Effect of silver deposits on the photocatalytic activity of titanium dioxide for the removal of 2-chlorophenol in water
Dmitry
Shchukin
*ab,
Elena
Ustinovich
c,
Dmitry
Sviridov
c and
Pierre
Pichat
b
aInstitute for Micromanufacturing, Louisiana Tech University, Ruston, LA 71272, USA. E-mail: shchukin@latech.edu
bLaboratoire Photocatalyse, Catalyse et Environnement, CNRS UMR (IFoS), Ecole Centrale de Lyon, BP 163, 69134 Ecully cédex, France
cBelarusian State University, 220050 Minsk, Belarus
First published on 27th August 2003
Abstract
The effect of Ag photodeposition on the rate of photocatalytic degradation of 2-chlorophenol (2-CP) in aqueous suspensions of Degussa P 25 titanium dioxide has been studied using HPLC. For Ag contents between 5 × 10−6 and 1.6 × 10−4 wt%, the rate was slowest in the absence of Ag and increased with increasing Ag content. For a Ag content of 6 × 10−4 wt%, the degradation rate was about 50% faster than with bare TiO2, and decreased for a Ag content of 7.2 × 10−4 wt% and beyond, eventually becoming slower than unmodified TiO2. Bare TiO2 has been compared with the 6 × 10−4 wt% Ag–TiO2 sample in terms of the variation of chlorohydroquinone (CHQ) and chloro-1,4-benzoquinone (CBQ) concentrations during 2-CP degradation, and also for the degradation of CHQ and CBQ separately. On the basis of these experiments, it is tentatively suggested that the net effect of Ag on 2-CP removal results from opposite influences, viz. the Ag+ cation-induced oxidation of CHQ to CBQ and the Ag0 particle-induced recombination of photoproduced charges, both influences being assumed to depend on the Ag particle size, itself related to the Ag content.
1 Introduction
The presence of harmful organic (especially aromatic) pollutants in wastewater effluents causes serious environmental problems, and purification of contaminated waters is one of the most interesting challenges in catalysis today. Over recent years, much attention has been directed towards heterogeneous photocatalytic detoxification, where water pollutants are degraded by irradiating aqueous suspensions of powdered semiconductor oxides, primarily focusing on TiO2 as a highly active and durable photocatalyst.1–5 In many cases, complete photocatalytic mineralisation of organic compounds in aqueous media has been reported. However, the quantum efficiency of photomineralisation on bare titania is relatively low because the photodegradation process, which involves interfacial transfer of photogenerated electrons and holes created within the semiconductor particles, competes with the recombination of these charge carriers. Therefore improvements in the photocatalytic activity of titania are required. Many studies have been devoted to increasing TiO2 photocatalytic activity by depositing noble metals on the surface. In particular, Ag nanoparticles were found to slightly improve the dehydrogenation and the oxidation of 2-propanol,6 as well as the degradation of chloroform,7 urea,7 1,4-dichlorobenzene,8 phenol9 and malic acid.10 Beneficial effects from Ag metal deposits were observed only at low silver contents.6In this study, the photocatalytic degradation of 2-chlorophenol (2-CP), one of the standard test aromatic pollutants used in TiO2 photocatalysis, has been investigated over Ag-loaded titania. Particular attention has been paid to the effect of the silver content on the formation and subsequent elimination of some aromatic intermediate products.
2 Experimental
The photochemical reactor was a 100 mL Pyrex cylindrical flask with an optical quartz window of 3.6 cm in diameter at the bottom. The radiation from a high pressure mercury lamp (Philips HPK 125 W) was filtered through a cuvette (thickness = 2.2 cm) full of water and a Corning 0.52 filter transmitting wavelengths above ca. 340 nm. The total radiant flux entering the photoreactor was approximately 40 mW cm−2. All photocatalytic tests were performed under continuous magnetic stirring. For each irradiation experiment, 40 mL of aerated aqueous 2-CP solution (10−3 mol L−1, pH 4.5) containing TiO2 (2 g L−1) was used. To equilibrate the starting system, the suspension of photocatalyst and 2-CP was first kept in darkness for 1.5 h before starting the irradiation. The percentage of 2-CP adsorbed on the TiO2 particles was < 4%.The concentrations of 2-CP and its intermediate products were measured by means of HPLC instrumentation comprising a LDC Constametric 3000 isocratic pump and a LDC Spectro Monitor D UV detector set at 254 nm. Separation of 2-CP and its aromatic intermediate products was achieved with a reverse-phase column (length 25 cm, inner diameter 4.6 mm) packed with Spherisorb 50DS2. The mobile phase was a ternary mixture of methanol (35 vol%), water (55 vol%) and acetonitrile. The intermediate products were identified by comparing their relative retention times with that of external standards.
The photoelectrochemical measurements were carried out by irradiating a stirred aqueous suspension of TiO2 at 365 nm under potentiostatic conditions, employing a standard three-electrode polarisation device. A platinum flag having an area of 1 cm2 was used as the collector electrode. Photocurrents were measured at +1 V vs. a Ag/AgCl reference electrode.
Silver nitrate, 2-CP, hydroquinone (HQ), chloro-2,4-dihydroxybenzene (chlorohydroquinone; CHQ), catechol (CT), chloro-1,4-benzoquinone (CBQ), hydroxy-1,4-benzoquinone (HBQ) and 1,2,4-trihydroxybenzene (hydroxyhydroquinone; HHQ) were supplied by Aldrich; all chemicals were of reagent grade and were used as received. The water employed in the experiments was purified using a three-stage Millipore Milli-Q Plus 185 system and had a resistivity higher than 18 MΩ cm.
Degussa P25 (∼80 wt% anatase, ∼20 wt% rutile, average particle size ca. 30 nm) titanium dioxide was used as the photocatalyst. A specific BET surface area of 51 m2 g−1 was determined with a Micromeretics Gemini II 2375 surface area analyser. To prepare Ag–TiO2 samples, 50 mL of a 10−3 mol L−1 AgNO3 solution containing 100 mg of P25 TiO2 was irradiated with UV light for 2 s to 4 min; the suspension was then filtered off, thoroughly washed and dried at 333 K. The amount of silver deposited was measured by conventional chemical analysis of the solid. The deposition of silver produced a slight increase in the surface area (up to 58 m2 g−1)
3 Results and discussion
Fig. 1 shows the influence of silver loading on the photocatalytic removal of 2-CP. The deposition of ∼5 × 10−6 wt% of silver resulted in a ca. 3-fold decrease in the 2-CP removal rate. At higher silver contents, the rate increased, but remained lower than that observed with bare TiO2 for a loading of 1.6 × 10−4 wt% (plot 3 in Fig. 1). It reached a maximum at 6 × 10−4 wt% (referring to the Ag contents studied here); this maximum corresponds to a ca. 1.5-fold higher 2-CP removal rate with respect to bare TiO2. The photocatalytic activity diminished by a factor of about five on increasing the Ag content from 6 × 10−4 to 9 × 10−4 wt% [Fig. 1(b)]. | ||
| Fig. 1 (a) Photocatalytic removal of 2-CP as a function of time over bare TiO2 (1), 5 × 10−6 wt% Ag–TiO2 (2), 1.6 × 10−4 wt% Ag–TiO2 (3) and 6.0 × 10−4 wt% Ag–TiO2 (4). (b) Photocatalytic removal of 2-CP after 90 min irradiation as a function of Ag loading. | ||
It has been shown previously using the HPLC analysis9 that the primary aromatic intermediate products of 2-CP in irradiated TiO2 aqueous suspensions are CHQ and, to a lesser extent, CT.11 CHQ is further oxidised to CBQ and, subsequently, to HBQ.11 These quinones can act as electron scavengers,12 competing with molecular oxygen for photogenerated electrons. As a result, back reaction to CHQ and HHQ occurs, producing the so-called photocatalytic short-circuit effect,13 which, at least in part, might account for the relatively low yields for the TiO2-photosensitised degradation of chlorophenols and other phenols.11,13–15
The variations in the concentrations (divided by the concentration of 2-CP that has decomposed at that particular time) of two main aromatic intermediate products (CHQ and CBQ) during 2-CP degradation are shown in Fig. 2. The kinetic curves show that the 6 × 10−4 wt% Ag loading results in a shift of the quinone–hydroquinone equilibrium towards the more oxidised product. This observation clearly illustrates the greater oxidising power of this Ag-loaded sample, a fact which is further confirmed by the higher 2-CP removal rate (Fig. 1) using this catalyst.
![Normalised CHQ and CBQ concentrations versus irradiation time over bare TiO2
(1, 1′), 6 × 10−4 wt% and Ag–TiO2
(2, 2′). Initial concentration of 2-CP: [2-CP]0
= 1 m mol L−1.](/image/article/2004/PP/b306077b/b306077b-f2.gif) | ||
| Fig. 2 Normalised CHQ and CBQ concentrations versus irradiation time over bare TiO2 (1, 1′), 6 × 10−4 wt% and Ag–TiO2 (2, 2′). Initial concentration of 2-CP: [2-CP]0 = 1 m mol L−1. | ||
In an effort to better understand the net favourable effect of Ag loading at 6 × 10−4 wt% on the degradation rate of 2-CP, analogous experiments were performed for the photocatalytic degradation of CHQ (Fig. 3) and CBQ (Fig. 4), separately, under the same experimental conditions as for 2-CP decomposition. Fig. 3 confirms the oxidation of CHQ to CBQ.13 Any occurrence of the back reaction cannot be inferred from this experiment. Fig. 3 also shows that this Ag–TiO2 specimen is more active than TiO2 for CHQ degradation, as for 2-CP decomposition. The photocatalytic degradation of CBQ yielded more informative results. Fig. 4 very clearly indicates that, in spite of the oxidation conditions, CBQ is reduced to CHQ within the first few minutes of irradiation under our conditions. The concentrations measured after 5 min of irradiation indicate that this reduction is even almost quantitative over bare TiO2. Also, the aforementioned short-circuit effect is evidenced by the nearly constant CBQ concentration between 5 and 30 min when using bare TiO2 as the photocatalyst. The presence of Ag deposits produces a decrease in the CBQ reduction rate, as is clearly shown by the initially lower rate of CBQ removal, as well as by the lower concentrations of CHQ over the entire duration of the experiment. The greater oxidising power of this Ag-loaded sample is also corroborated by the lower value of the sum of the concentrations of CBQ and CHQ during the whole experiment.
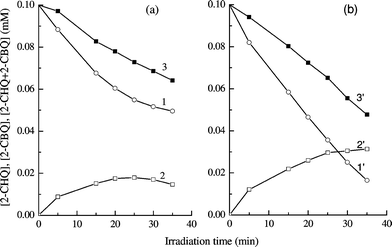 | ||
| Fig. 3 Photocatalytic degradation of CHQ in bare TiO2 (a) and 6 × 10−4 wt% Ag–TiO2 (b) suspensions. Comparison of CHQ elimination (1, 1′), CBQ concentration (2, 2′) and the sum of the concentrations of CHQ and CBQ (3, 3′). Initial concentration of CHQ: 0.1 mmol L−1; TiO2 concentration: 2 g L−1. | ||
 | ||
| Fig. 4 Photocatalytic degradation of CBQ in bare TiO2 (a) and 6 × 10−4 wt% Ag–TiO2 (b) suspensions. Comparison of CBQ elimination (1, 1′), CHQ concentration (2, 2′) and the sum of the concentrations of CHQ and CBQ (3, 3′). Initial concentration of CBQ: 0.1 mmol L−1; TiO2 concentration: 2 g L−1. | ||
The Ag deposits can play at least two roles. As in photographic development,16 Ag+ cations can effect the oxidation of hydroquinones (here CHQ) into quinones (here CBQ) and, accordingly, intervene positively in the degradation pathway of 2-CP (Fig. 5). The Ag metallic particles can attract the charge carriers and act as recombination centres, therefore decreasing the rate of photocatalytic removal of 2-CP and its intermediate degradation products. Fig. 1 shows that the net effect resulting from these opposite influences very much depends on the amount of silver deposited on the TiO2. When the amount is very small, that is, when the irradiation time of TiO2 in the AgNO3 solution is very short, tiny silver particles are formed. It has been proposed17 that “shallow” surface states are thus created in TiO2, which increases the rate of recombination of the photoproduced charges.18 We tentatively suggest that the increase in the Ag particle size for increasing Ag contents19 leads to deeper surface states related to these particles and thereby diminishes the charge recombination rate. For 6.0 × 10−4 wt% of silver, the net favourable effect reflects the dominant influence of the Ag-induced oxidation of CHQ (Fig. 3). For [Ag] ≥ 6.0 × 10−4 wt% the net unfavourable effect principally arise from an increased difficulty for bigger Ag metallic particles to supply Ag+ cations.
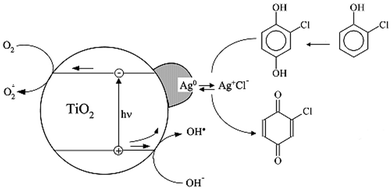 | ||
| Fig. 5 Schematic illustration of 2-CP photocatalytic degradation on Ag-modified TiO2 photocatalyst. | ||
4 Conclusion
This study shows that, at least in the particular case of benzenic pollutants, Ag deposits can enhance the photocatalytic activity of TiO2 for water purification. The Ag content must be finely adjusted and, presumably, the deposition method also plays a role. The stability over time also needs to be investigated. These results suggest a way of improving photocatalytic water treatment.Previous conclusions12,13 on the role of quinones in the photocatalytic degradation of benzenic pollutants are supported by this work. Our interpretations remain tentative, as further work is necessary to characterise the Ag–TiO2 samples.
Acknowledgement
D. G. S. gratefully acknowledges the support of INTAS (grant YSF00-161) which, in particular, allowed him to carry out the degradation experiments in Lyon.References
- P. Pichat, Heterogeneous photocatalysis, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, 1997, vol. 4, pp. 2111–2122 Search PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Environmental application of semiconductor catalysts, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- D. Bahnemann, J. Cunningham, M. A. Fox, E. Pelizzetti, N. Serpone and P. Pichat, Photocatalytic treatment of waters, in Aquatic and Surface Photochemistry, ed. G. R. Helz, R. G. Zepp and D. G. Crosby, Lewis Publishing, Boca Raton, 1994, pp. 261–316 Search PubMed.
- A. Mills and S. Le Hunte, An overview of semiconductor photocatalysis, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- A. Sclafani, M.-N. Mozzanega and P. Pichat, Effect of silver deposits on the photocatalytic activity of titanium dioxide samples for the dehydrogenation or oxidation of 2-propanol, J. Photochem. Photobiol., A, 1991, 59, 181–189 CrossRef CAS.
- M. M. Kondo and W. F. Jardim, Photodegradation of chloroform and urea using Ag-loaded titanium dioxide as catalyst, Water Res., 1991, 25, 823–829 CrossRef CAS.
- J.-M. Herrmann, H. Tahiri, Y. Ait-Ichou, G. Lassaletta, A. R. González-Elipe and A. Fernández, Characterization and photocatalytic activity in aqueous medium of TiO2 and Ag–TiO2 coatings on quartz, Appl. Catal., B., 1997, 13, 219–228 CrossRef CAS.
- J.-C. D'Oliveira, G. Al-Sayyed and P. Pichat, Photodegradation of 2- and 3-chlorophenol in titanium dioxide aqueous suspensions, Environ. Sci. Technol., 1990, 24, 990–996.
- W. Lee, H.-S. Shen, K. Dwight and A. Wold, Effect of silver on the photocatalytic activity of TiO2, J. Solid State Chem., 1993, 106, 288–294 CrossRef CAS.
- R. M. Alberici and W. F. Jardim, Photocatalytic degradation of phenol and chlorinated phenols using Ag–TiO2 in a slurry reactor, Water Res., 1994, 28, 1845–1849 CrossRef CAS.
- C. Richard, Photocatalytic reduction of benzoquinone in aqueous ZnO or TiO2 suspensions, New J. Chem., 1994, 18, 443–445 Search PubMed.
- J. Theurich, M. Lindner and D. W. Bahnemann, Photocatalytic degradation of 4-clorophenol in aerated aqueous titanium dioxide suspensions: a kinetic and mechanistic study, Langmuir, 1996, 12, 6368–6376 CrossRef CAS.
- K. Okamoto, Y. Yamamoto, M. Tanaka and A. Itaya, Heterogeneous photocatalytic decomposition of phenols over TiO2 powder, Bull. Chem. Soc. Jpn., 1985, 58, 2015–2022 CAS.
- J.-C. D'Oliveira, C. Minero, E. Pelizzetti and P. Pichat, Photodegradation of dichlorophenols and trichlorophenols in TiO2 aqueous suspensions: kinetic effects of the positions of the Cl atoms and identification of the intermediates, J. Photochem. Photobiol., A, 1993, 72, 261–269 CrossRef CAS.
- Young Ku, Ren-Ming Leu, and Kuen-Chyr Lee, Decomposition of 2-chlorophenol in aqueous solution by UV irradiation with the presence of titanium dioxide, Water Res., 1996, 30, 2569–2579 CrossRef CAS.
- W. E. Lee and E. R. Brown, The developing agents and their reactions, in The Theory of the Photographic Process, ed. T. H. James, MacMillan, New York, 1977, pp. 291–331 Search PubMed.
- A. I. Kulak, A. I. Kokorin and D. V. Sviridov, Electrolyte electroreflectance study of TiO2 films modified with metal nanoparticles, J. Mater. Res., 2001, 16, 2357–2361 CAS.
- E. S. Brandt, Mechanistic studies of silver image stability. 1: redox chemistry of oxygen and hydrogen peroxide at clean and adsorbate-covered silver electrodes, Photogr. Sci. Eng., 1984, 28, 1–12 Search PubMed.
- J. M. Herrmann, J. Disdier and P. Pichat, Photocatalytic deposition of silver on powder titania. Consequences for the recovery of silver, J. Catal., 1988, 113, 72–74 CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |