Synthetic anisomycin analogues activating the JNK/SAPK1 and p38/SAPK2 pathways
Received 15th September 2003, Accepted 21st November 2003
First published on 2nd December 2003
Abstract
The synthesis of C(4)H and C(4)Me analogues of the JNK/p38 pathway activator anisomycin, based upon an aldol or Claisen construction of the C(3)–C(4) bond, has been demonstrated. The relative activation of the JNK/SAPK1 and p38/SAPK2 pathways in RAW macrophages by these analogues, and their synthetic precursors, has been assessed using immunoblot assays against phosphorylated c-Jun and MAPKAP-K2. These studies demonstrate that some of the synthetic C(4) analogues are also potent activators of these stress kinase pathways.
Introduction
The pyrrolidine antibiotic anisomycin (1) has long been known to act as a peptidyl transferase inhibitor, binding to the 60S ribosomal subunit in eukaryotes, and has been shown to exhibit selective action against protozoa and several strains of fungi.1 Since its introduction a little over 10 years ago2,3 as a tool for the activation of stress kinase pathways at “sub-inhibitory” levels,† the precise target(s) of the pyrrolidine antibiotic in mammalian, insect, or yeast cells that triggers the activation of these signalling pathways has not been elucidated. Biochemical and genetic studies demonstrate that the stress-activated kinase (SAPK) pathways regulate processes such as cellular responses to stress and infection, as well as cell repair/apoptosis.3 Hence, studies into the mechanism of initiation and regulation of these pathways may provide new leads for therapeutic targets.Whilst the cellular targets of anisomycin are not currently known, biochemical and genetic studies have yielded information as to its downstream effects on the stress-activated mitogen-activated protein (MAP) kinases.4 Thus treatment of mammalian cells with anisomycin at sub-inhibitory, as well as inhibitory levels is known to strongly activate the protein kinases JNK/SAPK1 (c-Jun NH2-terminal kinase/stress-activated protein kinase) and p38/SAPK2.3
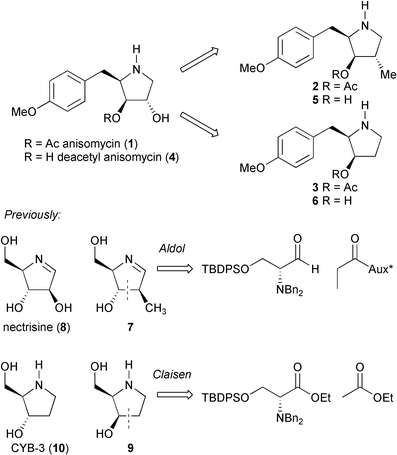
Following our successful synthesis of anisomycin (1) utilizing a highly diastereoselective syn glycolate aldol reaction with a tyrosine-derived α-dibenzylamino aldehyde,5 we targeted our efforts towards the production of a range of anisomycin derivatives for biological testing. Previous reports have shown that anisomycin's activity against a range of tumour cell lines6 is largely unaffected by modest changes to the substitutent at the C(4) position. We hypothesised that the C(4)Me (2) and C(4)H (3) analogues of anisomycin (Fig. 1) might show enhanced levels of activation of the JNK/SAPK1 and p38/SAPK2 pathways due to their more lipophilic nature. Similarly, the C(4)Me and C(4)H analogues of deacetyl anisomycin (4)
(5 and 6 respectively) might allow us to address the level to which the reduced activity of deacetyl anisomycin (4)‡ is associated with the loss of interactions with this ester functionality rather than changes in the molecule's polarity.
 |
| | Fig. 1 Analogues of anisomycin, nectrisine and CYB-3. | |
Results and discussion
Analogue preparation
We have recently reported the synthesis of the C(4)Me derivative 7 of nectrisine (8) using boron mediated aldol chemistry,7 and the epimer 9 of the natural product CYB-3 (10) using Claisen chemistry (Fig. 1).8 We were therefore confident that similar synthetic approaches would allow us to access the C(4)Me, and C(4)H analogues of anisomycin. Our previous synthesis of anisomycin (1) itself, made use of a ‘mismatched’ glycolate aldol reaction for the key C(3)–C(4) bond construction.5 Adaptation of this approach, with the propionate derivative of Evans oxazolidinone 11 in place of the previous protected glycolate equivalent, allowed the synthesis of aldol adduct 12 from aldehyde 13§ in excellent yield (87%) and as a single diastereomer (Scheme 1). Borohydride reduction of aldol adduct 12 to give diol 14, and selective tosylation of the primary alcohol resulted in spontaneous cyclisation to give the pyrrolidinium tosylate salt. This salt was converted to its chloride counterpart 15 by purification through Dowex Cl− exchange resin in excellent yield (83%). Selective mono-deprotection5 of the salt allowed facile acylation of the free hydroxyl in 16 to give the benzyl protected precursor 17 of the C(4)Me analogue. Protected pyrrolidine 17 was subjected to hydrogenolysis in the presence 1 M hydrochloric acid to allow the isolation of the C(4)Me analogue 2 as its hydrochloride salt in quantitative yield (13 steps, 36% overall yield from D-tyrosine). The C(4)Me analogue 5 of deacetyl anisomycin was obtained from the pyrrolidinium salt 15 directly in quantitative yield by exhaustive debenzylation and treatment with Dowex HO− exchange resin (11 steps, 47% overall yield from D-tyrosine).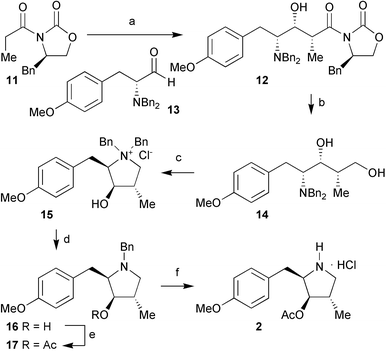 |
| | Scheme 1 Reagents and conditions:
(a)
(i) Bu2BOTf, NEt3, CH2Cl2, 0 °C, 15 min; (ii)
13, −78 °C → 0 °C, 5 h (87%); (b) LiBH4, CH3OH, THF, 0 °C → rt, 18 h (82%); (c)
(i) TsCl, DMAP, CH2Cl2, 0 °C, 18 h; (ii) Dowex Cl−
(83%); (d) H2, 5% Pd/C, K2CO3, CH3OH, rt, 20 min (93%); (e) Ac2O, NEt3, CH2Cl2, rt, 18 h (82%); (f) H2, 20% Pd(OH)2/C, 1 M HCl–Et2O, CH3OH, rt, 18 h (100%). | |
In tackling the synthesis of the C(4)H analogue of anisomycin using a Claisen approach, we were aware of the potential for epimerisation at the α-stereocentre in ester 18,¶ during conversion via acid 19 to the more reactive acylimidazole intermediate 20
(Scheme 2).8 To our relief, chiral HPLC analysis of the syn
β-hydroxy ester diastereomer 22
(obtained after sodium cyanoborohydride reduction of β-keto ester 21) and comparison with traces from the corresponding racemic series, indicated that in the case of the tyrosine-derived ester there is no epimerisation during this reaction sequence (alcohol 22 >98% ee). Even more pleasingly, the cyanoborohydride conditions for ketone reduction which had been shown to give optimum diastereoselectivity in the synthesis of 9,8 resulted in even higher selectivity in this case (>40 : 1 ratio syn : anti reduction products). Subsequent lithium aluminium hydride reduction of the ester 22 to give the required diol precursor to the pyrrolidine ring proceeded cleanly and in excellent yield (95%). However, monoprotection of the primary alcohol functionality in this diol was not possible using toluene sulfonyl chloride (TsCl) due to competing reaction of the secondary hydroxyl which is significantly less hindered in this case than in diol 14. To counter this, the more sterically demanding triisopropylbenzene sulfonyl chloride (TIBSCl) was employed. Using this reagent, selective sulfonylation of the primary alcohol was achieved and in situ cyclisation to the pyrrolidinium tosylate salt was observed. Treatment with Dowex Cl− exchange resin facilitated purification of the pyrrolidinium salt 23
(81% from β-hydroxy ester 22).|| Monodebenzylation to give alcohol 24, acylation to give 25 and final benzyl deprotection were achieved as in the C(4)Me series allowing the isolation of the C(4)H analogue (3) as its hydrochloride salt (12 steps, 33% overall yield from D-tyrosine). Similarly, direct conversion of pyrrolidinium salt 23 to the C(4)H analogue 6 of deacetyl anisomycin was achieved in high yield (95%) by exhaustive debenzylation and treatment with Dowex HO− exchange resin (10 steps, 45% overall yield from D-tyrosine).
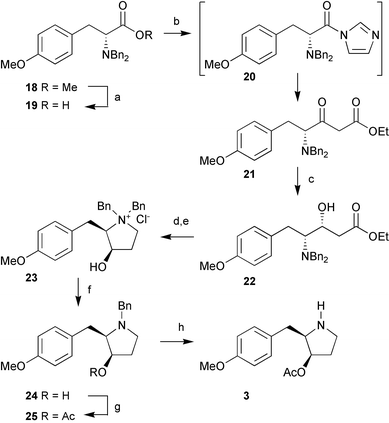 |
| | Scheme 2 Reagents and conditions:
(a) LiOH, THF–H2O, reflux, 18 h (95%); (b)
(i) CDI, THF, rt, 2.5 h, (ii) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OLi)OEt, −78 °C, 2 h (82%); (c) NaCNBH3, AcOH, Et2O, CH3OH, 0 °C → rt, 18 h (83 %); (d) LiAlH4, THF, −78 °C, 6 h (95%); (e)
(i) TIBSCl, DMAP, CH2Cl2, 0 °C, 18 h; (ii) Dowex Cl−
(85%); (f) H2, 5% Pd/C, K2CO3, CH3OH, rt, 20 min (80%); (g) Ac2O, NEt3, CH2Cl2, rt, 18 h (87%); (h) H2, 20% Pd(OH)2/C, 1 M HCl/–Et2O, CH3OH, rt, 20 h (100%). C(OLi)OEt, −78 °C, 2 h (82%); (c) NaCNBH3, AcOH, Et2O, CH3OH, 0 °C → rt, 18 h (83 %); (d) LiAlH4, THF, −78 °C, 6 h (95%); (e)
(i) TIBSCl, DMAP, CH2Cl2, 0 °C, 18 h; (ii) Dowex Cl−
(85%); (f) H2, 5% Pd/C, K2CO3, CH3OH, rt, 20 min (80%); (g) Ac2O, NEt3, CH2Cl2, rt, 18 h (87%); (h) H2, 20% Pd(OH)2/C, 1 M HCl/–Et2O, CH3OH, rt, 20 h (100%). | |
With C(4) anisomycin analogues 2 and 3, and C(4) deacetyl anisomycin analogues 5 and 6 in hand, and the corresponding N-benzyl derivatives of these (17, 25, 16 and 24 respectively) we were well-placed to begin investigation of the JNK/SAPK1 and p38/SAPK2 pathway activation by each of these. To this collection were added synthetic samples of anisomycin 1, deacetyl anisomycin 4, and their benzylated derivatives 26 and 27
(Fig. 2) prepared by our previously reported methodology,5 as well as a final synthetic sample of 3097-B1 (28) which has been isolated from the fermentation broths of Streptomyces strain SA3097 along with anisomycin,9 and which was prepared by a similar route.
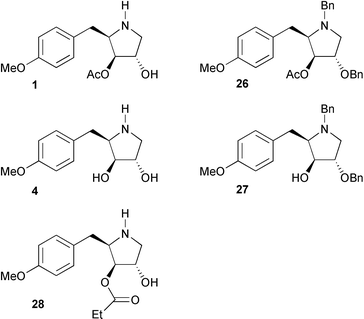 |
| | Fig. 2 Further analogues of anisomycin and deacetyl anisomycin. | |
In total this gave 13 synthetic samples for study in the activation of the stress activated protein kinase pathways. Using these aldol- and Claisen-based approaches more than 20 mg of each sample was available for the biological assays described.
SAPK pathway activation
Although the cellular target(s) of anisomycin is(are) not currently known, its downstream effects on the SAPKs have been determined (Fig. 3). Thus, treatment of mammalian cells with anisomycin at sub-inhibitory levels is known to strongly activate JNK/SAPK1 and p38/SAPK2, resulting in phosphorylation of their substrates, such as the nuclear transcription factor c-Jun and the protein kinase MAPKAP-K2 respectively.3 Commercial antibodies against both the phosphorylated and non-phosphorylated states of c-Jun and MAPKAP-K2 have recently become available, making it simple to assess the differential activation of each of these pathways by the current analogue set.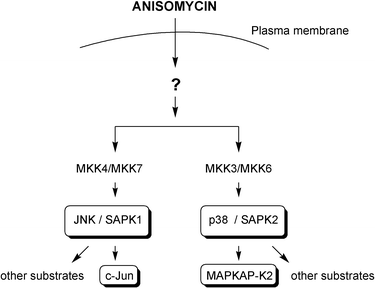 |
| | Fig. 3 SAPK pathway activation by anisomycin. | |
In order to assess the relative levels of pathway activation by each of the anisomycin analogues, murine RAW macrophage cell cultures were stimulated with the analogues for 30 minutes before cell lysis, and assessment of protein content by Bradford assay (Scheme 3). SDS PAGE separation of the lysates and transfer to a nitrocellulose membrane generated the appropriate protein displays for immunoblot assay. The immunoblot assay was carried out with antibodies that recognise all forms of the c-Jun protein (c-Jun), c-Jun phosphorylated at Ser63 (Ser63), all forms of MAPKAP-K2 and MAPKAP-K2 phosphorylated at Thr334 (Thr334) for each of the analogues.
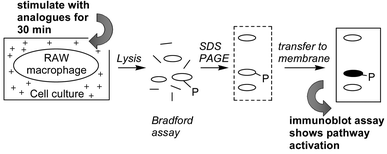 |
| | Scheme 3 Immunoblot assay of SAPK pathway activation. | |
The results of the immunoblot assays are shown in Figs 4 and 5. In each case the levels of pathway activation may be compared against those of commercially available anisomycin (Sigma) and a blank in which the analogues, which were (dissolved in DMSO) were replaced by the equivalent volume of DMSO (see Experimental).
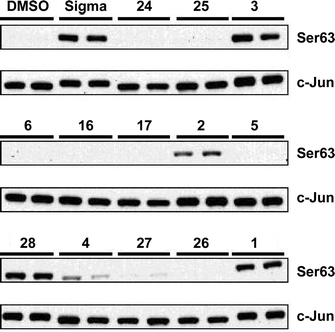 |
| | Fig. 4 Effect of anisomycin and its analogues on the phosphorylation of c-Jun in RAW macrophages. The cells were exposed to anisomycin (Sigma) or the anisomycin analogues indicated, each dissolved in DMSO, or to DMSO alone as a control. The cells were lysed and an aliquot (20 µg of lysate protein) was denatured in SDS, subjected to electrophoresis on a 10% polyacrylamide gel, transferred to a nitrocellulose membrane and immunoblotted with an antibody that recognises c-Jun phosphorylated at Ser63 (Ser63), or with an antibody that recognises phosphorylated and unphosphorylated c-Jun equally well (c-Jun). Further details are given in the Experimental. | |
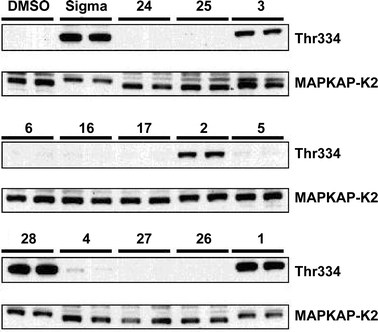 |
| | Fig. 5 Effect of anisomycin and its analogues on the phosphorylation of MAPKAP-K2 in RAW macrophages. The experiment was carried out as described in the legend to Fig. 4, except that the gels were immunoblotted with an antibody that recognises MAPKAP-K2 phosphorylated at Thr334 (Thr334), or with an antibody that recognises the phosphorylated and unphosphorylated forms of MAPKAP-K2 equally well. Further details are given in the Experimental. | |
These assays indicate that the C(4)–H analogue 3 is a particularly good activator showing similar levels of activation to the natural product itself, whilst the C(4)–Me analogue 2 is less effective. Acylation is essential for SAPK activation, although both acetate [anisomycin (1)] and propionate [3097-B1 (28)] esters show similar levels of activity. Deacetyl anisomycin 4 shows a dramatic reduction in both JNK/SAPK1 and p38/SAPK2 pathway activation. O- or N-benzylation reduces activation further, and the only analogue to show any pathway activation in this category is 27
(very weak activation of the JNK/SAPK1 pathway).
The activation of the S6 kinase pathway,10 by these analogues has also been tested using immmunoblot assays against ribosomal protein S6 phosphorylated at Ser235 and has been shown to give similar results.
Conclusions
We have demonstrated rapid and efficient routes to the chemical synthesis of C(4) analogues of anisomycin (1) and deacetyl anisomycin (4). Several of these analogues show similar, or only slightly reduced levels of activation of JNK/SAPK1 and p38/SAPK2 pathways relative to the natural product itself. Since differential acylation of the C(3) and C(4) hydroxyl groups has led to lengthy syntheses of anisomycin analogues in the past, the discovery of an active C(4)H analogue is of particular interest for future studies. These results also suggest ways of coupling anisomycin to insoluble supports to generate the matrices required for affinity purification of the anisomycin receptors from cell extracts.Experimental
Analogue preparation
All reactions involving air- or water-sensitive reagents were carried out under an atmosphere of argon using flame- or oven-dried glassware. Unless otherwise noted, starting materials and reagents were obtained from commercial suppliers and were used without further purification. THF was distilled from Na-benzophenone ketyl immediately prior to use. Toluene, CH2Cl2, and Et3N were distilled from calcium hydride. Anhydrous methanol, DMF, and acetonitrile were used as supplied by Acros. Unless otherwise indicated, organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure using a rotary evaporator. Purification by flash column chromatography was carried out using Merck Kieselgel 60 silica gel as the stationary phase. Chiral HPLC was performed using a Waters instrument equipped with a UV detector and a Chiracel OD-H column (internal diameter 4.6 mm, column length 250 mm, flow rate 0.5 cm3 min−1). Preparative HPLC was carried out using a Gilson instrument equipped with an RI detector and a Spherisorb column (internal diameter 25 mm, column length 250 mm, flow rate 9 cm3 min−1). All solvents for use in HPLC analysis were vacuum filtered and degassed prior to use. IR spectra were measured on a Biorad FTS-7 or Perkin-Elmer Paragon 1000 FT-IR spectrometer as thin films unless otherwise stated. 1H and 13C NMR spectra were measured on a Varian Gemini 200, Bruker AC250, Bruker AM360 or Varian Inova 600 spectrometer; J values are in Hz. Melting points were determined on a Gallenkamp Electrothermal Melting Point apparatus and are uncorrected. Optical rotations were measured on an AA-1000 polarimeter with a path length of 1.0 dm, at the sodium D-line at room temperature. Elemental analysis was carried out on a Perkin-Elmer 2400 CHN Elemental Analyser. Fast atom bombardment (FAB) mass spectra were obtained using a Kratos MS50TC mass spectrometer at The University of Edinburgh.(2′R,3′R,4R,4′R)-3-(4′-N,N-Dibenzylamino-3′-hydroxy-5′-(4-methoxyphenyl)-2′-methyl-1′-oxopentyl)-4-phenylmethyloxazolidin-2-one 12. To a solution of propionate equivalent 11
(0.202 g, 0.858 mmol) in CH2Cl2
(3 cm3) at 0 °C was added, dropwise, dibutylboron triflate (0.92 cm3, 1.0 M in CH2Cl2, 0.92 mmol) followed by triethylamine (0.10 g, 0.13 cm3, 0.95 mmol). The solution was stirred at 0 °C for 15 min, then cooled to −78 °C and a −78 °C solution of aldehyde 13
(0.200 g, 0.557 mmol) in CH2Cl2
(3 cm3) was added dropwise via cannula. The reaction mixture was stirred at −78 °C for 1 hour, and was allowed to warm to 0 °C over a period of 4 h. The reaction was quenched at 0 °C by the addition of methanol (5 cm3) followed by pH 7 phosphate buffer (1 cm3). Hydrogen peroxide (30% aq. solution; 5 cm3) in methanol (5 cm3) was added dropwise to the solution and the mixture was stirred and allowed to warm to room temperature over ca. 1 hour. The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 20 cm3); the combined organic phase was washed sequentially with saturated aq. sodium bicarbonate (25 cm3) and brine (25 cm3) then dried, and concentrated under reduced pressure. The residue was chromatographed using preparative HPLC [hexane : EtOAc (7 : 3)] to give 12
(0.144 g, 87%) as an oil, Rf
[hexane : EtOAc (7 : 3)] 0.40; Rt
[hexane : EtOAc (7 : 3)]
= 11.08 minutes; [α]D
−48.7 (c 2.00, CHCl3); νmax
(neat)/cm−1 3370, 2929, 1773, 1703, 1612, 1511; δH
(200 MHz; CDCl3) 7.36–6.90 (17H, m), 6.87 (2H, d, J 8.7), 4.65–4.58 (2H, m), 4.22–4.09 (2H, m), 3.91 (1H, dd, J 9.0, 2.5), 3.87 (2H, d, J 12.8), 3.81 (3H, s), 3.72 (1H, qd, J 6.9, 2.5), 3.40 (2H, d, J 12.8), 3.33 (1H, dd, J 13.3, 3.0), 3.13–2.97 (2H, m), 2.85–2.76 (1H, m), 2.67 (1H, dd, J 13.3, 10.0), 0.61 (3H, d, J 6.9); δC
(62.9 MHz, CDCl3) 175.3, 158.5, 153.8, 139.0 (2C), 136.0, 132.3, 130.7 (2C), 129.9 (2C), 129.5 (4C), 129.3 (2C), 129.0 (4C), 127.8 (2C,), 127.7, 114.4 (2C), 70.5, 66.6, 61.6, 56.4, 55.7, 54.3 (2C), 40.6, 38.1, 31.2, 8.7; m/z
(FAB) 593 ([M + H]+, 59%), 307 (16), 154 (100), 136 (70), 121 (16), 91 (51); HRMS (FAB)
(Found: [M + H]+, 593.3018. C37H41N2O5 requires m/z, 593.3015).
(2S,3R,4R)-4-N,N-Dibenzylamino-3-hydroxy-5-(4-methoxyphenyl)-2-methylpentan-1-ol 14. To a solution of aldol adduct 12
(0.281 g, 0.475 mmol) in THF (20 cm3) at 0 °C was added methanol (0.076 g, 0.096 cm3, 2.4 mmol) and LiBH4
(0.052 g, 1.1 mmol). The solution was warmed to room temperature and stirred for 18 hours then re-cooled to 0 °C and quenched by the addition of 1 M aqueous sodium hydroxide (2 cm3). The organic phase was separated, and the aqueous phase was extracted with CH2Cl2
(3 × 20 cm3); the combined organic phase was washed with brine (30 cm3), dried, and concentrated under reduced pressure. The residue was chromatographed on silica gel [hexane : EtOAc (7 : 3)] to give the diol 14
(0.162 g, 82%) as an oil, Rf
[hexane : EtOAc (7 : 3)] 0.10; [α]D
−31.72 (c 1.95, CHCl3); νmax
(neat)/cm−1 3397, 3027, 2933, 1611, 1583, 1512; δH
(250 MHz; CDCl3) 7.34–7.10 (10H, m), 7.13 (2H, d, J 8.7), 6.88 (2H, d, J 8.7), 4.74 (1H, s), 3.90–3.78 (1H, m), 3.88 (2H, d, J 12.7), 3.82 (3H, s), 3.68 (1H, dd, J 10.6, 4.2), 3.58 (1H, dd, J 10.6, 6.0), 3.36 (2H, d, J 12.7), 3.07–2.97 (1H, m), 3.02 (1H, dd, J 17.2, 9.1), 2.67 (1H, s), 2.55 (1H, dd, J 17.2, 8.4), 1.52 (1H, m), 0.39 (3H, d, J 6.8); δC
(62.9 MHz, CDCl3) 158.0, 138.4 (2C), 131.7, 130.0 (2C), 129.0 (4C), 128.3 (4C), 127.2 (2C), 113.9 (2C), 71.6, 67.6, 60.3, 55.1, 53.5 (2C), 35.8, 31.7, 8.2; m/z
(FAB) 420 ([M + H]+, 67%), 330 (66), 298 (62), 240 (21), 208 (15), 121 (75), 91 (100); HRMS (FAB)
(Found: [M + H]+, 420.2538. C27H34NO3 requires m/z, 420.2539).
(2R,3R,4S)-1,1-Dibenzyl-3-hydroxy-2-(4-methoxybenzyl)-4-methylpyrrolidinium chloride 15. To a solution of the diol 14
(0.125 g, 0.297 mmol) in CH2Cl2
(10 cm3) at 0 °C was added DMAP (0.163 g, 1.34 mmol) and TsCl (0.170 g, 0.892 mmol). The solution was stirred for 18 hours then diluted with CH2Cl2
(25 cm3) and water (25 cm3). The organic phase was separated and washed with 1% aqueous hydrochloric acid (2 × 25 cm3) then dried and concentrated under reduced pressure. The residue was chromatographed on silica gel [CH2Cl2 : MeOH (100 : 0)
→
(95 : 5)] to give a white foam. The salt obtained was subjected to ion exchange chromatography (Dowex Cl−; prepared by treatment of Dowex 1-X2 with 1% aqueous hydrochloric acid followed by flushing with methanol until the eluent returned to pH 7) eluting with methanol to give the chloride salt 15
(0.108 g, 83%) as an amorphous solid, Rf
[CH2Cl2 : MeOH (95 : 5)] 0.05; [α]D
−41.3 (c 3.08, CHCl3); νmax
(neat)/cm−1 3182, 2960, 1612, 1513; δH
(360 MHz; CDCl3) 7.76 (2H, d, J 7.0), 7.44–7.30 (10H, m), 6.83 (1H, d, J 3.6), 6.69 (2H, d, J 8.6), 5.78 (1H, d, J 13.4), 5.24 (1H, d, J 13.0), 5.09 (1H, d, J 13.0), 4.14 (1H, d, J 13.4), 3.95–3.92 (2H, m), 3.69 (3H, s), 3.66–3.56 (2H, m), 3.31–3.28 (1H, m), 3.26–3.04 (1H, m), 2.56 (1H, br t, J 11.1), 0.91 (3H, d, J 6.8); δC
(62.9 MHz; CDCl3) 158.2, 133.4 (2C), 133.2 (2C), 130.5 (3C), 130.2, 129.1 (2C), 129.0 (2C), 127.8, 127.4, 126.8, 113.9 (2C), 74.4, 74.3, 61.2, 60.6, 59.9, 55.0, 38.7, 26.8, 17.0; m/z
(FAB) 402 ([M]+, 66%), 312 (65), 190 (63), 121 (76), 91 (100); HRMS (FAB)
(Found: [M]+, 402.2433. [C27H32NO2]+Cl− requires m/z, 402.2433).
(2R,3R,4S)-1-Benzyl-3-hydroxy-2-(4-methoxybenzyl)-4-methylpyrrolidine 16. To a solution of the chloride salt 15
(0.0782 g, 0.178 mmol) in methanol (5 cm3) was added 5% Pd/C (0.0078 g) and potassium carbonate (0.074 g, 0.54 mmol). The mixture was exposed to a hydrogen atmosphere (1 atm) and stirred vigorously for 20 minutes. The suspension was filtered through a pad of Celite and concentrated under reduced pressure. To the residue was added CH2Cl2
(25 cm3) and water (25 cm3). The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 20 cm3); the combined organic phase was washed with saturated aq. sodium bicarbonate (50 cm3) then dried and concentrated under reduced pressure. The residue was chromatographed on silica gel [hexane : EtOAc (7 : 3)] to give pyrrolidine 16
(0.0518 g, 93%) as an oil, Rf
[hexane : EtOAc (1 : 1)] 0.35; [α]D
−78.3 (c 1.5, CHCl3); νmax
(neat)/cm−1 3419, 2954, 1611, 1583, 1512, 1246; δH
(250 MHz; CDCl3) 7.33–7.23 (5H, m), 7.26 (2H, d, J 8.7), 6.85 (2H, d, J 8.7), 4.10 (1H, d, J 12.7), 3.78 (3H, s), 3.50 (1H, dd, J 4.7, 2.6), 3.16 (1H, d, J 12.7), 3.13 (1H, dd, J 9.4, 7.9), 2.94 (1H, dd, J 13.4, 8.9), 2.87 (1H, dd, J 13.4, 5.3), 2.57 (1H, dt, J 8.9, 5.0), 2.35 (1H, br s), 2.03–1.94 (1H, m), 1.67 (1H, dd, J 9.4, 7.9), 0.91 (3H, d, J 7.1); δc
(62.9 MHz; CDCl3) 157.8, 138.6, 131.4, 130.1 (2C), 128.8 (2C), 128.1 (2C), 126.9, 113.7 (2C), 78.8, 69.3, 60.2, 58.2, 55.1, 40.1, 32.9, 18.0; m/z
(FAB) 312 ([M + H]+, 73%), 190 (65), 121 (41), 91 (100); HRMS (FAB)
(Found: [M + H]+, 312.1963. C20H26NO2 requires m/z 312.1964).
(2R,3R,4S)-3-Acetoxy-1-benzyl-2-(4-methoxybenzyl)-4-methylpyrrolidine 17. To a solution of the alcohol 16
(0.0322 g, 0.104 mmol) in CH2Cl2
(5 cm3) was added freshly distilled acetic anhydride (0.021 g, 0.020 cm3, 0.21 mmol), and triethylamine (0.021 g, 0.030 cm3, 0.21 mmol). The solution was stirred for 18 hours before being diluted with CH2Cl2
(10 cm3) and saturated aq. sodium bicarbonate (20 cm3). The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 10 cm3); the combined organic phase was dried and concentrated under reduced pressure. The residue was chromatographed on silica gel [hexane : EtOAc (7 : 3)] to give the ester 17
(0.0300g, 82%) as a colourless oil, Rf
[hexane : EtOAc (7 : 3)] 0.30; [α]D
−73.96 (c 0.70, CHCl3); νmax
(neat)/cm−1 2959, 2789, 1734, 1612, 1512; δH
(250 MHz; CDCl3), 7.31–7.22 (5H, m), 7.10 (2H, d, J 8.6), 6.80 (2H, d, J 8.6), 4.62–4.58 (1H, m), 3.96 (1H, d, J 12.9), 3.77 (3H, s), 3.24 (1H, d, J 12.9), 3.12 (1H, dd, J 9.3, 7.5), 2.94–2.80 (2H, m), 2.83 (1H, dd, J 14.2, 8.9), 2.11–2.03 (1H, m), 2.06 (3H, s), 1.80 (1H, t, J 9.3), 0.99 (3H, d, J 7.0); δc
(62.9 Hz; CDCl3) 170.8, 157.9, 138.3, 131.1, 129.8 (2C), 129.1(2C), 128.2 (2C), 127.0, 113.8 (2C), 80.9, 67.1, 60.0, 58.8, 55.2, 38.3, 33.8, 21.2, 17.1; m/z
(FAB) 354 ([M + H]+, 39%), 352 (52), 294 (34), 232 (63), 172 (28), 121 (75), 91 (100); HRMS (FAB)
(Found: [M + H]+, 354.2069. C22H28NO3 requires m/z 354.2069).
(2R,3R,4S)-3-Acetoxy-2-(4-methoxybenzyl)-4-methylpyrrolidine hydrochloride 2. To a solution of the pyrrolidine 17
(0.0407 g, 0.115 mmol) and Pearlman's catalyst [0.0407 g; 20% Pd(OH)2/C] in methanol (5 cm3) was added hydrochloric acid (1 M in ether; 0.23 cm3, 0.23 mmol). The solution was exposed to a hydrogen atmosphere (1 atm) and stirred vigorously for 18 hours. The mixture was then filtered through a pad of Celite and concentrated under reduced pressure to give the hydrochloride salt 2
(0.0341 g, 100%) as a colourless oil, [α]D
+20.0 (c 1.35, CH3OH); νmax
(neat)/cm−1 3398, 2934, 1740, 1612, 1582, 1514; δH
(250 MHz; CD3OD), 7.33 (2H, d, J 7.9), 7.01 (2H, d, J 7.9), 5.05 (1H, br s), 4.15 (1H, br s), 3.87 (3H, s), 3.73 (1H, m), 3.19–3.04 (3H, m), 2.63 (1H, br s), 2.26 (3H, s), 1.26 (3H, d, J 6.7); δc
(62.9 MHz; CD3OD) 170.2, 159.4, 130.0 (2C), 127.8, 114.4 (2C), 78.5, 62.9, 54.7, 50.1, 39.3, 31.4, 19.8, 16.3; m/z
(FAB) 264 ([M + H]+, 100%), 204 (60), 142 (19), 121 (49); HRMS (FAB)
(Found: [M + H]+, 264.1593. [C15H22NO3]+Cl− requires m/z 264.1600).
(2R,3R,4S)-3-Hydroxy-2-(4-methoxybenzyl)-4-methylpyrrolidine 5. A solution of the chloride salt 15
(0.044 g, 0.10 mmol) and Pearlman's catalyst [0.044 g; 20% Pd(OH)2/C] in methanol (5 cm3) was exposed to a hydrogen atmosphere (1 atm) and stirred vigorously for 24 hours. The mixture was then filtered through a pad of Celite and concentrated under reduced pressure to give (2R,3R,4S)-3-hydroxy-2-(4-methoxybenzyl)-4-methyl-pyrrolidine hydrochloride (0.026 g, 100%) as a white solid, mp 191–192 °C; [α]D
+33.3 (c 0.27, CH3OH); νmax
(KBr)/cm−1 3356, 2956, 2920, 1614, 1577, 1516; δH
(250 MHz; CD3OD) 7.40 (2H, d, J 8.1), 7.03 (2H, d, J 8.1), 4.05 (1H, br s), 3.90 (3H, s), 3.84–3.83 (1H, m), 3.74 (1H, dd, J 11.4, 7.5), 3.27 (1H, dd, J 14.2, 6.1), 3.05 (1H, dd, J 14.2, 8.6), 2.97 (1H, dd, J 11.4, 3.2), 2.24–2.12 (1H, m), 1.19 (3H, d, J 7.3); δc
(62.9 MHz; CD3OD) 157.9, 128.7 (2C), 127.4, 112.9 (2C), 74.5, 63.1, 53.2, 48.6, 39.7, 30.1, 15.1; m/z
(FAB) 222 ([M + H]+, 68%), 206 (47), 134 (52), 121 (40), 91 (68); HRMS (FAB)
(Found: [M + H]+, 222.1494. [C13H20NO2]+Cl− requires m/z 222.1494).The HCl salt was subjected to ion-exchange chromatography (Dowex OH−; prepared by treating Dowex 1-X2 with 1 M aqueous NaOH, followed by methanol until the pH of the eluent returned to 7). Eluting with methanol gave the free base 5
(quantitative recovery) as a white solid, Rf
[CH2Cl2 : MeOH (95 : 5)] 0.05; mp 108–110 °C; [α]D
+5.7 (c 0.35, CH3OH); νmax
(KBr)/cm−1 3420, 3260, 2951, 1612, 1584, 1512; δH
(250 MHz; CD3OD) 7.28 (2H, d, J 8.7), 6.92 (2H, d, J 8.7), 3.84 (3H, s), 3.71 (1H, dd, J 2.4, 1.7), 3.37 (1H, dd, J 11.1, 7.8), 3.19–3.11 (1H, m), 2.99 (1H, dd, J 13.6, 6.8), 2.74 (1H, dd, J 13.6, 7.6), 2.38 (1H, dd, J 11.1, 5.7), 2.24–2.15 (1H, m), 1.07 (3H, d, J 7.2); δc
(62.9 MHz; CD3OD) 157.7 (1C, Q), 131.2 (1C, Q), 129.1 (2C, CH), 112.9 (2C, CH), 77.9 (1C, CH), 63.1 (1C, CH), 53.7 (1C, CH3), 50.9 (1C, CH2), 41.7 (1C, CH), 33.3 (1C, CH2), 16.5 (1C, CH3); m/z
(FAB) 222 ([M + H]+, 100%), 121 (17), 91 (95), 57 (66); HRMS (FAB)
(Found: [M + H]+, 222.1495. C13H20NO2 requires m/z 222.1494).
(2R)-2-Dibenzylamino-3-(4-methoxyphenyl)propanoic acid 19. To a solution of methyl ester 18
(1.97 g, 5.06 mmol) in tetrahydrofuran : water (30 cm3
[4 : 1]) was added lithium hydroxide (1.06 g, 25.3 mmol). The solution was heated to reflux and held there for 18 hours, and then acidified to pH 2. The organic phase was separated, and the aqueous phase was extracted with CH2Cl2
(3 × 25 cm3). The combined organics were washed with water (20 cm3), dried, and then concentrated under reduced pressure to give acid 19
(1.81 g, 95%) as a foam, which was used in the Claisen condensation reaction without further purification, Rf
[hexane : EtOAc (7 : 3)] 0.30; [α]D
+25.8 (c 0.6, CHCl3)
[lit.(ent-19),11
[α]D
−31.4 (c 1.0, CHCl3)]; νmax
(neat)/cm−1 3029, 2934, 2835, 1704, 1612, 1513; δH
(360 MHz; CDCl3) 9.25 (1H, br s), 7.29–7.25 (10H, m), 7.11 (2H, d, J 8.4), 6.82 (2H, d, J 8.4), 4.00–3.82 (4H, m), 3.86–3.83 (1H, m), 3.78 (3H, s), 3.30 (1H, dd, J 14.5, 6.3), 3.17 (1H, dd, J 14.5, 8.5); δc
(62.9 MHz; CDCl3) 175.5, 158.2, 137.3 (2C), 130.2 (2C), 129.8, 128.9 (4C), 128.4 (4C), 127.5 (2C), 113.7 (2C), 62.9, 55.2, 54.4 (2C), 33.3; m/z
(FAB) 367 ([M + H]+, 83%), 330 (22), 254 (40), 154 (38), 91 (100); HRMS (FAB)
(Found: [M + H]+, 376.1903. C24H26NO3 requires m/z 376.1913). All spectroscopic data in good agreement with the literature data.11 Ethyl (4R)-4-dibenzylamino-5-(4-methoxyphenyl)-3-oxo-pentanoate 21. To a solution of the acid 19
(1.81 g, 4.83 mmol) in THF (20 cm3) was added carbonyldiimidazolide (1.56 g, 9.65 mmol). The mixture was stirred for 2.5 hours, and then cooled to −78 °C. Meanwhile a separate flask containing ethyl acetate (1.49 g, 1.65 cm3, 16.9 mmol) was cooled to −78 °C, and lithium hexamethyldisilazide, (15.9 cm3, 1.06 M in THF, 16.9 mmol) was added. The solution was stirred at −78 °C for 20 minutes and then transferred to the flask containing the imidazole via cannula. The combined solution was stirred at −78 °C for 2 hours and then quenched by the addition of 1% HCl (20 cm3) and diluted with CH2Cl2
(20 cm3). The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 75 cm3); the combined organics were washed sequentially with water (100 cm3) and brine (100 cm3) then dried, and concentrated under reduced pressure. The residue was chromatographed on silica gel [hexane : EtOAc (95 : 5)] to give 21
(1.76 g, 82%) as a white solid, Rf
[hexane : EtOAc (10 : 1)] 0.30; mp 83–85 °C; [α]D
+75.3 (c 1.0, CHCl3); νmax
(KBr)/cm−1 2936, 1745, 1716, 1612, 1584, 1513; δH
(360 MHz; CDCl3) 7.36–7.19 (10H, m), 7.05 (2H, d, J 8.7), 6.78 (2H, d, J 8.7), 4.01 (2H, q, J 7.2), 3.83 (2H, d, J 13.5), 3.76 (3H, s), 3.65 (1H, d, J 15.8), 3.59 (1H, dd, J 9.3, 3.7), 3.54 (2H, d, J 13.5), 3.34 (1H, d, J 15.8), 3.13 (1H, dd, J 13.6, 9.3), 2.89 (1H, dd, J 13.6, 3.7), 1.09 (3H, t, J 7.2); δc
(62.9 MHz; CDCl3) 202.2, 167.1, 157.8, 138.6 (2C), 130.9, 130.3 (2C), 128.9 (4C), 128.4 (4C), 127.3 (2C), 113.7 (2C), 68.3, 60.9, 55.1, 54.4 (2C), 46.6, 27.4, 13.8; (Found: C, 75.19; H, 6.83; N, 3.11. C28H31NO4 requires C, 75.51; H, 6.97; N, 3.15%).
Ethyl (3R,4R)-4-dibenzylamino-3-hydroxy-5-(4-methoxyphenyl)pentanoate 22. To a solution of β-keto ester 21
(1.54, 3.47 mmol) in ether (30 cm3) was added methanol (5.21 g, 6.59 cm3, 163 mmol). The solution was adjusted to pH 4 by the addition of a few drops of acetic acid, and then cooled to 0 °C. Sodium cyanoborohydride (2.15 g, 34.6 mmol) was added cautiously and the mixture was allowed to warm to room temperature. The solution was stirred for 18 hours and then quenched by the addition of a saturated aqueous solution of ammonium chloride (20 cm3). The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 75 cm3). The combined organics were dried, and concentrated under reduced pressure. The remaining residue was chromatographed on silica gel [hexane : EtOAc (9 : 1)] to give the syn diastereomer 22
(1.28 g, 83%) and a small amount of the anti diastereomer (0.0307 g, 2%) both as oils. Data for (3R,4R) major syn diastereomer 22, Rf
[hexane : EtOAc (10 : 1)] 0.40; [α]D
−36.0 (c 2.2, CHCl3); νmax
(neat)/cm−1 3591, 3025, 2933, 1731, 1611, 1581, 1511; δH
(360 MHz; CDCl3) 7.35–7.25 (10H, m), 7.11 (2H, J 8.5), 6.88 (2H, d, J 8.5), 4.11–4.05 (2H, m), 4.08 (2H, q, J 7.2), 4.01–3.98 (1H, m), 3.81 (3H, s), 3.44 (2H, d, J 13.4), 3.11–3.09 (1H, m), 2.82–2.74 (2H, m), 2.39 (1H, dd, J 15.7, 9.6), 2.12 (1H, br d, J 15.7), 1.21 (3H, t, J 7.2); δc
(62.9 MHz; CDCl3) 172.7, 157.9, 139.1 (2C), 131.7, 129.9 (2C), 128.9 (4C), 128.3 (4C), 127.1 (2C), 113.9 (2C), 67.8, 62.9, 60.4, 55.1, 54.3 (2C), 39.7, 29.9, 14.0; m/z
(FAB) 448 ([M + H]+, 97%), 330 (88), 326 (89), 121 (37), 91 (100); HRMS (FAB)
(Found: [M + H]+, 448.2480. C28H34NO4 requires m/z 448.2488); Chiral HPLC (R enantiomer)
Rt
= 16.0 minutes, (S enantiomer)
Rt
= 24.4 minutes [hexane–propan-2-ol (19 : 1)], >98% ee.Data for (3S,4R) minor anti diastereomer, Rf
[hexane : EtOAc (10 : 1)] 0.35; [α]D
−25.9 (c 0.81, CHCl3); νmax
(neat)/cm−1 3501, 2934, 2833, 1728, 1612, 1584, 1512; δH
(200 MHz; CDCl3) 7.39–7.25 (10H, m), 7.20 (2H, d, J 8.8), 6.92 (2H, d, J 8.8), 4.33–4.25 (1H, m), 4.18 (2H, q, J 7.0), 3.90 (3H, s), 3.82 (2H, d, J 13.7), 3.68 (2H, d, J 13.7), 3.19–2.97 (2H, m), 3.10 (1H, dd, J 9.0, 6.3), 2.80 (1H, dd, J 16.4, 2.7), 2.38 (1H, dd, J 16.4, 9.8), 1.32 (3H, t, J 7.0); δc
(62.9 MHz; CDCl3) 173.4, 158.0, 139.7 (2C), 133.1, 130.5 (2C), 128.9 (4C), 128.4 (4C), 127.1 (2C), 113.9 (2C), 69.2, 63.0, 60.8, 55.5, 54.8 (2C), 39.7, 31.5, 14.3; m/z
(FAB) 448 ([M + H]+, 88%), 330 (96), 326 (96), 121 (78), 91 (100); HRMS (FAB)
(Found: [M + H]+, 448.2489. C28H34NO4 requires m/z 448.2488).
(3R,4R)-4-Dibenzylamino-3-hydroxy-5-(4-methoxyphenyl)pentan-1-ol. A solution of ethyl ester 22
(0.697 g, 1.56 mmol) in THF (30 cm3) was cooled to −78 °C and lithium aluminium hydride (7.8 cm3, 1.0 M in THF, 7.8 mmol) was added. The solution was stirred at −78 °C for 6 hours then quenched by the addition of 1 M aqueous sodium hydroxide (10 cm3). The resulting mixture was allowed to warm to room temperature and diluted with CH2Cl2
(20 cm3). Saturated aq. sodium potassium tartrate (40 cm3) was added and the biphasic mixture was stirred vigorously for 15 hours by which time two clear phases were apparent. The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 50 cm3). The combined organics were dried, and concentrated under reduced pressure. The remaining residue was chromatographed on silica gel [hexane : EtOAc]
(7 : 3 → 1 : 1)] to give the title compound (0.598 g, 95%) as an oil, Rf
[hexane : EtOAc (3 : 1)] 0.35; [α]D
−38.3 (c 1.7, CHCl3); νmax
(neat)/cm−1 3388, 3028, 2933, 2835, 1612, 1583, 1512; δH
(250 MHz; CDCl3) 7.39–7.18 (10H, m), 7.12 (2H, d, J 8.7), 6.88 (2H, d, J 8.7), 4.80 (1H, s), 3.90 (2H, d, J 13.2), 3.82 (3H, s), 3.77 (1H, dd, J 8.9, 2.7), 3.65 (2H, br s), 3.39 (2H,d, J 13.2), 3.08 (1H, dd, J 14.3, 6.4), 2.89 (1H, td, J 6.4, 2.7), 2.60 (1H, dd, J 14.3, 6.4), 1.82 (1H, s), 1.65–155 (1H, m), 1.35–1.23 (1H, m); δc
(62.9 MHz; CDCl3) 158.0, 138.5 (2C), 131.8, 129.9 (2C), 128.9 (4C), 128.4 (4C), 127.2 (2C), 113.9 (2C), 70.5, 63.8, 61.2, 55.1, 53.6 (2C), 35.6, 31.1; m/z
(FAB) 406 ([M + H]+, 69%), 330 (71), 284 (69), 181 (45), 91 (100); HRMS (FAB)
(Found: [M + H]+, 406.2387. C26H32NO3 requires m/z 406.2382).
(2R,3R)-1,1-Dibenzyl-3-hydroxy-2-(4-methoxybenzyl)-pyrrolidinium chloride 23. To a solution of (3R,4R)-4-dibenzylamino-3-hydroxy-5-(4-methoxyphenyl)pentan-1-ol (0.587 g, 1.45mmol) in CH2Cl2
(20 cm3) at 0 °C was added DMAP (0.292 g, 2.39 mmol) and triisopropylbenzene sulfonyl chloride (TIBSCl)
(0.483 g, 1.59 mmol). The solution was stirred for 18 hours before being diluted with CH2Cl2
(20 cm3) and water (25 cm3). The organic phase was separated and washed with 1% HCl (2 × 30 cm3) then dried and concentrated under reduced pressure. The residue was chromatographed on silica gel [CH2Cl2 : MeOH (100 : 0)
→
(95 : 5)] to give a white foam. The salt obtained was subjected to ion exchange chromatography [Dowex Cl−; prepared by treating Dowex 1-X2 with 1% aqueous hydrochloric acid followed by flushing with methanol until the eluent returned to pH 7] eluting with methanol to give the chloride salt 23
(0.520 g, 85%) as an amorphous solid, Rf
[CH2Cl2 : CH3OH (95 : 5)] 0.05; [α]D
−55.2 (c 0.75, CHCl3); νmax
(neat)/cm−1 3198, 1611, 1584, 1513; δH
(360 MHz; CDCl3) 7.78–7.29 (10H, m), 7.33 (2H, d, J 8.5), 6.74 (1H, s), 6.67 (2H, d, J 8.5), 5.72 (1H, br d, J 13.5), 5.12 (1H, d, J 13.1), 5.07 (1H, d, J 13.1), 4.34 (1H, br s), 4.28 (1H, d, J 13.5), 3.97 (1H, br d, J 12.0), 3.92–3.86 (1H, m), 3.69 (3H, s), 3.56 (1H, br t, J 12.0), 3.39 (1H, br t, J 10.5), 3.10 (1H, dt, 11.3, 8.3), 2.63 (1H, dt, 14.6, 7.3), 1.95–1.87 (1H, m); δc
(62.9 MHz; CDCl3) 158.9, 134.1 (2C), 133.6 (2C), 131.2 (2C), 131.1, 130.8, 129.8 (2C), 129.6 (2C), 128.6, 128.0, 127.9, 114.5 (2C), 76.7, 68.2, 63.1, 62.4, 56.3, 55.5, 31.9, 28.1; m/z
(FAB) 388 ([M]+, 98%), 296 (48), 210 (24), 176 (71), 91 (100); HRMS (FAB)
(Found: [M]+, 338.2267. [C26H30NO2]+Cl− requires m/z 388.2277).
(2R,3R)-1-Benzyl-3-hydroxy-2-(4-methoxybenzyl)pyrrolidine 24. To a solution of the chloride salt 23
(0.269 g, 0.634 mmol) in methanol (10 cm3) was added 5% Pd/C (0.027 g) and potassium carbonate (0.263 g, 1.90 mmol). The mixture was exposed to a hydrogen atmosphere (1 atm) and stirred vigorously for 20 minutes. The suspension was filtered through a pad of Celite and concentrated under reduced pressure. To the residue was added CH2Cl2
(25 cm3) and water (25 cm3). The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 25 cm3); the combined organic phase were washed with water (25 cm3) and then dried and concentrated under reduced pressure. The residue was chromatographed on silica gel [hexane : EtOAc (1 : 1)] to give pyrrolidine 24
(0.150 g, 80%) as an oil, Rf
[hexane : EtOAc (1 : 1)] 0.35; [α]D
−104.0 (c 1.18, CHCl3); νmax
(neat)/cm−1 3418, 2934, 1611, 1583, 1512; δH
(250 MHz; CDCl3) 7.41–7.26 (7H, m), 6.89 (2H, d, J 8.7), 4.20 (1H, d, J 13.0), 3.96 (1H, br s), 3.83 (3H, s), 3.26 (1H, d, J 13.0), 3.05 (1H, dt, J 8.8, 3.7), 2.98–2.86 (2H, m), 2.49 (1H, ddd, J 8.5, 6.0, 3.7), 2.20–2.06 (1H, m), 1.99 (1H, ddd, 13.7, 6.4, 3.7), 1.75–1.63 (1H, m); δc
(62.9 MHz; CDCl3) 158.4, 139.4, 131.9, 130.7 (2C), 129.3 (2C), 128.7 (2C), 127.4, 114.2 (2C), 72.5, 71.3, 58.3, 55.7, 51.9, 33.4, 32.7; m/z
(FAB) 298 ([M + H]+, 90%), 176 (93), 154 (62), 91 (100); HRMS (FAB)
(Found: [M + H]+, 298.1805. C19H24NO2 requires m/z 298.1805).
(2R,3R)-3-Acetoxy-1-benzyl-2-(4-methoxybenzyl)pyrrolidine 25. To a solution of the alcohol 24
(0.0643 g, 0.216 mmol) in CH2Cl2
(5 cm3) was added freshly distilled acetic anhydride (0.044 g, 0.041 cm3, 0.43 mmol), and triethylamine (0.044 g, 0.061 cm3, 0.43 mmol). The solution was stirred for 18 hours and then quenched by the addition of saturated aq. sodium bicarbonate (20 cm3). The organic phase was separated and the aqueous phase was extracted with CH2Cl2
(3 × 20 cm3); the combined organic phase was dried and concentrated under reduced pressure. The residue was chromatographed on silica gel [hexane : EtOAc (7 : 3)] to give the ester 25
(0.0638 g, 87%) as a colourless oil, Rf
[hexane : EtOAc (1 : 1)] 0.60; [α]D
−123.1 (c 0.26, CHCl3); νmax
(neat)/cm−1 2935, 2791, 1733, 1612, 1581, 1513; δH
(250 MHz; CDCl3) 7.40–7.33 (5H, m), 7.22 (2H, d, J 8.7), 6.86 (2H, d, J 8.7), 5.01 (1H, ddd, J 8.8, 5.1, 3.4), 4.12 (1H, d, J 13.1), 3.39 (3H, s), 3.36 (1H, d, J 13.1), 2.97 (1H, dd, J 8.7, 1.8), 2.94 (1H, dd, J 13.3, 5.1), 2.78 (1H, dd, J 13.3, 9.5,), 2.66 (1H, dt, J 9.5, 5.1, C2H), 2.17 (2H, m), 2.14 (3H, s), 1.69–1.54 (1H, m); δc
(62.9 MHz; CDCl3) 171.1, 158.4, 138.8, 131.5, 130.2 (2C), 129.6 (2C), 128.7 (2C), 127.5, 114.3 (2C), 75.2, 68.8, 58.8, 55.7, 52.1, 33.8, 31.1, 21.7; m/z
(FAB) 340 ([M + H]+, 70%), 280 (28), 218 (52), 121 (43), 91 (100); HRMS (FAB)
(Found: [M + H]+, 340.1916. C21H26NO3 requires m/z 340.1913).
(2R,3R)-3-Acetoxy-2-(4-methoxybenzyl)pyrrolidine hydrochloride 3. To a solution of the pyrrolidine 25
(0.0527 g, 0.155 mmol) and Pearlman's catalyst [0.0527 g; 20% Pd(OH)2/C] in methanol (5 cm3) was added hydrochloric acid (1 M in ether; 0.31 cm3, 0.31 mmol]. The solution was exposed to a hydrogen atmosphere (1 atm) and stirred vigorously for 20 hours. The mixture was then filtered through a pad of Celite and concentrated under reduced pressure to give hydrochloride 3
(0.0450 g, 100%) as a pale brown oil, [α]D
−46.3 (c 0.56, CHCl3); νmax
(neat)/cm−1 3398, 2934, 1741, 1613, 1581, 1515; δH
(250 MHz; CD3OD) 7.32 (2H, d, J 8.4), 7.00 (2H, d, J 8.4), 5.40 (1H, br s), 4.02 (1H, br s), 3.86 (3H, s), 3.54 (2H, br q, J 8.6), 3.18 (1H, dd, J 14.1, 6.2), 3.06 (1H, dd, J 14.1, 8.4), 2.52–2.44 (1H, m), 2.31–2.18 (1H, m), 2.27 (3H, s); δc
(62.9 MHz; CD3OD) 169.3, 158.6, 129.1 (2C), 127.1, 113.6 (2C), 72.4, 64.1), 53.8, 42.4, 30.7, 29.9, 19.0; m/z
(FAB) 250 ([M + H]+, 100%), 190 (36), 128 (22), 121 (32), 91 (41); HRMS (FAB)
(Found: [M + H]+, 250.1442. [C14H20NO3]+Cl− requires m/z 250.1443).
(2R,3R)-3-Hydroxy-2-(4-methoxybenzyl)pyrrolidine 6. A solution of the chloride salt 23
(0.243 g, 0.574 mmol) and Pearlman's catalyst [0.243 g; 20% Pd(OH)2/C] in methanol (7 cm3) was exposed to a hydrogen atmosphere (1 atm) and stirred vigorously for 24 hours. The mixture was then filtered through a pad of Celite and concentrated under reduced pressure to give (2R,3R)-3-hydroxy-2-(4-methoxybenzyl)pyrrolidine hydrochloride (0.133 g, 95%) as a white solid, mp 261–263 °C; [α]D
+15.2 (c 1.12, CH3OH); νmax
(KBr)/cm−1 3330, 3302, 2955, 1614, 1556, 1516; δH
(250 MHz; CD3OD) 7.35 (2H, d, J 8.7), 6.99 (2H, d, J 8.7), 4.39–4.36 (1H, m), 3.87 (3H, s), 3.67 (1H, ddd, J 8.5, 6.6, 2.8), 3.55 (1H, ddd, J 11.5, 9.9, 8.2), 3.57–3.23 (1H, m), 3.24 (1H, dd, J 14.2, 6.6), 3.02 (1H, dd, J 14.2, 8.5), 2.32 −2.10 (2H, m); δc
(62.9 MHz; CD3OD) 158.4, 129.2 (2C), 128.0, 113.4 (2C), 69.1, 66.0, 53.8, 42.3, 32.2, 30.8; m/z
(FAB) 208 ([M + H]+, 97%), 154 (100), 136 (85), 121 (15); HRMS (FAB)
(Found: [M + H]+, 208.1340. [C12H18NO2]+Cl− requires m/z 208.1338).The HCl salt was subjected to ion-exchange chromatography (Dowex OH−; prepared by treating Dowex 1-X2 with 1 M aqueous NaOH, followed by methanol until the pH of the eluent returned to 7). Eluting with methanol gave the free base 6
(quantitative recovery) as a white solid, Rf
[CH2Cl2 : MeOH (95 : 5)] 0.05; mp 127–128 °C; [α]D
−30.9 (c 1.36, CH3OH); νmax
(KBr)/cm−1 3426, 3290, 2961, 1612, 1583, 1512; δH
(250 MHz; CD3OD) 7.23 (2H, d, J 8.7), 6.87 (2H, d, J 8.7), 4.06 (1H, ddd, J 4.9, 3.1, 1.5), 3.79 (3H, s), 3.17 (1H, ddd, J 11.1, 8.5, 7.5), 2.96 (1H, dd, J 7.1, 3.2), 2.90 (1H, dd, J 7.1, 2.5), 2.79 (1H, ddd, J 11.1, 9.7, 4.7), 2.85–2.67 (1H, m), 2.01 (1H, dddd, J 13.7, 9.7, 7.5, 4.9), 1.83 (1H, dddd, J 13.7, 8.5, 4.7, 1.5); δc
(62.9 MHz; CD3OD) 158.6, 132.1, 130.0 (2C), 113.8 (2C), 71.9, 66.2, 54.6, 43.6, 35.0, 34.1; m/z
(FAB) 208 ([M + H]+, 100%), 190 (11), 121 (27), 91 (69); HRMS (FAB)
(Found: [M + H]+, 208.1333. C12H18NO2 requires m/z 208.1338).
SAPK pathway activation
Cell culture and stimulation. 3c Murine RAW 264.7 macrophages were purchased from the European Tissue Culture Collection and maintained in a 95% air/5% CO2 37 °C atmosphere. Cells were cultured in Dulbecco's Modified Eagles' Medium (DMEM) containing 10% heat inactivated foetal calf serum, 100 U ml−1 penicillin, 100 µg ml−1 streptomycin and 2 mM L-glutamine. Anisomycin was dissolved at 10 mg ml−1 in DMSO. Cells were stimulated for 30 min by the addition of 1 µl of 10 mg ml−1 anisomycin or an anisomycin analogue in DMSO, or 1 µl of DMSO as control, to each 1 ml of culture medium. The final concentration of DMSO in the culture medium was therefore always 0.1%
(v/v). Cell lysis. 3c After stimulation, the media was aspirated, the cells from each dish washed with ice cold PBS and then lysed in 250 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 0.5 mM sodium orthovanadate, 5 mM sodium β-glycerophosphate, 25 mM sodium fluoride, 2.5 mM sodium pyrophosphate, 0.135 M sucrose, 0.5%
(v/v) Triton X-100, 5%
(v/v) glycerol, 0.05%
(v/v) 2-mercaptoethanol, 0.5 µM microcystin-LR and ‘Complete’ protease inhibitor cocktail (one tablet per 100 ml)] and 1%
(w/v) SDS. Protein concentrations were determined according to Bradford. Immunoblotting. 3cSamples were denatured in SDS, run on polyacylamide gels and transferred to nitrocellulose membranes. The membranes were then incubated for 1 h at room temperature in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.2%
(v/v) Tween and 5%
(w/v) skimmed milk powder. Antibodies that recognise c-Jun and MAPKAP-K2 phosphorylated at Thr334 were purchased from Cell Signalling Technologies (Hitchin, UK), while an antibody that recognises all forms of MAPKAP-K2 was raised in sheep against a peptide corresponding to the C-terminus of MAPKAP-K2 (MTSALATMRVDYEQIK). Antibodies were added to 10 ml of the above buffer except that the skimmed milk was replaced by 5%
(w/v) BSA. Antibodies were then incubated with the membranes on a shaker at 4 °C overnight. The membranes were then washed four times with buffer (five min per wash) to remove the primary antibodies, and secondary antibodies were incubated with the membranes for 1 h at room temperature. After washing six times with buffer to remove the secondary antibodies (5 min per wash), immunoreactive proteins were visualised via enhanced chemiluminescence (ECL) reagent according to the manufacturer's instructions. Acknowledgements
We thank the BBSRC (Studentship 99/A1/B/05153 to EMR), Pfizer (Summer Studentship to KSA), the MRC (Studentship to SM) and the MRC and The Royal Society (PC) for financial support of this work.References and notes
- A. Jimènez and D. Vázquez, Anisomycin and Related Antibiotics, in Antibiotics, ed. F. E. Hahn, 1979, Springer-Verlag, New York, vol. 5, part 2, pp. 1–19 Search PubMed.
- L. C. Mahadevan and D. R. Edwards, Nature, 1991, 349, 747–748 CrossRef CAS.
-
(a) E. Cano, C. A. Hazzalin and L. C. Mahadevan, Mol. Cell. Biol., 1994, 14, 7352–7362 CAS;
(b) E. Cano, Y. N. Doza, R. BenLevy, P. Cohen and L. C. Mahadevan, Oncogene, 1996, 12, 805–812 CAS;
(c) S. Morton, R. J. Davis, A. McLaren and P. Cohen, EMBO J., 2003, 15, 3876–3886 CrossRef.
- Z. Chen, T. B. Gibson, F. Robinson, L. Silvestro, G. Pearson, B. Xu, A. Wright, C. Vanderbilt and M. H. Cobb, Chem. Rev., 2001, 101, 2449–2476 CrossRef CAS.
- A. N. Hulme and E. M. Rosser, Org. Lett., 2002, 4, 265–267 CrossRef CAS.
- O. Schwardt, U. Veith, C. Gaspard and V. Jäger, Synthesis, 1999, 1473–1490 CrossRef CAS.
- A. N. Hulme and C. H. Montgomery, Terahedron Lett., 2003, 44, 7649–7653 Search PubMed.
- A. N. Hulme and K. S. Curley, J. Chem. Soc., Perkin Trans. 1, 2002, 1083–1091 RSC.
- Y. Hosoya, T. Kameyama, H. Naganawa, Y. Okami and T. Takeuchi, J. Antibiot., 1993, 46, 1300–1302 CAS.
- J. Blenis, J. K. Chung, E. Erikson, D. A. Alcorta and R. L. Erikson, Cell Growth Differ., 1991, 2, 279–285 CAS.
- J. Bonjoch, J. Catena, D. Terricabras, J. Fernandez, M. Lopez-Canet and N. Valls, Tetrahedron: Asymmetry, 1997, 8, 3143–3151 CrossRef CAS.
Footnotes |
| † At “sub-inhibitory” levels (typically 25–50 ng mL−1) anisomycin activates the kinase pathways but does not inflict any toxic effects on cells through protein synthesis inhibition and may even cause a slight stimulation of protein synthesis, presumably due to its gene-inducing effect. |
| ‡ Deacetylation of anisomycin markedly inhibits its ability to bind to ribosomes (104-fold reduction in potency), arrest translation, activate JNK/SAPK1 and p38/SAPK2 and induce apoptosis. |
| § Aldehyde 13 is available in 7 steps (80% overall yield) from D-tyrosine (ref. 5). |
| ¶ Ester 18 is available in 5 steps (91% overall yield) from D-tyrosine. |
| || Confirmation of the relative stereochemistry achieved during the cyanoborohydride reduction of 21 was obtained at this juncture by preparation of the hexafluorophosphate salt of 23
(Dowex PF6−, 70% from the intermediate diol) and X-ray crystallographic analysis. |
|
| This journal is © The Royal Society of Chemistry 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.