Total synthesis of (−)-microcarpalide, a novel microfilament disrupting metabolite†
Paolo Davoli*, Alberto Spaggiari, Luca Castagnetti and Fabio Prati*
Dipartimento di Chimica, Università di Modena e Reggio Emilia, via Campi 183, I-41100 Modena, Italy. E-mail: davoli.chemistry@unimo.it; prati.fabio@unimo.it
First published on 6th November 2003
Abstract
The stereoselective total synthesis of (−)-microcarpalide, a recently discovered 10-membered lactone of fungal origin displaying a remarkable disrupting action on actin microfilaments, was accomplished by using ring-closing metathesis (RCM) as the key step for the formation of the medium-sized ring. The diene ester required for the macrocyclization reaction was assembled via DCC-mediated esterification of two suitable partners, each bearing a terminal alkene group. The alcohol fragment was synthesized from n-bromohexane through a seven-step sequence entailing two consecutive stereoselective homologations of chiral boronic esters as strategic transformations for the sequential insertion of the two stereocentres with the final S absolute configuration, using (+)-pinanediol as the chiral director; final elaboration to the desired C11 framework envisaged treatment with an allyl Grignard reagent and oxidative cleavage of the boronic scaffold. In contrast, the acidic fragment was prepared in ten steps from D-tartaric acid, whose C4 backbone was elongated to the required C7 skeleton by means of two distinct Swern–Wittig oxidation–homologation sequences.
Introduction
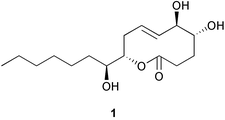
Secondary metabolites from endophytic fungi have been receiving a great deal of attention in recent years, and a number of peculiar structures with specific bioactivities have been discovered so far.1,2Along this line, microcarpalide (1) has been recently characterised as a new secondary metabolite produced by an endophytic fungus (as yet unidentified) isolated from the bark of the tropical tree Ficus microcarpa L.3 Bioassay-guided purification of fermentation broths using immunofluorescence microscopy to test anticytoskeletal activity led to the isolation of a new substance displaying a remarkable disrupting action on actin microfilaments, to which the structure 1 was assigned (Fig. 1).3
 | ||
| Fig. 1 | ||
Microcarpalide represents a novel alkyl-substituted nonenolide structurally related to a family of phytotoxins such as achaetolide,4 pinolidoxin,5 lethalotoxin,6 putaminoxins7 and herbarumins,8 from which it differs in the hydroxylation pattern and the double bond position within the 10-membered lactone, as well as in the longer side chain at C-10. At concentrations of 0.5–1 µg mL−1, microcarpalide was found to disrupt actin microfilaments in approximately 50% of A-10 cells (from rat smooth muscle);3 moreover, it displayed a weak cytotoxicity to mammalian cells,3 thus making it an attractive tool for studying cell motility and metastasis, and a potential lead structure to develop new anticancer drugs.
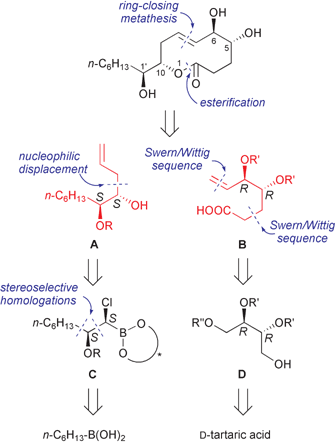
Owing to such a peculiar biological activity and attracted by the structural potential of microcarpalide for structure–activity relationship studies, we decided to challenge its total synthesis. In this respect, aiming also at shedding light on the mechanism of action of this novel fungal metabolite, the synthetic route ought to be versatile enough to allow the preparation of analogues thereof. With this in mind, the retrosynthetic approach outlined in Scheme 1 was devised.
 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Firstly, the macrocyclic framework of microcarpalide was retrosynthetically disassembled into fragments A and B (Scheme 1) by means of two key disconnections, namely a ring-closing metathesis (RCM)9 for the construction of the oxecin ring, and an esterification reaction for assembling the two alkene fragments. Both fragments contain two stereogenic carbon atoms and bear a terminal alkene group that is required for the RCM macrocyclization.
Fragment A represents a dihydroxy substituted 1-undecene with S absolute configuration at both adjacent stereocentres (C-10 and C-1′ in 1) and can be ultimately disconnected to the C6 alkyl boronic acid (Scheme 1). In fact, boronic ester chemistry provides a highly stereoselective method for the sequential insertion of stereocentres into the C–B bond.10 In particular, chain elongation by means of two strategic stereoselective homologations of the corresponding (+)-pinanediol ester would allow installation of the C-1′ and C-10 atoms with the desired stereochemistry under the guide of the same chiral director, thus affording chloro derivative C. Final insertion of the allylic appendage via SN2 displacement and oxidative removal of the boronic ester would then lead to the required C11 skeleton of A, providing at the same time the free hydroxy group at C-10 to be used for the coupling reaction with partner B.
Fragment B is a 4,5-disubstituted 6-heptenoic acid bearing two contiguous hydroxy groups in a threo fashion, whose R absolute configurations can be matched by the pattern displayed by D-tartaric acid. Retrosynthetically, the C7 backbone was traced back to a suitably protected D-threitol D (Scheme 1) by use of two different Swern–Wittig oxidation–homologation sequences (each one with an appropriate phosphorus ylide), which would allow chain elongation at both termini of the C4 chiral synthon D. In turn, threitol derivative D can be easily prepared from D-tartaric acid, as reported in the literature for the L-enantiomer.
Results and discussion
Synthesis of fragment A
For our synthesis we envisaged using the stereoselective homologation of chiral boronic esters developed by Matteson and coworkers10 to install the two adjacent stereocentres with the required S absolute configuration. In particular, (+)-pinanediol was chosen as the chiral auxiliary to direct the stereochemical outcome of the homologation reaction; in fact, the (+) isomer is known to induce the S configuration when used as the chiral director.10,11The synthetic approach to fragment A commenced from commercially available n-bromohexane (2), which was converted into the corresponding Grignard derivative; reaction between n-hexylmagnesium bromide and trimethyl borate in diethyl ether at −78 °C, followed by acidic work-up, furnished boronic acid 3 on a multigram scale in 72% yield (Scheme 2);12 acid 3 undergoes spontaneous dehydration to form the corresponding trimeric boroxine, as shown by MS analysis. Esterification with the chiral auxiliary (+)-pinanediol in dry Et2O at rt readily afforded the corresponding boronate 4 (71% yield).10 The presence of a carbon–boron bond was confirmed on the basis of the 13C NMR spectrum displaying a diagnostic broad signal at 10.5 ppm for C-1, which could be detected only in highly concentrated samples. Chiral boronate 4 is an air-stable liquid that can be easily handled and stored over time on the lab shelf.10 Hence, the stage was set to exploit Matteson's asymmetric homologation for inserting the first stereocentre. Addition of in situ-generated (dichloromethyl)lithium to chiral boronic ester 4 at −100 °C in THF13 provided α-chloro derivative 5 in good yield (64%) and high diastereoisomeric excess (d.e. ≥ 98%) (Scheme 2). The presence of a new signal at 3.48 ppm in the 1H NMR spectrum indicated the successful insertion of a chlorine-bearing carbon atom into the C–B bond of 4, whereas the diastereoselectivity of the homologation was determined by means of the diagnostic signal of the Hendo proton of the pinanyl moiety (1.19 ppm, doublet).11 Since (+)-pinanediol is reported to direct the formation of (S)-α-chloroboronic esters,10,11 the S absolute configuration could be assigned to 1-chloroheptaneboronate 5. Subsequently, exposure to a solution of (benzyloxy)lithium (prepared by titration of benzyl alcohol with n-butyllithium in the presence of 1,10-phenanthroline as indicator) in THF resulted in the SN2 nucleophilic displacement of the chlorine atom,11 thus affording the corresponding 1-benzyloxy derivative 6 in 70% yield (Scheme 2). Compound 6 bears a protected hydroxy group with R absolute configuration,14 which corresponds to the desired 1′S stereochemistry in 1. More interestingly, benzyl ether 6 could be also obtained directly from 4 through a one-pot procedure that avoided the isolation of the α-chloroboronic ester 5 and, gratifyingly, proceeded with higher overall yield (55%).
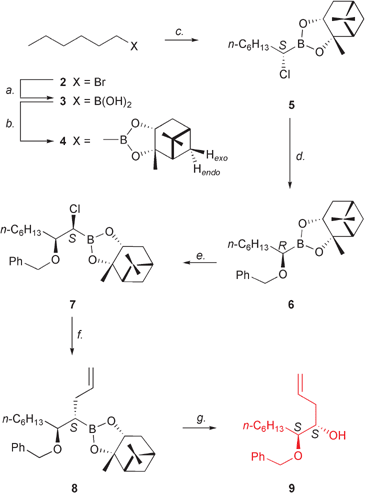 | ||
| Scheme 2 Synthesis of fragment A. Reagents and conditions: a. Mg, Et2O, reflux, then trimethyl borate, −78 °C → rt, 72%; b. (1S,2S,3R,5S)-(+)-pinanediol, Et2O, rt, 71%; c. (dichloromethyl)lithium, THF, −100 °C → rt, 64%; d. benzyl alcohol, n-BuLi, THF, −78 °C → rt, then reflux, 70%; e. (dichloromethyl)lithium, ZnCl2, THF, −100 °C → rt, 61%; f. allylmagnesium bromide, THF, −78 °C → rt, 74%; g. H2O2, NaOH, THF, 0 °C → 45 °C, 90%. | ||
By close analogy, the second asymmetric centre was introduced sequentially through stereoselective homologation of α-benzyloxy boronic ester 6. Addition of (dichloromethyl)lithium at −100 °C in THF,13 followed by treatment with zinc chloride (1 M solution in Et2O)10 provided 2-benzyloxy-1-chloroboronate 7 in good yield (61%) (Scheme 2). In the absence of zinc chloride, the homologation reaction was found to display incomplete conversion and proceeded in lower yield, albeit with excellent diastereoselectivity. In contrast, addition of zinc chloride resulted in better conversions and yield, without affecting d.e. NMR spectroscopy confirmed the insertion of a new carbon atom (δH 3.65 ppm, doublet; δC 46.1 ppm, broad signal due to the C–B bond) and the Hendo proton was used again as diastereotopic marker to assess d.e., which was found to be greater than 98%. By use of the same chiral director, the S absolute configuration was installed.
The desired C11 backbone of fragment A was then constructed by exploiting the ease of reaction of α-halo boronic esters with C-nucleophiles, namely Grignard reagents.10 In the case of 1-chlorooctaneboronate 7, an allylic appendage was required for such a purpose. Accordingly, a solution of 7 in THF was reacted with allylmagnesium bromide at −78 °C, affording compound 8 in 74% yield (Scheme 2). Since nucleophilic displacements on α-chloroboronates are known to occur with SN2 mechanism,14 alkene 8 has opposite configuration at the boron-bearing C atom with respect to 7 (even though the same S chiral descriptor is used for priority reasons). Hence, boronic ester 8 already features the two adjacent asymmetric centres with the correct final stereochemistry required by 1. Finally, oxidative removal of the boronic scaffold by treatment with basic hydrogen peroxide in THF,10,11,15 which is reported to occur with retention of configuration,16 yielded the desired alcohol 9 (90%) with S,S absolute configuration (Scheme 2), thus releasing the free hydroxy function required for the coupling reaction with fragment B and meanwhile retaining the benzylic protection at the adjacent position (the C-1′-to-be in 1) to be cleaved off only at the end of the overall sequence.
Synthesis of fragment B
For the preparation of the acidic partner B, we devised using D-tartaric acid as the starting chiral synthon, to which a number of transformations described in the literature for the corresponding L-enantiomer were applied.Firstly, D-(−)-tartaric acid (10) was refluxed with 2,2-dimethoxypropane in the presence of p-toluenesulfonic acid to yield the corresponding dimethyl ester acetonide 11 in excellent yield (Scheme 3).17 Subsequent lithium aluminium hydride reduction in diethyl ether gave D-(−)-threitol acetonide 12 (87% yield),18 which was treated with sodium hydride in THF and reacted with tert-butyldimethylsilyl chloride at rt to afford the monosilylated derivative 13 on a multigram scale in excellent yield (91%) (Scheme 3).19
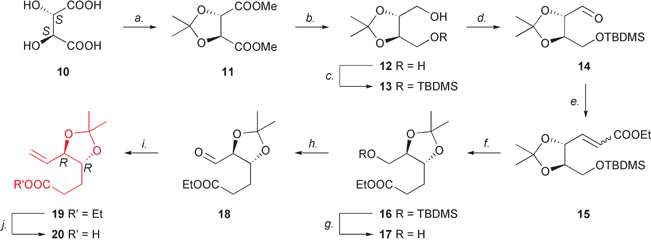 | ||
| Scheme 3 Synthesis of fragment B. Reagents and conditions: a. 2,2-dimethoxypropane, p-TsOH, reflux, 91%; b. LAH, Et2O, reflux, 87%; c. TBDMSCl, NaH, THF, 91%; d. Swern oxidation (oxalyl chloride–DMSO, CH2Cl2, triethylamine, −70 °C), 90%; e. (ethoxycarbonylmethylene)triphenylphosphorane, DMF, rt, 94%; f. H2, Pd/C, EtOH, rt, 99%; g. TBAF, THF, rt, 82%; h. Swern oxidation (oxalyl chloride–DMSO, CH2Cl2, triethylamine, −72 °C), 76%; i. methyltriphenylphosphonium bromide, n-BuLi, THF, −20 °C → rt, 42%; j. KOH, THF–MeOH–water, rt, 81%. | ||
Alcohol 13 was oxidised to aldehyde 14 under Swern conditions at −70 °C;20–221H NMR analysis of the crude residue revealed the presence of the diagnostic proton at 9.78 ppm as a doublet, thus confirming the identity of the product, which was used without further purification for the next Wittig reaction. Homologation of 14 with (ethoxycarbonylmethylene)triphenylphosphorane in anhydrous DMF at rt22 smoothly afforded alkene 15 as an 85 : 15 trans–cis mixture in 85% combined yield (over two steps) (Scheme 3); separation of the geometric isomers was not required since the newly formed double bond was to be hydrogenated in the following step. Catalytic hydrogenation of alkene 15 with 10% palladium on charcoal in ethanol under H2 atmosphere furnished hexanoate 16 in quantitative yield.23 Subsequently, removal of the TBDMS protecting group with tetra-n-butylammonium fluoride in dry THF at rt24 provided alcohol 17 in excellent yield (82%), thus releasing the hydroxy function at C-6 to be used for the final chain elongation, yet again by means of a Swern–Wittig sequence. Oxidation of 17 in the presence of oxalyl chloride–DMSO in dry CH2Cl2 at −72 °C, followed by treatment with triethylamine,20–22 gave aldehyde 18 (76%) which was immediately homologated with methylenetriphenylphosphorane in THF at −20 °C to provide 6-heptenoate derivative 19 in 42% yield (Scheme 3).25 Batty and Crich used buffered pyridinium chlorochromate (PCC) oxidation to prepare the (4S,5R) enantiomer of the L-threitol-derived aldehyde 18 in order to avoid migration of the isopropylidene group to the 5,6 site, which they experienced on the erythro analogue using other oxidation methods, including Swern's.25 In contrast, however, we did not observe any 5,6-isopropylidene shift during Swern oxidation of either alcohol 13 or 17, and both could be converted into the desired aldehyde 14 and 18, respectively. Saponification of the ester 19 with KOH in THF–MeOH–water (2 : 2 : 1) afforded the desired free acid 20 in 81% yield,25 which was directly employed for the next coupling step with alcohol 9.
Completion of the total synthesis
With the two alkene partners in hand, assembly of the dienic substrate for the crucial macrocyclization reaction via ring-closing metathesis (RCM)9 was performed by coupling alcohol 9 to acid 20 with DCC in the presence of DMAP in diethyl ether at rt, to give ester 21 in 85% yield (Scheme 4). A highly-diluted solution (0.45 mM) of diene 21 in anhydrous degassed dichloromethane was then refluxed for 48 h under argon atmosphere in the presence of Grubbs' catalyst I (17 mol %) (Fig. 2),9 to afford the 10-membered lactone 22 in excellent yield (92%) as a mixture of two geometric isomers in a 67 : 33 E–Z-ratio (Scheme 4). The desired trans-oxecin (E)-22 could be separated by column chromatography from the cis-analogue (Z)-22; the stereochemistry of the newly formed double bond was unambiguously assigned by comparison of the J7–8 coupling constants (15.6 against 10.3 Hz, respectively).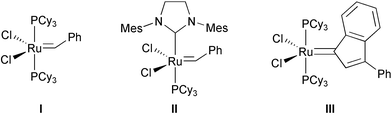 | ||
| Fig. 2 Catalysts for ring-closing metathesis (RCM): I Grubbs' catalyst; II 2nd generation Grubbs' catalyst; III Fürstner's catalyst. | ||
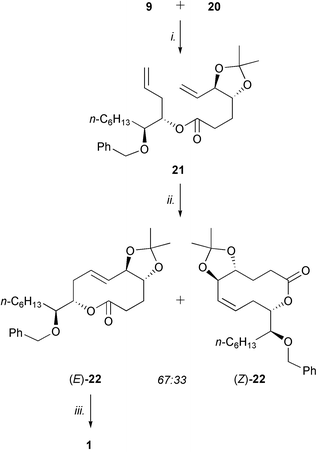 | ||
| Scheme 4 Completion of the total synthesis. Reagents and conditions: i., DCC, DMAP, Et2O, rt, 85%; ii., Grubbs' catalyst I, CH2Cl2, reflux, 92%; iii., TiCl4, CH2Cl2, 0 °C, 66%. | ||
The same stereoselectivity in the RCM cyclization has been also reported by Marco and coworkers in the course of the first total synthesis of microcarpalide,26 by using a closely related diene ester that only differed in the protection at 1′-OH, whereas formation of the unwanted (Z)-olefin resulted upon exposure to second-generation Grubbs' catalyst II (Fig. 2).
The lack of stereocontrol in the synthesis of medium-sized rings by RCM has been repeatedly addressed in the literature (for leading references, see ref. 27) and still represents a major drawback of this reaction. Mixtures of (E) and (Z) cycloalkenes are usually formed and, in addition, 8- to 11-membered rings are sensitive to the reverse process (ROM or ROMP) because of their intrinsic ring strain.27 Therefore, a reliable and general method of controlling the geometry of the newly formed double bond in RCM macrocyclizations has been called for. In this respect, second-generation Grubbs' catalyst II is known to favour the formation of the thermodynamically more stable (Z)-isomers.27
Within 10-membered lactones, for instance, in the total synthesis of pinolidoxin by Liu and Kozmin, the presence of an acetonide group spanning the two hydroxy functions in positions α and β to one of the alkene appendages was found to result in the stereoselective formation of the undesired (Z)-olefin in the key RCM step with catalyst II, whereas removal of the acetonide prior to metathetic ring closure determined a loss of stereocontrol in double bond formation.28 Similarly, but opposite in sense, excellent stereoselectivity in RCM was reported by Fürstner's group in the course of their recent total synthesis of herbarumins and pinolidoxin,27 a family of phytotoxic nonenolides related to 1, by using the newly developed ruthenium indenylidene catalyst III29 (Fig. 2). In particular, the stereocontrol of the key RCM macrocyclization was successfully reversed by simply switching to ruthenium complex III, resulting in the selective formation of the desired (E)-oxecin in a 10 : 1 ratio with the (Z)-isomer, which, in contrast, was formed exclusively when carbene II was employed as the catalyst.27
Prompted by such promising performances, we also exposed diene 21 to a catalytic amount of Fürstner's complex III,29 under similar reaction conditions. Although compound 21 was successfully metathesized to lactone 22, unfortunately the reaction afforded a mixture of (E) and (Z) isomers in about 2 : 1 ratio, respectively, as determined on the basis of 1H NMR analysis, by using the signal belonging to H-7 [for (E)-olefin: 5.32 ppm, doublet of doublets; for (Z): 5.50 ppm, triplet] as diagnostic marker.
In contrast, the most recent total synthesis of microcarpalide by Gurjar et al. featured an RCM reaction with Grubbs' catalyst I which proceeded with excellent stereoselectivity (10 : 1), yielding almost exclusively the required (E)-alkene from a diene ester similar to 21, except for the presence of two benzyl groups protecting the hydroxyls at positions 5 and 6, as well as for a methoxyethoxymethyl substituent at 1′-OH.30 Hence, the nature of the diene substrate that is subjected to the RCM reaction, and especially of its appendages, seems to play an important role in determining the stereochemical outcome of the metathetic process, at least for 10-membered lactones, thus making it difficult to draw up general rules for controlling selectivity in RCM reactions, even within a given catalyst.
Treatment of (E)-22 with titanium tetrachloride in CH2Cl2 at 0 °C30–31 (Scheme 4) resulted in the simultaneous removal of both protective groups, affording a single product whose spectral data matched perfectly those reported by Hemscheidt3 for the natural compound. Like natural microcarpalide, when dissolved in acetonitrile synthetic 1 appears as a mixture of two slowly interconverting conformers in a 76 : 24 ratio, as determined from the 1H NMR spectrum by using signals which displayed suitable separation, namely those for H-10 (4.84 ppm for the major conformer and 4.63 ppm for the minor one) and, in addition, 5-OH (3.09 and 3.19 ppm, respectively). This conformer ratio is identical to the 3.5 : 1 value described in the literature for the natural 1 in the same solvent.3
At present, only two total syntheses of microcarpalide have been disclosed in the literature so far.26,30 Like ours, both rely on the construction of the 10-membered skeleton by means of a ring-closing metathesis (RCM) macrocyclization using Grubbs' catalyst I as the crucial step, thus requiring the synthesis of two alkene partners, namely A and B (Scheme 1). Subunit B was always prepared from the chiral pool, by starting either from D-tartaric acid26 or D-mannose;30 the latter, however, required a cumbersome and unduly lengthy sequence, thus making the tartrate-derived one more attractive and applicable, even though a more straightforward and, possibly, enantioselective approach would be desirable for future developments. As far as partner A is concerned, it was previously synthesized by means of diastereoselective addition of an allyltin reagent to an (R)-glycidol-derived aldehyde as the key step26 or, later,30 through asymmetric dihydroxylation of a suitable alkene precursor, followed by a long sequence of transformations. In contrast, here a completely different and original route to fragment A was followed, which fully exploited the stereoselective homologation of chiral boronic esters10 as a powerful tool to introduce sequentially stereocentres with high diastereoselectivity and desired absolute stereochemistry by use of the same chiral auxiliary.
Conclusions
In summary, a new stereoselective total synthesis of the microfilament disrupting agent microcarpalide (1), a secondary metabolite produced by an endophytic fungus, has been accomplished. Formation of the required 10-membered lactone was achieved by means of ring-closing metathesis (RCM) of a suitable diene ester that was assembled by coupling the two advanced fragments A and B (i.e.9 and 20, respectively). Fragment A has been synthesized in 7 steps from n-bromohexane through two consecutive stereoselective homologations of chiral boronic esters 4 and 6 as key transformations, which allowed the sequential introduction of the two asymmetric centres with the final S absolute configuration, using (+)-pinanediol as the chiral auxiliary. Final elaboration entailed insertion of the allylic appendage on derivative 7 by nucleophilic displacement of the chlorine atom, followed by oxidative removal of the boronic scaffold to reveal alcohol 9. In turn, fragment B has been prepared in 10 steps from D-(−)-tartaric acid. The required C7 backbone of 20 has been constructed from the C4 starting synthon using two different Swern–Wittig oxidation–homologation sequences as key steps, each one with a suitable phosphorus ylide. Hence, D-tartrate-derived monosilylated threitol 13 was oxidized and homologated to afford alkene 15, which was hydrogenated and exposed to TBAF, thus revealing alcohol 17. Swern oxidation and Wittig methylenation furnished ester 19, whose saponification yielded 6-heptenoic acid 20. Completion of the total synthesis entailed a DCC-mediated coupling between partners 9 and 20, followed by RCM using Grubbs' catalyst I, providing the desired 10-membered macrocyclic framework with good stereoselectivity (67 : 33 E–Z) and excellent yield. Finally, cleavage of protective groups afforded a single product whose spectroscopic properties were identical with those of the natural compound 1.With a view to future structure–activity relationship (SAR) studies, the synthetic route to 1 herein disclosed appears flexible enough to allow the preparation of microcarpalide analogues, by simply varying the starting materials and/or the chiral director employed. In particular, our total synthesis features an original enantioselective approach to fragment A that allows the asymmetric insertion of suitably labelled atoms in specific positions by means of Matteson's stereoselective homologation, thus making available labelled derivatives which could be of value for shedding light on the mechanism of action of this novel fungal metabolite endowed with such a peculiar biological activity. Further applications of this methodology toward the preparation of analogues for SAR studies are being currently pursued in our laboratory and will be reported in due course.
Experimental
General
1H and 13C NMR spectra were recorded in CDCl3 solution (unless otherwise noted) on a Bruker DPX 200 or AMQ 400 MHz spectrometer; chemical shifts are reported in δ values from TMS as internal standard; coupling constants (J) are given in Hz. For mass spectral determinations a Finnigan MAT SSQ A and a Hewlett Packard HP5989A mass spectrometer were used (EI, 70 eV). Elemental analyses were performed with a Carlo Erba Elemental Analyzer 1110. Optical rotations were recorded in chloroform (unless stated otherwise) at 20 °C on a Perkin-Elmer 241 polarimeter and are in 10−1 deg cm2 g−1. All organic solvents were dried and distilled by standard methods prior to use and all reactions requiring anhydrous conditions were carried out using oven-dried and argon-flushed glassware. Chromatographic purification of compounds was performed on silica gel (particle size 0.05–0.20 mm). Analytical TLC was performed on pre-loaded (0.25 mm) glass supported silica gel plates (Merck, Kieselgel 60, F254). Compounds were visualized by exposure to UV light, I2 vapours and by dipping the plates in 1% Ce(SO4)2·4H2O, 2.5% (NH4)Mo7O24·4H2O in 10% sulfuric acid followed by heating on a hot plate. (1S,2S,3R,5S)-(+)-Pinanediol and all other reagents were obtained from Aldrich. Grubbs' catalyst I was purchased from Strem Chemicals Ltd., whereas dichloro(3-phenyl-1H-inden-1-ylidene)bis(tricyclohexylphosphine)ruthenium(IV) (Fürstner's catalyst, III) was prepared according to Fürstner et al.29Synthesis of fragment A
Synthesis of fragment B
(2E,4R,5R)-(15). 1H NMR (200 MHz): δ 0.07 (6H, s, 2 × tBuMe2Si), 0.90 (9H, s, 3 × Me2tBuSi), 1.29 (3H, t, J 7.1, CH2CH3), 1.42 (6H, s, 2 × Me2C), 3.69–3.86 (3H, m, H-5 and 2 × H-6), 4.20 (2H, q, J 7.1, CH2CH3), 4.45–4.56 (1H, m, H-4), 6.12 (1H, dd, J 15.7, 1.6, H-2), 6.94 (1H, dd, J 15.7, 5.1, H-3). 13C NMR: δ −4.13, −4.07, 15.6, 19.6, 27.2, 28.1, 28.3, 61.8, 64.1, 79.2, 82.1, 111.2, 123.3, 146.0, 176.7. MS, m/z: 329 ([M − 15]+, 13%), 299 (7.3), 281 (5.9), 241 (4.0), 229 (100), 199 (20), 183 (34), 155 (34), 117 (36), 109 (51), 89 (41), 84 (29), 75 (74), 59 (21), 43 (18).
(2Z,4R,5R)-(15). 1H NMR (200 MHz): δ 0.057 (3H, s, tBuMe2Si), 0.061 (3H, s, tBuMe2Si), 0.88 (9H, s, 3 × tBuMe2Si), 1.28 (3H, t, J 7.1, CH2CH3), 1.43 (6H, s, 2 × Me2C), 3.69–3.86 (3H, m, H-5 and 2 × H-6), 4.17 (2H, q, J 7.1, CH2CH3), 4.76–4.84 (1H, m, H-4), 5.91 (1H, dd, J 11.7, 1.0, H-2), 6.18 (1H, dd, J 11.7, 8.6, H-3). 13C NMR: δ −4.0, 27.3, 28.4, 28.5, 61.7, 65.2, 75.0, 83.6, 111.3, 124.2, 146.9, (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O not seen). MS, m/z: 329 ([M − 15]+, 4.3%), 287 (26), 269 (24), 241 (17), 229 (91), 199 (9.2), 183 (68), 155 (34), 117 (52), 109 (100), 89 (15), 84 (23), 75 (62), 59 (20), 43 (18).CH2), 5.38 (1H, ddd, J 17.1, 1.5, 0.8, CHCH2), 5.81 (1H, ddd, J 17.1, 10.1, 7.2, CHCH2). 13C NMR: δ 15.6, 28.2, 28.3, 28.6, 32.0, 61.8, 80.9, 83.8, 110.2, 120.6, 136.4, 174.5. MS, m/z: 213 ([M − 15]+, 21%), 195 (0.6), 171 (9.9), 125 (100), 115 (5.5), 98 (66), 83 (34), 69 (32), 55 (21), 43 (61). (Found: C, 63.2; H, 8.7. C12H20O4 requires: C, 63.1; H, 8.8%).CH2), 5.38 (1H, ddd, J 17.2, 1.5, 0.9, CHCH2), 5.81 (1H, ddd, J 17.2, 10.2, 7.3, CHCH2). 13C NMR: δ 26.5, 26.9, 27.1, 30.3, 79.4, 82.4, 108.9, 119.1, 135.0, 178.7. MS, m/z: 200 (M+, 0.1%), 185 (64), 167 (1.4), 144 (5.1), 125 (100), 98 (93), 83 (57), 69 (47), 55 (28), 43 (84). (Found: C, 60.2; H, 7.8. C10H16O4 requires: C, 60.0; H, 8.0%).
O not seen). MS, m/z: 329 ([M − 15]+, 4.3%), 287 (26), 269 (24), 241 (17), 229 (91), 199 (9.2), 183 (68), 155 (34), 117 (52), 109 (100), 89 (15), 84 (23), 75 (62), 59 (20), 43 (18).CH2), 5.38 (1H, ddd, J 17.1, 1.5, 0.8, CHCH2), 5.81 (1H, ddd, J 17.1, 10.1, 7.2, CHCH2). 13C NMR: δ 15.6, 28.2, 28.3, 28.6, 32.0, 61.8, 80.9, 83.8, 110.2, 120.6, 136.4, 174.5. MS, m/z: 213 ([M − 15]+, 21%), 195 (0.6), 171 (9.9), 125 (100), 115 (5.5), 98 (66), 83 (34), 69 (32), 55 (21), 43 (61). (Found: C, 63.2; H, 8.7. C12H20O4 requires: C, 63.1; H, 8.8%).CH2), 5.38 (1H, ddd, J 17.2, 1.5, 0.9, CHCH2), 5.81 (1H, ddd, J 17.2, 10.2, 7.3, CHCH2). 13C NMR: δ 26.5, 26.9, 27.1, 30.3, 79.4, 82.4, 108.9, 119.1, 135.0, 178.7. MS, m/z: 200 (M+, 0.1%), 185 (64), 167 (1.4), 144 (5.1), 125 (100), 98 (93), 83 (57), 69 (47), 55 (28), 43 (84). (Found: C, 60.2; H, 7.8. C10H16O4 requires: C, 60.0; H, 8.0%).DCC-mediated coupling and final steps of the total synthesis
(5R,6R,7E,10S)-22. [α]D −37.9 (c 3.0). 1H NMR (200 MHz): δ 0.90 (3H, t, J 6.5, 3 × H-7′), 1.27 (8H, br envelope, H-3′ to H-6′), 1.41 (6H, s, 2 × Me2C), 1.51–1.80 (2H, m, 2 × H-2′), 1.87–2.71 (6H, m, 2 × H-3, H-4 and H-9), 3.47 (1H, m, H-1′), 3.64 (1H, m, H-5), 3.92 (1H, t, J 9.2, H-6), 4.56 (1H, d, J 11.6, CH2Ph), 4.65 (1H, d, J 11.6, CH2Ph), 4.95 (1H, m, H-10), 5.32 (1H, dd, J 15.6, 9.2, H-7), 5.78 (1H, ddd, J 15.6, 11.0, 4.7, H-8), 7.29–7.40 (5H, m, arom). 13C NMR: δ 14.0, 22.5, 25.4, 25.6, 26.9, 27.1, 29.4, 29.7, 31.7, 34.2, 72.5, 73.4, 79.8, 80.4, 84.4, 108.8, 127.8, 127.9, 128.4, 129.2, 130.3, 138.2, 171.8. MS, m/z: 430 (M+, 0.22%), 415 (0.33), 373 (0.60), 328 (1.1), 298 (0.71), 237 (3.3), 220 (1.8), 205 (4.3), 203 (7.0), 179 (1.8), 123 (6.2), 113 (14), 91 (100), 85 (23), 79 (6.2), 65 (3.1), 55 (5.5). (Found: C, 72.4; H, 9.0. C26H38O5 requires: C, 72.5; H, 8.9%).
(5R,6R,7Z,10S)-22. [α]D +4.5 (c 1.6). 1H NMR (200 MHz): δ 0.89 (3H, t, J 6.5, 3 × H-7′), 1.27 (8H, br envelope, H-3′ to H-6′), 1.40 (3H, s, Me2C), 1.42 (3H, s, Me2C), 1.50–1.77 (2H, m, 2 × H-2′), 2.01–2.77 (6H, m, 2 × H-3, H-4 and H-9), 3.49 (1H, m, H-1′), 3.66 (1H, ddd, J 10.2, 9.5, 2.3, H-5), 4.52 (1H, dd, J 9.5, 8.0, H-6), 4.58 (1H, d, J 11.6, CH2Ph), 4.66 (1H, d, J 11.6, CH2Ph), 5.10 (1H, ddd, J 11.8, 4.4, 2.2, H-10), 5.50 (1H, t, J 10.3, H-7), 5.74 (1H, dt, J 10.3, 7.0, H-8), 7.28–7.44 (5H, m, arom). 13C NMR: δ 14.0, 22.5, 25.4, 26.8, 27.0, 29.3, 29.6, 30.5, 31.7, 32.1, 72.6, 72.8, 77.1, 79.7, 81.5, 107.6, 127.7, 128.4, 130.3, 130.9, 138.0, 176.6. MS, m/z: 430 (M+, 0.69%), 415 (2.3), 373 (0.30), 328 (1.1), 298 (0.60), 265 (1.7), 237 (3.0), 220 (1.7), 205 (3.9), 203 (6.3), 179 (2.5), 123 (5.1), 113 (13), 91 (100), 85 (16), 79 (6.6), 65 (3.1), 55 (6.6). (Found: C, 72.7; H, 9.1. C26H38O5 requires: C, 72.5; H, 8.9%).
Acknowledgements
We are grateful to the Università di Modena e Reggio Emilia (“Progetto Giovani Ricercatori” to P.D.) for financial support. Spectroscopic assistance from the staff at Centro Interdipartimentale Grandi Strumenti (Università di Modena) has been greatly appreciated throughout the project. We thank Stefania Morandi and Chiara Danieli for critically examining NMR assignments of pinanediol boronates. We are also grateful to Dr Claudia Zucchi for kindly providing Fürstner's catalyst III.References
- (a) J. Gusman and M. Vanhaelen, Recent Res. Dev. Phytochem., 2000, 4, 187–206 Search PubMed; (b) R.-X. Tan and W.-H. Zou, Nat. Prod. Rep., 2001, 18, 448–459 RSC; (c) B. Schulz, C. Boyle, S. Draeger, A. K. Rommert and K. Krohn, Mycol. Res., 2002, 106, 996–1004 Search PubMed.
- For recent examples: (a) D. B. Stierle, A. A. Stierle and T. Bugni, J. Org. Chem., 2003, 68, 4966–4969 CrossRef CAS; (b) G. Y. Chen, Y. C. Lin, L. Wen, L. L. P. Vrijmoed and E. B. G. Jones, Tetrahedron, 2003, 59, 4907–4909 CrossRef CAS; (c) J. A. Findlay, G. Q. Li, J. D. Miller and T. O. Womiloju, Can. J. Chem., 2003, 81, 284–292 CrossRef CAS; (d) A. Abdel-Lateff, C. Klemke, G. M. König and A. D. Wright, J. Nat. Prod., 2003, 66, 706–708 CrossRef CAS; (e) P. Kongsaeree, S. Prabpai, N. Sriubolmas, C. Vongvein and S. Wiyakrutta, J. Nat. Prod., 2003, 66, 709–711 CrossRef CAS; (f) B. Köpke, R. W. S. Weber and H. Anke, Phytochemistry, 2002, 60, 709–714 CrossRef; (g) G. Strobel, E. Ford, J. Worapong, J. K. Harper, A. M. Arif, D. M. Grant, P. C. W. Fung and R. M. W. Chau, Phytochemistry, 2002, 60, 179–183 CrossRef CAS; (h) S. F. Brady, S. M. Bondi and J. Clardy, J. Am. Chem. Soc., 2001, 123, 9900–9901 CrossRef CAS; (i) J. Y. Li, J. K. Harper, D. M. Grant, B. O. Tombe, B. Bashyal, W. M. Hess and G. A. Strobel, Phytochemistry, 2001, 56, 463–468 CrossRef CAS.
- A. S. Ratnayake, W. Y. Yoshida, S. L. Mooberry and T. Hemscheidt, Org. Lett., 2001, 3, 3479–3481 CrossRef CAS.
- B. Bodo, L. Molho, D. Davoust and D. Molho, Phytochemistry, 1983, 22, 447–451 CrossRef CAS.
- (a) A. Evidente, R. Lanzetta, R. Capasso, M. Vurro and A. Bottalico, Phytochemistry, 1993, 34, 999–1003 CrossRef CAS; (b) A. Evidente, R. Capasso, M. A. Abouzeid, R. Lanzetta, M. Vurro and A. Bottalico, J. Nat. Prod., 1993, 56, 1937–1943 CrossRef CAS.
- A. Arnone, G. Assante, M. Montorsi, G. Nasini and E. Ragg, Gazz. Chim. Ital., 1993, 123, 71–73 CAS.
- (a) A. Evidente, R. Lanzetta, R. Capasso, A. Andolfi, A. Bottalico, M. Vurro and M. C. Zonno, Phytochemistry, 1995, 40, 1637–1641 CrossRef CAS; (b) A. Evidente, R. Lanzetta, R. Capasso, A. Andolfi, M. Vurro and M. C. Zonno, Phytochemistry, 1997, 44, 1041–1045 CrossRef CAS; (c) A. Evidente, R. Capasso, A. Andolfi, M. Vurro and M. C. Zonno, Phytochemistry, 1998, 48, 941–945 CrossRef CAS.
- (a) J. F. Rivero-Cruz, G. García-Aguirre, C. M. Cerda-García-Rojas and R. Mata, Tetrahedron, 2000, 56, 5337–5344 CrossRef; (b) J. F. Rivero-Cruz, M. Macías, C. M. Cerda-García-Rojas and R. Mata, J. Nat. Prod., 2003, 66, 511–514 CrossRef CAS.
- Reviews: (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS; (b) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef CAS; (c) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef CAS.
- (a) D. S. Matteson, Acc. Chem. Res., 1988, 21, 294–300 CrossRef CAS; (b) D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS; (c) D. S. Matteson, J. Organomet. Chem., 1999, 581, 51–65 CrossRef CAS.
- D. S. Matteson, K. M. Sadhu and M. L. Peterson, J. Am. Chem. Soc., 1986, 108, 810–819 CrossRef CAS.
- H. R. Snyder, J. A. Kuck and J. R. Johnson, J. Am. Chem. Soc., 1938, 60, 105–111 CrossRef CAS.
- (a) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588–7590 CrossRef CAS; (b) D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 1529–1535 CrossRef CAS.
- (a) M. M. Midland, A. R. Zolopa and R. L. Halterman, J. Am. Chem. Soc., 1979, 101, 248–249 CrossRef CAS; (b) D. S. Matteson, Acc. Chem. Res., 1970, 3, 186–193 CrossRef CAS.
- (a) D. S. Matteson and R. Ray, J. Am. Chem. Soc., 1980, 102, 7590–7591 CrossRef CAS; (b) D. S. Matteson, R. Ray, R. R. Rocks and D. J. Tsai, Organometallics, 1983, 2, 1536–1543 CrossRef CAS.
- (a) H. C. Brown, in Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 317–409 Search PubMed; (b) G. W. Kabalka, R. J. Newton, Jr. and J. Jacobus, J. Org. Chem., 1978, 43, 1567–1569 CrossRef CAS.
- M. Carmack and C. H. Kelley, J. Org. Chem., 1968, 33, 2171–2173 CrossRef CAS.
- P. W. Feit, J. Med. Chem., 1964, 7, 14–17 CrossRef CAS.
- J. Taunton, J. L. Collins and S. L. Schreiber, J. Am. Chem. Soc., 1996, 118, 10412–10422 CrossRef CAS.
- H. Iida, N. Imazaki and C. Kibayashi, J. Org. Chem., 1987, 52, 3337–3342 CrossRef CAS.
- S. Clough, M. E. Raggatt, T. J. Simpson, C. L. Willis, A. Whiting and S. K. Wrigley, J. Chem. Soc., Perkin Trans. 1, 2000, 2475–2481 RSC.
- P. Schnurrenberger, E. Hungerbühler and D. Seebach, Liebigs Ann. Chem., 1987, 733–744 Search PubMed.
- During a preliminary approach to fragment B, diol 12 was treated with NaH and benzyl bromide in DMF at −40 °C to afford the corresponding monobenzyl derivative.21 In fact, a benzyl protection seemed convenient since it could have been removed en route, during the hydrogenation step that was planned to follow the first Wittig homologation. Surprisingly and unexpectedly, however, cleavage of the benzyl group by catalytic hydrogenation was found rather troublesome, even under higher pressures. Hence, annoyingly, the problem was circumvented by switching to a different protecting group for diol 12, namely TBDMS, and the synthetic sequence had to be lengthened by one step, i.e., deprotection with TBAF.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190–6191 CrossRef CAS.
- (a) D. Batty and D. J. Crich, J. Chem. Soc., Perkin Trans. 1, 1992, 3193–3204 RSC; (b) D. Batty and D. J. Crich, Tetrahedron Lett., 1992, 33, 875–878 CrossRef CAS.
- J. Murga, E. Falomir, J. García-Fortanet, M. Carda and J. A. Marco, Org. Lett., 2002, 4, 3447–3449 CrossRef CAS.
- A. Fürstner, K. Radkowski, C. Wirtz, R. Goddard, C. W. Lehmann and R. Mynott, J. Am. Chem. Soc., 2002, 124, 7061–7069 CrossRef.
- D. Liu and S. A. Kozmin, Org. Lett., 2002, 4, 3005–3007 CrossRef CAS.
- A. Fürstner, O. Guth, A. Düffels, G. Seidel, M. Liebl, B. Gabor and R. Mynott, Chem.-Eur. J., 2001, 7, 4811–4820 CrossRef CAS.
- M. K. Gurjar, R. Nagaprasad and C. V. Ramana, Tetrahedron Lett., 2003, 44, 2873–2875 CrossRef CAS.
- E. J. Corey, J. L. Gras and P. Ulrich, Tetrahedron Lett., 1976, 11, 809–812 CrossRef.
- M. F. Lappert, Chem. Rev., 1956, 56, 959–1064 CrossRef CAS.
- H. C. Brown, N. G. Bhat and V. Somayaji, Organometallics, 1983, 2, 1311–1316 CrossRef CAS.
- L. J. Rubin, H. A. Lardy and H. O. L. Fischer, J. Am. Chem. Soc., 1952, 74, 425–428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of synthetic microcarpalide (1). See http://www.rsc.org/suppdata/ob/b3/b308709c/ |
| This journal is © The Royal Society of Chemistry 2004 |