORR catalysts, owing to their tunable active sites and high catalytic efficiency.13–15 Extensive efforts have been made to elucidate the nature of active sites in M–N–C catalysts and optimize their local environments, achieving rapid performance improvements.16–18 Although the precise identity of active species remains under debate, recent studies have suggested that precise atomic-level control over metal coordination is crucial for developing highly active catalysts.19–21 The strong electronegativity of nitrogen atoms surrounding metal centers often induces suboptimal free energy for intermediate adsorption, thereby limiting reaction activity.21 To address this issue, incorporating additional non-metal heteroatoms such as S and P has been demonstrated as an effective strategy to modify the coordination configuration and enhance catalytic activity.21–23 The introduction of these heteroatoms alters electron distribution and atomic electronegativity, inducing charge redistributions and ultimately improving catalytic performance.24–26 However, this process inevitably introduces additional defects throughout the carbon framework, increasing susceptibility to corrosion under operating conditions.27 Thus, balancing activity enhancement with structural stability remains a key challenge for non-metal heteroatom-incorporated M–N–C catalysts.28
Beyond the incorporation of non-metal heteroatoms, catalysts with multimetallic active sites offer additional opportunities to enhance ORR performance.29 These catalysts not only exhibit high atomic utilization efficiency but also benefit from synergistic interactions between adjacent metal sites, which optimize intermediate adsorption and reaction pathways. For instance, binuclear Fe–Fe and Co–Co sites with nitrogen coordination have been shown to facilitate O2 adsorption and O–O bond cleavage, leading to improved ORR activity.30,31 Given that catalytic efficiency is closely linked to the electron density around metal active sites, incorporating a second metal site with higher electronegativity can further modulate the electronic properties and enhance catalytic performance.23
In this work, we report the synthesis and investigation of atomically dispersed Fe–Cu dual-metal sites embedded in a N-doped porous carbon framework (denoted as FeCu–NC) as an ORR catalyst. The porous structure and atomic-level dispersion of Fe–Cu sites facilitate efficient mass transfer and maximize active site utilization. The FeCu–NC catalyst demonstrates remarkable ORR activity under both alkaline and acidic conditions, outperforming its counterparts and rivaling commercial Pt/C. In addition to its superior performance, the FeCu–NC catalyst is composed of earth-abundant and low-cost elements, offering a clear economic advantage over commercial Pt/C. Experimental and theoretical analyses reveal that the cooperative interaction between Fe and Cu atoms optimizes intermediate adsorption free energy, significantly enhancing catalytic performance. Furthermore, FeCu–NC demonstrates satisfactory performance as a cathode catalyst in zinc–air batteries and proton exchange membrane fuel cells (PEMFCs), underscoring its potential for advancing energy technologies.
Results and discussion
Fig. 1a shows a schematic illustration of the FeCu–NC synthesis process. A zinc-based zeolitic imidazolate framework (ZIF-8) was utilized to encapsulate and immobilize Fe and Cu ions within its micropores through a host–guest strategy.32 Following heat treatment under a nitrogen atmosphere, ZIF-8 was transformed into nitrogen-enriched porous carbon. During pyrolysis, volatile Zn can be removed, while Fe and Cu interacted with the organic ligands, resulting in the formation of atomically dispersed metal centers on the carbon substrate. The use of ZIF-8 as a confinement scaffold provides a highly adaptable platform for embedding various transition metal precursors, owing to its high nitrogen content and well-defined microporous structure. These features suggest that this strategy can be applied to other bimetallic systems simply by substituting the metal precursors. The intrinsic microporous structure of ZIF-8, combined with the formation of mesopores induced by Zn evaporation, gives rise to a hierarchical porous carbon matrix. This hierarchical structure not only provides abundant anchoring sites for metal atoms, enabling relatively high metal loading. It also enhances the accessibility and exposure of active sites, thereby improving their utilization efficiency during electrocatalysis, consistent with previous findings that mesoscale porous structures can effectively enhance catalyst-electrolyte interactions and active site accessibility.33 To investigate Fe–Cu interatomic interactions, control samples with varying metal contents were synthesized by a similar method (see the Experimental section for details), and they are denoted as Fe–NC (containing only Fe), Cu–NC (containing only Cu), Fe2–NC (with double the Fe content of Fe–NC), Cu2–NC (with double the Cu content of Cu–NC), and NC (metal-free). Notably, no characteristic peaks associated with crystalline metal phases were detected in the X-ray diffraction (XRD) patterns of the pyrolyzed materials (Fig. S1, ESI†), suggesting the absence of large metal-based particles. Instead, two broad peaks at approximately 23° and 44° were ascribed to amorphous carbon with abundant defects, further supported by the Raman spectrum of FeCu–NC, which exhibited a pronounced D-band peak (Fig. S2, ESI†).34 Moreover, the ID/IG value increased from 1.01 for NC to 1.06 for FeCu–NC, indicating that metal atom incorporation into the carbon framework introduced extra defects, potentially influencing the electronic properties of the active sites.35,36 | ||
| Fig. 1 (a) Schematic illustration of the FeCu–NC synthesis process. (b) and (c) SEM images of FeCu–NC at different magnifications. (d) HAADF-STEM image and corresponding EDS element mapping of FeCu–NC. (e) and (f) Aberration-corrected HAADF-STEM images of FeCu–NC, with bimetallic pairs indicated by red circles and isolated metal atoms by green circles. (g) Intensity profiles obtained from two pairs in (f), labelled as A and B. | ||
Scanning electron microscopy (SEM) analysis revealed that ZIF-8 nanocrystals exhibited a well-defined rhombic dodecahedron shape with an average size of ∼400 nm (Fig. S3, ESI†). After high-temperature pyrolysis, the resulting products with different metal contents retained a similar morphology to that of ZIF-8 (Fig. S4, ESI†), while the diameter was decreased to approximately 300 nm, likely due to thermal stress effects.37 Transmission electron microscopy (TEM) analysis further verified the absence of metallic and oxide nanoparticles in FeCu–NC, in agreement with XRD results (Fig. S5, ESI†). Beyond the inherent porosity of ZIF-8, the rapid evaporation of Zn during pyrolysis facilitated the formation of additional pores, acting as an effective porogen.38 The high-resolution SEM image revealed that the carbon substrate derived from ZIF-8 was rich in micropores (Fig. 1c), which enhance mass transport and provide anchoring sites for metal atoms. Nitrogen adsorption–desorption analysis further demonstrated the high specific surface area of FeCu–NC (792.5 m2 g−1), attributed to its porous nanostructure (Fig. S6, ESI†).
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), combined with energy-dispersive X-ray spectroscopy (EDS) elemental mapping, demonstrated the uniform distribution of C, N, Fe, and Cu throughout the entire FeCu–NC architecture (Fig. 1d). To gain deeper insight into metal dispersion, aberration-corrected HAADF-STEM was employed. The resulting image (Fig. 1e) revealed numerous bright spots evenly distributed across the carbon support, corresponding to Fe and Cu atoms. An enlarged image further confirmed the successful formation of bimetallic pairs in FeCu–NC, highlighted by red circles (Fig. 1f), while isolated metal atoms were marked with green circles. Intensity profiles indicated that the distance between two sites in a bimetallic pair was approximately 2.3–2.4 Å, suggesting close proximity conducive to interatomic interactions (Fig. 1g). The differing intensities of the two peaks in each pair suggest the presence of distinct metal atoms, further supporting the identification of these pairs as Fe–Cu. To further evaluate the distances between metal atoms, statistical measurements were conducted based on multiple dual-metal pairs observed in the HAADF-STEM image (Fig. S7, ESI†). Most of the measured distances clustered around 2.3 Å, which is consistent with the Fe–Cu separation observed from the intensity profiles. However, it should be noted that HAADF-STEM is a projection-based imaging technique, and the measured two-dimensional distances may be significantly smaller than the actual distances. In some cases, very short projected distances may result in overlapping atomic contrast, making it difficult to distinguish individual atoms. Conversely, some bright spot pairs with projected distances greater than 3 Å may not originate from true dual-metal sites, but rather from two atoms that are spatially distant in three dimensions yet appear adjacent due to projection overlap.
Given the limitations of TEM in precisely determining the chemical states of atomically dispersed metal species, X-ray photoelectron spectroscopy (XPS) analysis was performed to investigate the valence states and elemental composition of the synthesized materials. XPS survey spectra (Fig. S8a, ESI†) confirmed the presence of C, N, and O in FeCu–NC, Fe–NC, and Cu–NC, along with minor amounts of metal elements, consistent with EDS mapping results (Fig. 1d). The detected oxygen likely originated from doping within the carbon matrix and the adsorption of atmospheric oxygen species due to the high surface area.39 XPS analysis revealed that Fe and Cu atomic concentrations in FeCu–NC were 0.59 at% and 0.46 at%, respectively (Table S1, ESI†), comparable to those in Fe–NC (0.61 at% Fe) and Cu–NC (0.47 at% Cu), highlighting the effectiveness of the host–guest approach employed for metal incorporation. Inductively coupled plasma optical emission spectrometry further quantified the Fe and Cu contents in FeCu–NC as 1.95 wt% and 2.14 wt%, respectively, with Fe–NC containing 2.33 wt% Fe and Cu–NC containing 2.04 wt% Cu. The relatively low single-atom loading is a well-recognized challenge in this field. In future work, we can improve the utilization of active sites further by constructing hierarchically porous architectures through chemical etching, such as tannic acid treatment, as demonstrated in our previous study.
The N 1s spectra for the three samples were analyzed and deconvoluted into four peaks located at 398.5, 399.5, 401.0, and 403.4 eV, assigned to pyridinic N, metal–N, graphitic N, and oxidized N, respectively (Fig. S8b, ESI†).40 The presence of metal–N confirmed the coordination of Fe and Cu with nitrogen species, forming atomically dispersed metal sites. Pyrrolic N, which is typically unstable at high temperatures, was absent in the final product due to its transformation into pyridinic N during pyrolysis.41 Notably, FeCu–NC exhibited a higher metal–N content than Fe–NC and Cu–NC (Fig. S8b, ESI†), likely due to the higher loading of metal sites in the bimetallic catalyst. Additionally, FeCu–NC had a higher total N doping level (5.19 at%) compared to Fe–NC (4.16 at%) and Cu–NC (4.62 at%), which can be ascribed to the enhanced nitrogen incorporation into the carbon framework promoted by the presence of more metal species.42 The relatively low metal content in these samples resulted in weak intensity signals for metal-related peaks (Fig. S8c and d, ESI†). High-resolution Fe 2p and Cu 2p spectra provided insights into the oxidation states of the metal species. For FeCu–NC, the Fe 2p spectrum exhibited a binding energy shift toward higher values compared to Fe–NC, whereas the Cu 2p spectrum showed a shift to lower values relative to Cu–NC. This trend suggests that Cu atoms, owing to their higher electronegativity, withdraw electrons from neighboring Fe atoms, which is likely to benefit ORR activity of Fe sites.43
X-ray absorption spectroscopy (XAS) was conducted to investigate the atomic structure and coordination environment of the synthesized samples. For both Fe–NC and FeCu–NC, the Fe K-edge normalized X-ray absorption near-edge structure (XANES) spectra exhibited absorption edges similar to that of iron(II) phthalocyanine (FePc) (Fig. 2a), suggesting that Fe atoms are in a positively charged state in both samples, in agreement with previously reported single-atom Fe sites on carbon substrates.44 As shown in the inset of Fig. 2a, the adsorption edge for FeCu–NC was shifted to higher energy compared to Fe–NC, suggesting a higher Fe oxidation state in the bimetallic catalyst. To quantitatively assess the oxidation state, we calculated the average valence states of the samples based on the absorption threshold energies (E0) derived from the first derivative (Fig. S9a, ESI†). The results revealed a higher Fe oxidation state in FeCu–NC than in Fe–NC, likely due to Fe–Cu atomic interactions, aligning with the XPS analysis.45 Similarly, Fig. 2b displays the Cu K-edge XANES profiles, showing that the Cu valence states in FeCu–NC and Cu–NC were similar to that of copper(II) phthalocyanine (CuPc). The absorption threshold for FeCu–NC was shifted to a lower energy compared to Cu–NC, suggesting a reduction in the Cu oxidation state in the presence of Fe (Fig. 2b and Fig. S9b, ESI†). The XANES results suggest that, in FeCu–NC, electrons are partially transferred from Fe to Cu in FeCu–NC, resulting in a higher Fe oxidation state and a lower Cu oxidation state in the bimetallic catalyst compared to the monometallic counterparts.
 | ||
| Fig. 2 (a) and (b) Normalized XANES spectra at the (a) Fe K-edge and (b) Cu K-edge, with insets showing enlarged views of the regions enclosed by dashed boxes. (c) and (d) Fourier-transformed EXAFS spectra at the (c) Fe K-edge and (d) Cu K-edge. (e) and (f) EXAFS fitting curves at the (e) Fe K-edge and (f) Cu K-edge for FeCu–NC, with insets showing the proposed Fe–Cu pair structure (gray, blue, red, and green spheres represent C, N, Fe, and Cu atoms, respectively). (g) WT-EXAFS spectra of different samples at the Fe K-edge or Cu K-edge. | ||
The Fourier-transformed Fe K-edge extended X-ray absorption fine structure (EXAFS) data showed a main peak at 1.4 Å for both FeCu–NC and Fe–NC, similar to that of FePc, which corresponds to Fe–N coordination in the first coordination shell (Fig. 2c).46 A secondary peak at approximately 2.1 Å, corresponding to Fe–metal backscattering, appeared in the FeCu–NC spectrum, confirming the formation of dual-metal pairs. Similarly, the EXAFS spectra at the Cu K-edge showed a prominent peak around 1.4 Å for Cu–NC, FeCu–NC, and CuPc, which could be typically assigned to Cu–N coordination (Fig. 2d).47 The FeCu–NC spectrum also showed a small peak near 2.1 Å, confirming Cu–metal scattering in the bimetallic system. However, the intensity of the Fe–metal and Cu–metal peaks in the FeCu–NC EXAFS spectra was much lower compared to those observed in the spectra of Fe and Cu foils, indicating the atomic-level dispersion of metal species.47
To further understand the coordination environment of metal sites in FeCu–NC, EXAFS fitting using a quantitative least-squares approach was carried out (Fig. 2e and f), with the corresponding parameters shown in Table S2 (ESI†). The results revealed that the Fe–N and Cu–N coordination numbers are approximately four, while the metal–metal coordination number is around one. The metal–N and metal–metal bond lengths were approximately 2.0 Å and 2.4 Å, respectively. These results suggest that, in FeCu–NC, Fe and Cu form Fe–N4 and Cu–N4 coordination structures, with Fe and Cu atoms linked through two shared nitrogen atoms, as illustrated in the insets of Fig. 2e and f. Although the possibility of the formation of additional metal centers during thermal treatment cannot be entirely ruled out, precisely identifying their presence with the current characterization techniques remains a significant challenge.39 To further validate these configurations, we calculated the formation energies of these structures, showing that the aforementioned Fe–Cu pair configuration was thermodynamically more stable (Fig. S10, ESI†). Moreover, the experimentally measured Fe–Cu distance closely matched the calculated value, providing further support for the reliability of this structure in FeCu–NC (Fig. 1g and Fig. S7, ESI†). Moreover, wavelet transform (WT)-EXAFS was employed to explore the atomic configurations in these samples with high resolution in both K and R spaces.48 This method incorporates a slowly varying amplitude component and a rapidly oscillating phase term.49Fig. 2g shows the WT-EXAFS contour plots of FeCu–NC, Fe–NC, and Cu–NC, all exhibiting a single peak with maximum intensity near 4.0 Å−1, closely resembling the spectra of FePc and CuPc (Fig. S11, ESI†). In contrast, the WT-EXAFS plots of FeCu–NC, Fe–NC and Cu–NC lacked the prominent peak at around 8.0 Å−1 observed for Fe and Cu foils, which is typically attributed to metal–metal scattering. This absence further confirms the atomic-level dispersion of the metal species.
The ORR electrocatalytic performance of the synthesized catalysts was evaluated using a rotating disk electrode (RDE) in a three-electrode configuration with a 0.1 M KOH electrolyte. All potentials were referenced to the reversible hydrogen electrode (RHE). Fig. 3a shows the cyclic voltammetry (CV) curves of the catalysts. FeCu–NC displayed a distinct oxygen reduction peak at approximately 0.905 V in an O2-saturated electrolyte, whereas no noticeable peak appeared in an N2-saturated electrolyte, confirming its ability to effectively catalyze the ORR. Similar CV curves were observed for Fe–NC and Cu–NC in both O2- and N2-saturated electrolytes, but their oxygen reduction peaks occurred at more negative potentials (0.871 V for Fe–NC and 0.823 V for Cu–NC) compared to FeCu–NC, indicating that FeCu–NC possesses enhanced ORR catalytic activity compared with its monometallic counterparts. To further assess ORR performance, linear sweep voltammetry (LSV) measurements were performed using a RDE rotating at 1600 rpm (Fig. 3b and Fig. S12, ESI†). Compared to the metal-free NC catalyst, all metal-containing catalysts displayed significantly improved catalytic activity, confirming the crucial role of metal sites. Among the catalysts, FeCu–NC demonstrated the best performance, achieving an onset potential (Eonset) of 1.03 V and a half-wave potential (E1/2) of 0.918 V. These metrics surpassed those recorded for Fe–NC (Eonset = 0.98 V; E1/2 = 0.891 V), Cu–NC (Eonset = 0.92 V; E1/2 = 0.841 V), and even the commercial Pt/C catalyst (Eonset = 0.99 V; E1/2 = 0.889 V).
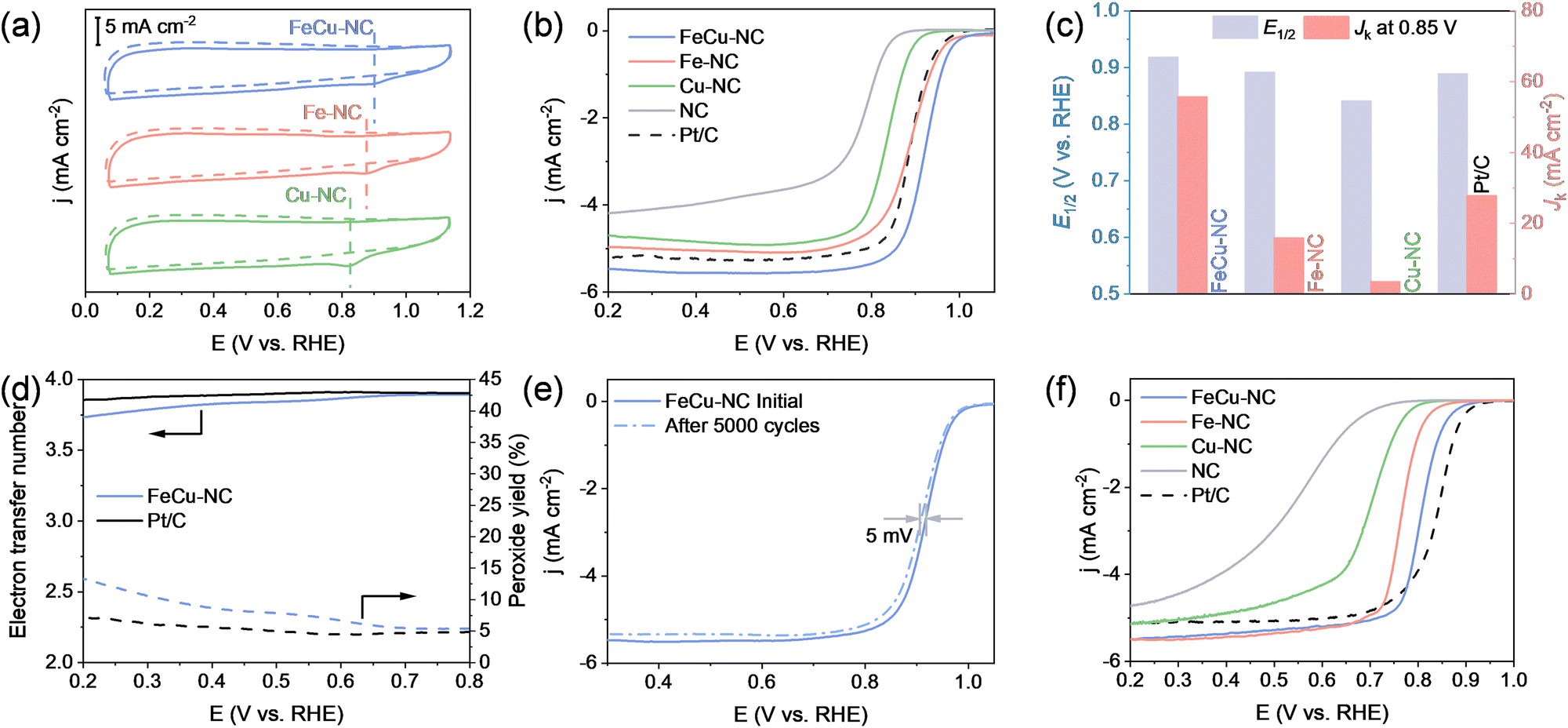 | ||
| Fig. 3 (a) CV profiles of various catalysts in O2-saturated (solid line) or N2-saturated (dash line) 0.1 M KOH solution. (b) ORR polarization curves of different samples in O2-saturated 0.1 M KOH solution. (c) E1/2 and Jk at 0.85 V of different samples in 0.1 O2-saturated M KOH solution. (d) Peroxide yield and the corresponding electron transfer number for FeCu–NC and Pt/C determined by RRDE measurements in O2-saturated 0.1 M KOH solution. (e) ORR polarization curves of FeCu–NC before and after the durability test in 0.1 M KOH solution. (f) ORR polarization curves of different samples in O2-saturated 0.1 M HClO4 solution. | ||
To examine the possibility that catalytic activity depends on active site density, additional monometallic catalysts, Fe2–NC and Cu2–NC, with metal contents similar to that of FeCu–NC were prepared. The catalytic activity for the ORR improved with increasing metal site content (Fig. S11, ESI†). However, the bimetallic FeCu–NC catalyst still outperformed both Fe2–NC and Cu2–NC, suggesting that the enhanced catalytic performance of FeCu–NC is not due to an increased active site density, but rather the improved intrinsic catalytic activity arising from the interactions between Fe and Cu atoms. As shown in Fig. 3c, FeCu–NC achieved a kinetic current density (Jk) of 55.7 mA cm−2 at 0.85 V, significantly higher than those of Fe–NC (15.8 mA cm−2) and Cu–NC (3.5 mA cm−2), and commercial Pt/C (27.7 mA cm−2), highlighting its superior reaction kinetics. Furthermore, the ORR activity of FeCu–NC outperformed that of the majority of other reported catalysts without precious metals, including both single-metal and dual-metal catalysts (Table S3, ESI†). The improved ORR performance of FeCu–NC can be attributed to the successful formation of Fe–Cu dual-atom sites. Unlike other transition metals, which tend to exhibit either overly strong or weak oxygen binding, Cu offers a moderate oxygen affinity. This enables more balanced adsorption and desorption of reaction intermediates. This helps regulate the local electronic environment around the Fe active sites, thereby promoting favorable ORR kinetics.
To investigate the ORR pathway, rotating ring-disk electrode (RRDE) tests were performed (Fig. S13, ESI†). The calculated number of electrons transferred for FeCu–NC ranged from 3.75 to 3.9 (Fig. 3d). This result suggests that FeCu–NC follows a nearly four-electron ORR pathway, similar to commercial Pt/C. In addition to high activity, FeCu–NC demonstrated exceptional durability and methanol tolerance. Fig. 3e presents LSV curves recorded before and after an accelerated degradation test (ADT), conducted via 5000 CV cycles between 0.6 and 1.0 V. The Eonset remained almost unchanged after 5000 cycles, and the E1/2 decreased by only 5 mV. In contrast, commercial Pt/C showed obvious activity degradation, with a 23 mV drop in E1/2 following 5000 CV cycles (Fig. S14, ESI†), highlighting the high stability of FeCu–NC. Moreover, FeCu–NC exhibited no noticeable change during the methanol exposure, while commercial Pt/C experienced a sudden decline in current, demonstrating the superb resistance of FeCu–NC to methanol crossover (Fig. S15, ESI†). FeCu–NC also exhibited high ORR activity under acidic conditions (Fig. 3f). In 0.1 M HClO4, it exhibited an E1/2 of 0.805 V, approaching that of commercial Pt/C (0.835 V) and significantly outperforming Fe–NC, Cu–NC and NC. This combination of high activity, durability, and methanol tolerance establishes FeCu–NC as a promising candidate for practical ORR applications.
To further investigate the mechanism underlying the high catalytic performance of FeCu–NC, first-principles density functional theory (DFT) simulations were performed. Building on the experimental results and formation energy evaluations, the FeCu–N6 configuration was proposed as a structural model for FeCu–NC, comprising Fe–N4 and Cu–N4 connected through two shared nitrogen atoms (Fig. S16a, ESI†). In comparison, structural models for Fe–NC and Cu–NC were constructed as Fe–N4 and Cu–N4 configurations, respectively (Fig. S16b and c, ESI†). The charge density difference maps for Fe–N4 and Cu–N4 (Fig. S17, ESI†) revealed symmetric electron interactions between the central metal atoms and their nitrogen ligands, consistent with the D4h symmetry of the planar coordination structure. In contrast, the asymmetric diatomic FeCu–N6 site exhibited symmetry-breaking charge transfer, inducing electron redistribution and charge polarization (Fig. 4a). Bader charge analysis revealed that Fe atoms lost 1.085 electrons in Fe–NC and 1.174 electrons in FeCu–NC, while Cu atoms lost 0.928 electrons in Cu–NC and 0.922 electrons in FeCu–NC. These results suggest that electron transfer occurs between adjacent metal atoms via nitrogen bridges in the bimetallic pair, aligning with the experimental findings from XPS and XAS measurements. It has been shown that such electron redistribution influences intermediate binding strength and catalytic selectivity.50,51
 | ||
| Fig. 4 (a) Bader charge analysis and visualization of charge density redistribution in FeCu–N6. (b) Spin density distribution and channel in FeCu–N6. (c) Density of states (DOS) of FeCu–N6, Fe–N4, and Cu–N4. (d) PDOS of d orbitals for metal sites in different models with corresponding d-band center positions indicated. (e) and (f) Analysis of the pCOHP between the Fe active sites in different models and oxygen atoms during (e) O2 adsorption and (f) *OH adsorption. (g) Adsorption energy of O2 and *OH on different metal active sites in various models. (h) and (i) Gibbs free energy diagrams for the ORR process in different models. | ||
Oxygen molecules exist in a triplet state in their paramagnetic ground state (Fig. S18, ESI†), making electron transfer in the ORR highly dependent on spin alignment, a critical factor influencing reaction kinetics.52 To explore the spin properties, the magnetic moments of metal sites in the three models were calculated (Table S4, ESI†). The magnetic moment of the Fe site increased from 1.913μB in Fe–N4 to 2.151μB in FeCu–N6, indicating a transition to a higher spin state, which can be attributed to electron redistribution from t2g to eg orbitals.53 Consequently, the electronic configuration changed, with a single unpaired electron occupying the eg orbital of the Fe site (t52ge1g) (Fig. S19, ESI†). The spin density map of FeCu–N6 (Fig. 4b) revealed that the higher Fe spin state, along with the elongation of Fe–N bonds, generated broader spin-polarized channels within the Fe active center, facilitating charge transfer during the ORR.41,52 Furthermore, the incorporation of Cu atoms eliminated the bandgap in FeCu–N6, with electronic states becoming occupied near the Fermi level, strongly supporting enhanced charge transfer mobility in FeCu–NC (Fig. 4c).
To examine adsorption behavior, the projected density of states (PDOS) was calculated for various metal sites, both in their original state and after oxygen adsorption (Fig. 4d and Fig. S20, ESI†). The results showed that the d-band center of Cu in Cu–N4 and FeCu–N6 was located far from the Fermi level, whereas the d-band center of Fe in Fe–N4 was much closer to it. The incorporation of Cu shifted the d-band center of Fe in FeCu–N6 downward, bringing it to a more favorable position. This downward shift increases electron occupancy in the antibonding orbitals, thereby weakening the bonding strength between the d–p orbitals, which could lead to the easier desorption of intermediates.54,55 Fig. S21 (ESI†) illustrates the orbital interactions between Fe in low or medium spin states and oxygen-containing intermediates (*OH and *O2) in detail. As the spin state increased, an unpaired electron occupied the dz2 orbital. This occupancy leads to greater electron population in antibonding orbitals after hybridization with *OH and *O2, thereby weakening the interaction strength between Fe and the intermediates.
To further quantify the bonding interactions between reaction intermediates (*OH and *O2) and metal sites, projected crystal orbital Hamilton population (pCOHP) analysis was conducted for the three proposed models. This method predicts the bonding and antibonding states, enabling a quantitative evaluation of the bonding strength between active sites and adsorbed intermediates and offering deeper insights into the nature of metal–intermediate interactions and their influence on catalytic performance.56 In FeCu–N6–O2, the calculated integrated pCOHP (ICOHP) value of the Fe–O bond was −2.94 eV, which was less negative than the ICOHP value of −3.58 eV in Fe–N4–O2 (Fig. 4e), suggesting a reduced adsorption strength of O2 at the Fe sites in FeCu–N6. For both FeCu–N6 and Cu–N4, the ICOHP values of the Cu–O bond were significantly lower than those for the Fe–O bond, suggesting that Cu sites have a much weaker capacity to adsorb O2 molecules, hindering their ability to capture O2 and initiate the ORR cycle (Fig. S22, ESI†).57 The electrostatic potential distribution (Fig. S23, ESI†) further demonstrated that the Fe atoms in FeCu–N6 had a higher electrostatic potential compared to surrounding regions. This indicates that the Fe sites in FeCu–N6 are more favorable for nucleophilic attack by reaction intermediates, making them effective active sites for electrocatalysis.58 In the ORR, *OH is the final reaction intermediate, and its binding affinity at active centers dictates how readily it is released, which is a critical step for the reaction pathway. The Fe–O bond in FeCu–N6–OH exhibited higher antibonding state occupancy compared to that in Fe–N4–OH, leading to a decrease in the ICOHP value from −2.92 eV to −2.61 eV (Fig. 4f). The weakened Fe–*OH interaction in FeCu–NC–OH facilitates *OH desorption, enabling efficient completion of the ORR cycle and enhancing catalytic performance.
Furthermore, we calculated the adsorption energies of *OH and O2 on the metal active sites (Fig. 4g). The results showed that both O2 and *OH exhibited significantly weaker adsorption on FeCu–N6 compared to Fe–N4, suggesting that the introduction of Cu atoms, through metal–nitrogen bridge coupling, synergistically mitigates the over-adsorption of O2 and *OH on Fe sites. At the Fe active sites, oxygen molecules undergo a typical ORR process, involving various reaction intermediates such as O2, *OOH, *O, and *OH (Fig. S24, ESI†).59Fig. 4h and i present the Gibbs free energy diagrams for the ORR on various metal sites at U = 0 and 1.23, respectively. Consistent with the previous analysis, Cu sites are unlikely to serve as active centers for the ORR because of their extremely weak oxygen adsorption. This is further supported by the Gibbs free energy diagrams, which show a substantially higher overpotential for the ORR at Cu sites compared to Fe sites. For the Fe active site, the incorporation of the heterometallic Cu atom to form the FeCu–N6 coordination structure weakened the adsorption of ORR intermediates, leading to an optimized free energy profile throughout the reaction process. The construction of the FeCu–N6 structure weakened the adsorption of *OH at the Fe active sites, shifting the rate-determining step of the ORR from the final step of *OH desorption to the initial step of *OOH generation. However, the overall reaction process becomes more energetically favorable due to a reduction in the energy barrier, effectively lowering the overpotential and improving the catalytic efficiency.
The remarkable electrocatalytic performance of FeCu–NC highlights its potential for oxygen-related electrochemical energy systems, such as zinc–air batteries and PEMFCs. A primary zinc–air battery was assembled using FeCu–NC as the cathode catalyst, with a Pt/C-based battery used for comparison. As shown in Fig. 5a, the zinc–air battery employing the FeCu–NC catalyst achieved an open-circuit voltage of 1.53 V, exceeding the value of 1.48 V recorded for the battery based on commercial Pt/C. The practical viability of FeCu–NC was further demonstrated by using a FeCu–NC-based zinc–air battery to successfully power a light-emitting diode panel (inset of Fig. 5a). Furthermore, as shown in Fig. 5b, the FeCu–NC-based battery demonstrated a rapid response and stable voltage across varying current densities. It demonstrated a higher discharge potential compared to its Pt/C-based counterpart at identical current densities, highlighting the superior catalytic performance of FeCu–NC. Upon restoring the original current density, the discharge voltage fully recovered, confirming the good rate performance. At a discharge current density of 20 mA cm−2, the specific capacity of the FeCu–NC-based battery, calculated based on the zinc consumption, reached 787 mA h g−1, surpassing 741 mA h g−1 achieved by a commercial Pt/C-based battery, demonstrating the superior zinc utilization efficiency. To evaluate the cycling stability of the zinc–air battery assembled with FeCu–NC as the cathode catalyst, we conducted a long-term galvanostatic discharge test at 10 mA cm−2, during which the zinc anode was mechanically refueled after each full discharge (Fig. S25, ESI†). After four discharge–refueling cycles over a total duration of nearly 96 h, the battery experienced only a slight mV voltage drop. Notably, the discharge voltage nearly fully recovered after each refueling, demonstrating good cycling stability. Fig. 5d presents the discharge profiles and corresponding power density curves for both batteries using different catalysts. The FeCu–NC-based battery exhibited a peak power density of 250.3 mW cm−2 at a current density of 403.6 mA cm−2, significantly outperforming the Pt/C-based battery, which delivered a maximum power density of 184.4 mW cm−2 at 296.5 mA cm−2.
 | ||
| Fig. 5 (a) Open-circuit voltage profiles of zinc–air batteries using FeCu–NC or Pt/C as the cathode catalyst (inset: a photograph of a light-emitting diode panel powered by an FeCu–NC-based zinc–air battery). (b) Discharge curves obtained at different current densities and (c) specific capacity curves at 20 mA cm−2 of zinc–air batteries using FeCu–NC or Pt/C as the cathode catalyst. (d) Discharge polarization curves and corresponding power density plots of zinc–air batteries using FeCu–NC or Pt/C as the cathode catalyst. (e) and (f) Polarization and power density curves of PEMFCs utilizing FeCu–NC or Pt/C as the cathode catalyst, recorded before and after 5000-cycle ADT. | ||
In addition, FeCu–NC was also explored as a noble-metal-free cathode catalyst in PEMFCs, with performance evaluated at 65 °C. Pt/C served as the catalyst for the anode. Fig. 5e and f present the polarization curves and the corresponding power density plots for PEMFCs using FeCu–NC or Pt/C as ORR electrocatalysts. Under H2–O2 conditions, the FeCu–NC-based PEMFC delivered an open-circuit voltage of approximately 0.86 V, similar to that of the benchmark Pt/C catalyst with a cathode platinum loading of 0.1 mgPt cm−2. The PEMFC using FeCu–NC at the cathode delivered a peak power density of 0.58 W cm−2 at 1.53 A cm−2, comparable to that of its Pt/C counterpart (0.77 W cm−2 at 1.88 A cm−2). The performance of the FeCu–NC-based cell at higher current densities became increasingly comparable to that of Pt/C, which can be attributed to the large specific surface area of FeCu–NC. The durability of the catalysts was assessed through an ADT employing square wave cycling from 0.6 to 0.9 V with 3.0 s hold at each potential. As shown in Fig. 5e, the FeCu–NC-based PEM25FC retained a peak power density of 0.54 W cm−2 at 1.47 A cm−2, which corresponds to 93% of its initial value after 5000 cycles, demonstrating remarkable stability. In contrast, the Pt/C-based PEMFC exhibited substantial performance degradation under the same conditions (Fig. 5f).
Conclusions
In conclusion, we successfully synthesized a dual-metal FeCu–NC catalyst with atomically dispersed Fe–Cu pairs on N-doped porous carbon frameworks. Experimental results demonstrated that the introduced Cu atoms were integrated into the coordination environment of Fe active sites, effectively modulating their electronic states. Theoretical calculations further revealed that the adjacent Cu atom induces asymmetric electronic distributions through metal–nitrogen bridge coupling, altering the spin state of Fe from low to medium. This adjustment optimized the adsorption and desorption behaviors of reaction intermediates, thereby accelerating the ORR kinetics and improving catalytic efficiency. The FeCu–NC catalyst demonstrated favorable ORR performance in both alkaline and acidic environments, outperforming Fe–NC and Cu–NC catalysts containing individual Fe or Cu single atoms. Furthermore, it exhibited satisfactory performance in practical applications, including zinc–air batteries and PEMFCs. This study presents a cost-effective and highly efficient electrocatalyst for the ORR, offering valuable insights into the relationship between the electronic structure of single-atom metal active sites and their catalytic performance. These findings provide valuable guidance for developing next-generation single-atom electrocatalysts for applications beyond the ORR.Author contributions
X. Ao conceived this work. X. Ao designed and conducted the experiments, analyzed the experimental data, and wrote the original manuscript. L. Li performed the theoretical calculations and contributed to data analyses. Y. Ding carried out the STEM tests. G. Nam collected the XAS data. B. Zhao revised the manuscript and supervised the project. C. Wang reviewed the manuscript and provided supervision. M. Liu supervised the entire project and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants No. 52272202, W2421027, 52102248, and 22479050), the Fundamental Research Funds for the Central Universities (xtr052024009), the National Key R&D Program of China (2021YFA1502400), and the Hightower Endowment through the Georgia Tech Foundation.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS.
- F. Liu, L. Shi, X. Lin, B. Zhang, Y. Long, F. Ye, R. Yan, R. Cheng, C. Hu and D. Liu, Sci. Adv., 2023, 9, eadg0366 CrossRef CAS.
- C. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS.
- X. Lin, X. Zhang, D. Liu, L. Shi, L. Zhao, Y. Long and L. Dai, Adv. Energy Mater., 2024, 14, 2303740 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- H. Ze, X. Chen, X.-T. Wang, Y.-H. Wang, Q.-Q. Chen, J.-S. Lin, Y.-J. Zhang, X.-G. Zhang, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2021, 143, 1318–1322 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Agnew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS.
- L. Liu, X. Rao, S. Zhang and J. Zhang, Chem, 2024, 10, 1994–2030 CAS.
- Y. Zhu, Y. Jiang, H. Li, D. Zhang, L. Tao, X.-Z. Fu, M. Liu and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202319370 CrossRef CAS.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Agnew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS.
- H. Yan, Y. Lin, H. Wu, W. Zhang, Z. Sun, H. Cheng, W. Liu, C. Wang, J. Li and X. Huang, Nat. Commun., 2017, 8, 1–11 CrossRef.
- Y. Pan, C. Zhang, Z. Liu, C. Chen and Y. Li, Matter, 2020, 2, 78–110 CrossRef.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS.
- Z. Wang, X. Jin, C. Zhu, Y. Liu, H. Tan, R. Ku, Y. Zhang, L. Zhou, Z. Liu, S.-J. Hwang and H. J. Fan, Adv. Mater., 2021, 33, 2104718 CrossRef CAS.
- X. Ao, H. Wang, X. Zhang and C. Wang, ACS Appl. Mater. Interfaces, 2025, 17, 2844–2862 CrossRef CAS.
- X. Jin, M. Chang, H. Sun, C.-W. Chang, M. G. Sendeku, Y. Li, M. Wang, J. Fang, Y. Li, Q. Zhu, B. Li, J. Yu, Y. Liu, Z. Chang, G. Zhang, Z. Zhuang, L. Bai, Q. Ma, Z. Feng, W. Liu, J. Li and X. Sun, J. Am. Chem. Soc., 2025, 147, 2689–2698 CrossRef CAS.
- M. Liu, J. Zhang, H. Su, Y. Jiang, W. Zhou, C. Yang, S. Bo, J. Pan and Q. Liu, Nat. Commun., 2024, 15, 1675 CrossRef CAS.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS.
- X. Wei, D. Zheng, M. Zhao, H. Chen, X. Fan, B. Gao, L. Gu, Y. Guo, J. Qin and J. Wei, Agnew. Chem., Int. Ed., 2020, 59, 14639–14646 CrossRef CAS.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS.
- H. Shen, E. Gracia-Espino, J. Ma, K. Zang, J. Luo, L. Wang, S. Gao, X. Mamat, G. Hu and T. Wagberg, Agnew. Chem., Int. Ed., 2017, 56, 13800–13804 CrossRef CAS.
- J. Liu, W. Chen, S. Yuan, T. Liu and Q. Wang, Energy Environ. Sci., 2024, 17, 249–259 RSC.
- W. Wang, Q. Jia, S. Mukerjee and S. Chen, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS.
- J. Liu, C. Fan, G. Liu and L. Jiang, Appl. Surf. Sci., 2021, 538, 148017 CrossRef CAS.
- X. Xie, Z. Zhai, L. Peng, J. Zhang, L. Shang and T. Zhang, Sci. Bull., 2023, 68, 2862–2875 CrossRef CAS.
- M. Xiao, H. Zhang, Y. Chen, J. Zhu, L. Gao, Z. Jin, J. Ge, Z. Jiang, S. Chen and C. Liu, Nano Energy, 2018, 46, 396–403 CrossRef CAS.
- N. Zhang, T. Zhou, J. Ge, Y. Lin, Z. Du, W. Wang, Q. Jiao, R. Yuan, Y. Tian and W. Chu, Matter, 2020, 3, 509–521 CrossRef.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao and S. Wei, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS.
- F. Gong, M. Liu, L. Gong, S. Ye, Q. Jiang, G. Zeng, X. Zhang, Z. Peng, Y. Zhang, S. Fang and J. Liu, Adv. Funct. Mater., 2022, 32, 2202141 CrossRef CAS.
- X. Ao, X. Xue, Z. Yang, Y. Yang and C. Wang, Mater. Today Energy, 2019, 12, 62–69 CrossRef.
- Y. Zhang, L. Guo, L. Tao, Y. Lu and S. Wang, Small Methods, 2019, 3, 1800406 CrossRef.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- J. S. Kang, J. Kang, D. Y. Chung, Y. J. Son, S. Kim, S. Kim, J. Kim, J. Jeong, M. J. Lee, H. Shin, S. Park, S. J. Yoo, M. J. Ko, J. Yoon and Y.-E. Sung, J. Mater. Chem. A, 2018, 6, 20170–20183 RSC.
- H. Li, Y. Wen, M. Jiang, Y. Yao, H. Zhou, Z. Huang, J. Li, S. Jiao, Y. Kuang and S. Luo, Adv. Funct. Mater., 2021, 31, 2011289 CrossRef CAS.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS.
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Nat. Commun., 2021, 12, 5589 CrossRef CAS.
- X. Ao, W. Zhang, Z. Li, J.-G. Li, L. Soule, X. Huang, W.-H. Chiang, H. M. Chen, C. Wang, M. Liu and X. C. Zeng, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Agnew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS.
- Y. Zhao, H.-C. Chen, X. Ma, J. Li, Q. Yuan, P. Zhang, M. Wang, J. Li, M. Li, S. Wang, H. Guo, R. Hu, K.-H. Tu, W. Zhu, X. Li, X. Yang and Y. Pan, Adv. Mater., 2024, 36, 2308243 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng and A. I. Rykov, Nat. Commun., 2018, 9, 1–12 CrossRef.
- Z. Zhang, J. Sun, F. Wang and L. Dai, Agnew. Chem., Int. Ed., 2018, 57, 9038–9043 CrossRef CAS.
- Z. Jiang, W. Sun, H. Shang, W. Chen, T. Sun, H. Li, J. Dong, J. Zhou, Z. Li and Y. Wang, Energy Environ. Sci., 2019, 12, 3508–3514 RSC.
- H. Funke, A. Scheinost and M. Chukalina, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094110 CrossRef.
- A. Grossmann, M. Holschneider, R. Kronland-Martinet and J. Morle, Adv. Electron. Electron Phys., 1987, 19, 298–306 Search PubMed.
- F. Gong, Z. Chen, C. Chang, M. Song, Y. Zhao, H. Li, L. Gong, Y. Zhang, J. Zhang, Y. Zhang, S. Wei and J. Liu, Adv. Mater., 2025, 37, 2415269 CrossRef CAS.
- F. Gong, M. Liu, S. Ye, L. Gong, G. Zeng, L. Xu, X. Zhang, Y. Zhang, L. Zhou, S. Fang and J. Liu, Adv. Funct. Mater., 2021, 31, 2101715 CrossRef CAS.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, 2003297 CrossRef CAS.
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Adv. Funct. Mater., 2019, 29, 1906174 CrossRef CAS.
- Z. Zhou, L. Zhao, J. Wang, Y. Zhang, Y. Li, S. Shoukat, X. Han, Y. Long and Y. Liu, Small, 2023, 19, 2302598 CrossRef CAS.
- C. Chang, X. Li, S. Wei, Y. Zhao, L. Gong, Y. Zhang, J. Liu and F. Gong, Adv. Energy Mater., 2025, 15, 2402825 CrossRef CAS.
- J. Yu, X. Yong and S. Lu, Energy Environ. Mater., 2024, 7, e12587 CrossRef CAS.
- X. Wang, X. Yang, C. Zhao, Y. Pi, X. Li, Z. Jia, S. Zhou, J. Zhao, L. Wu and J. Liu, Agnew. Chem., Int. Ed., 2023, 62, e202302829 CrossRef CAS.
- W. Xiao, K. Yoo, J. H. Kim and H. Xu, Adv. Sci., 2023, 10, 2303916 CrossRef CAS.
- G. Chen, Y. An, S. Liu, F. Sun, H. Qi, H. Wu, Y. He, P. Liu, R. Shi and J. Zhang, Energy Environ. Sci., 2022, 15, 2619–2628 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01457c |
| This journal is © The Royal Society of Chemistry 2025 |