Amphidinolides, bioactive macrolides from symbiotic marine dinoflagellates†
Jun'ichi
Kobayashi
* and
Masashi
Tsuda
Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
First published on 7th January 2004
Abstract
Covering: 1986–2003
This review covers the structures, synthesis, biosynthesis, and bioactivity of a series of cytotoxic macrolides, named amphidinolides, isolated from symbiotic marine dinoflagellates of the genus Amphidinium which were separated from inside cells of marine flatworms. The structures of long-chain polyketides such as colopsinols isolated from Amphidinium sp. are also described.
 Jun'ichi Kobayashi | Jun'ichi Kobayashi was born in 1949 at Hirosaki, Japan. He completed his B.S. degree in 1973, and his M.S. degree in 1975, at Hokkaido University, working with Professor Yoshihisa Mizuno on studies of nucleic acid synthesis. In 1975 he joined Mitsubishi-Kasei Institute of Life Sciences where he worked on the synthesis and conformational analyses of bioactive peptides. After receiving his Ph.D. from Hokkaido University in 1979, he initiated his research program at the University of Illinois with Professor K. L. Rinehart from 1982 to 1984. In 1989 he was appointed as a full professor at Hokkaido University, Graduate School of Pharmaceutical Sciences, where he still continues his research career. His main research interests are the search for bioactive substances from marine organisms, plants, and microorganisms and their application to the basic research of life sciences as well as the development of new drugs. |
 Masashi Tsuda | Masashi Tsuda was born in 1965 at Muroran, Japan. He received his B.S. in 1989 under the direction of Professor Ko Kaneko and his M.S. in 1991 under the direction of Professor Jun'ichi Kobayashi from Hokkaido University. From 1993 he worked as a research assistant in Professor Kobayashi's laboratory. He obtained his Ph.D. for studies on the structures and stereochemistry of marine natural products with unique ring systems from Hokkaido University in 1995 (under the supervision of Professor Jun'ichi Kobayashi). His research is focused on structural studies on bioactive substances from marine micro- and macroorganisms. |
1 Introduction
Marine microorganisms such as bacteria, cyanobacteria, dinoflagellates, etc. have attracted many natural product chemists as the real producers of marine toxins such as fish and algal poisons and bioactive substances isolated from marine invertebrates such as sponges, tunicates, and so on. Among marine microorganisms, dinoflagellates have been important sources of marine toxins, and have been investigated world-wide by natural product chemists. We have continued investigations on chemically interesting and biologically significant secondary metabolites from symbiotic marine dinoflagellates Amphidinium sp., which were separated from inside cells of Okinawan marine flatworms.This review covers the literature published on a series of cytotoxic macrolides, designated amphidinolides, and long-chain polyketides isolated from marine symbiotic dinoflagellates Amphidinium sp., and is a comprehensive review including our early reviews from 1993,1 1997,2 1999,3,4 and 2003.5 Synthetic work on amphidinolides up to 2000 was reviewed by Chakraborty and Das.6 Other reviews covering secondary metabolites from dinoflagellates have been published previously.7–12 In this review, topics include the isolation, structures, synthesis, biosynthesis, and bioactivity of amphidinolides, and cultivation and taxonomic studies of the dinoflagellates.
2 Culture of dinoflagellates Amphidinium sp. and isolation of amphidinolides
In our research on bioactive substances from marine organisms, we started the search for secondary metabolites from symbiotic marine microorganisms in 1986.13,14 When extracts of a number of microorganisms were subjected to several biological screens, the extracts of symbiotic dinoflagellates Amphidinium sp. collected by Professor Yamasu, University of the Ryukyus, were found to exhibit potent cytotoxic activity (70–90% inhibition at 3 µg mL−1) against murine lymphoma L1210 cells and human epidermoid carcinoma KB cells. The dinoflagellates Amphidinium sp. were isolated from the inner tissue of acoel flatworms Amphiscolops sp. living on algae or seaweeds in Okinawa coral reefs.15Large-scale cultures of the dinoflagellates Amphidinium sp. in our laboratory have been performed using 3-L flat-bottomed glass flasks containing 2 L of seawater medium enriched with 1–3% Provasoli's Erd-Schreiber (ES) supplement16,17 [NaNO3: 300 mg, sodium glycerophosphate: 50 mg; FeEDTA: 2.5 mg, metal solution (including BO3−, Mn2+, Zn2+, and Co2+): 25 mL, vitamin B12: 10 µg, vitamin B1: 0.5 mg, biotin: 5 µg, TRIS: 500 mg in 100 mL distilled water, pH 7.8]. Static incubation with 8000 luces illumination in a cycle of 16 h of light and 8 h of darkness is carried out for 2 weeks at 25 °C. The cultures were harvested by removal of the supernatant through suction and then centrifugation to yield algal cells ranging from 0.1–0.3 g per 1 L medium. Recently, 0.8 L glass tall-dishes containing 500 mL of seawater medium have been used instead of 3-L glass flasks. The latter method is easier for handling, and yields of the cells were improved to 0.7–1.0 g per 1 L medium.
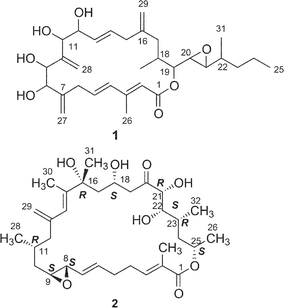
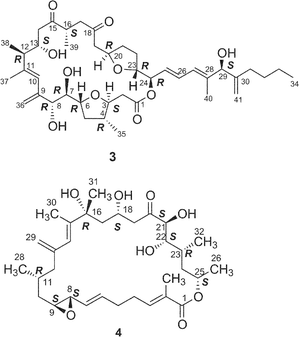
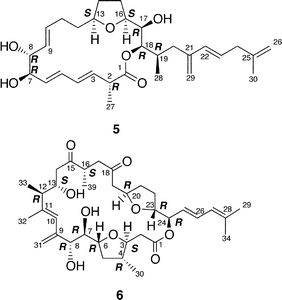
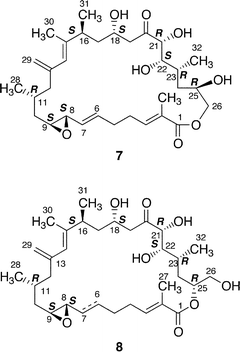
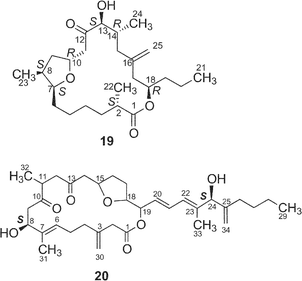
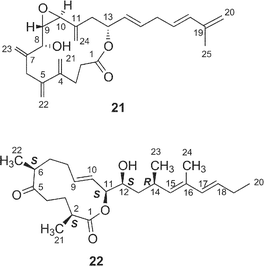
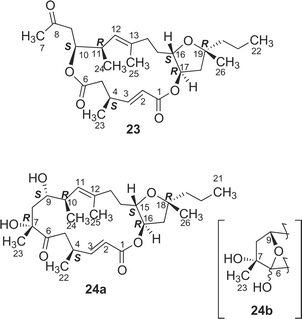
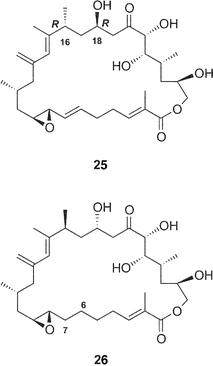
Harvested cells were extracted with methanol–toluene followed by partitioning between toluene and water. The toluene-soluble fractions were subjected to a systematic separation using silica gel column chromatography and C18 HPLC. In total, 34 cytotoxic macrolides, designated amphidinolides A–H (1–8), J–S (9–18), T1 (19), U (20)–Y (24), G2 (25), G3 (26), H2–H5 (27–30), and T2–T5 (31–34), have been isolated so far. From Amphidinium sp. (Y-5 strain, 25 µm in length and 20 µm in width) separated from the inside of a cell of a flatworm Amphiscolops sp. (500 µm in length and 220 µm in width, green color) collected off Chatan beach, Okinawa, 15 macrolides, amphidinolides A18 (1), B19,20 (2), C21 (3), D20 (4), E22 (5), J23 (9), K24 (10), M25 (12), N26 (13), O27 (14), P27 (15), Q28 (16), R29 (17), S29 (18), and V30 (21), and a linear polyketide, amphidinin A31 (35), have been isolated. Isolation yields and cytotoxicity of the amphidinolides are shown in Table 1. Two other collections of Amphidinium sp. (strain numbers Y-25 and Y-26) have been investigated. From extracts of the cultured cells of the Y-25 strain separated from a flatworm Amphiscolops breviviridis collected off Sunabe Beach, Okinawa, amphidinolides G32 (7), H32 (8), and L33 (11) were isolated, while from those of the Y-26 strain obtained from a flatworm Amphiscolops magniviridis collected at Zampa, Okinawa, amphidinolide F34 (6) was isolated together with amphidinolides B (2) and C (3). Recently, a variety of amphidinolides have been isolated from four strains (Y-56, Y-71, Y-72, and Y-42) of Amphidinium sp. obtained from different collections of Amphiscolops sp. flatworms as follows; 1) amphidinolides T135–37 (19), T3–T536,37 (32–34), and U38 (20) together with 2 and 3 from Y-56 strain separated from the Zampa collection of the flatworm, 2) amphidinolides T1 (19) and T236 (31) together with 2, 3, and 6 from Y-71 strain separated from the Sunabe collection, 3) amphidinolides G (7) and H (8) from Y-72 strain separated from the Zampa collection, and 4) amphidinolides G239 (25), G339 (26), H2–H539 (27–30), W40 (22), X41 (23), and Y42 (24) together with 7 and 8 from Y-42 strain separated from the Sunabe collection.
| Compd. | Lactone size |
Isolation yields (10−4
%)
Strain no.a |
Cytotoxicity
(IC50,b µg mL−1) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Y-5 | Y-25 | Y-26 | Y-42 | Y-56 | Y-71 | Y-72 | L1210c | KBd | ||
| a Amphidinium sp. b 50% inhibition concentration. c Murine lymphoma cell. d Human epidermoid carcinoma cells. e Macrodiolide. f Linear polyketide. | ||||||||||
| 1 | 20 | 20 | 2.0 | 5.7 | ||||||
| 2 | 26 | 10 | 0.8 | 17 | 0.00014 | 0.0042 | ||||
| 3 | 25 | 15 | 0.3 | 9 | 12 | 0.0058 | 0.0046 | |||
| 4 | 26 | 4 | 0.019 | 0.08 | ||||||
| 5 | 19 | 4 | 2.0 | 10 | ||||||
| 6 | 25 | 0.1 | 6 | 1.5 | 3.2 | |||||
| 7 | 27 | 20 | 8 | 46 | 0.0054 | 0.0059 | ||||
| 8 | 26 | 17 | 7 | 82 | 0.00048 | 0.00052 | ||||
| 9 | 15 | 60 | 2.7 | 3.9 | ||||||
| 10 | 19 | 0.3 | 1.65 | 2.9 | ||||||
| 11 | 27 | 2 | 0.092 | 0.1 | ||||||
| 12 | 29 | 4 | 1.1 | 0.44 | ||||||
| 13 | 26 | 9 | 0.00005 | 0.00006 | ||||||
| 14 | 15 | 1 | 1.7 | 3.6 | ||||||
| 15 | 15 | 2 | 1.6 | 5.8 | ||||||
| 16 | 12 | 0.5 | 6.4 | > 10 | ||||||
| 17 | 15 | 5 | 1.4 | 0.67 | ||||||
| 18 | 16 | 1 | 4.0 | 6.5 | ||||||
| 19 | 19 | 50 | 9.2 | 18 | > 20 | |||||
| 20 | 20 | 2 | 12 | > 20 | ||||||
| 21 | 14 | 0.5 | 3.2 | 7 | ||||||
| 22 | 12 | 90 | 3.9 | > 10 | ||||||
| 23 | 16e | 4 | 0.6 | 7.5 | ||||||
| 24 | 17 | 7 | 0.8 | 8.0 | ||||||
| 35 | f | 0.6 | 3.6 | 3.0 | ||||||
3 Amphidinolide A
Amphidinolide A is a 20-membered macrolide possessing a dienoate chromophore, three exo-methylenes, two 1,2-diols, three branched methyls, and an epoxide, and exhibits cytotoxicity against L1210 cells (IC50 2.0 µg mL−1) and KB cells (IC50 5.7 µg mL−1) (Table 1).18 Previously, we proposed the stereochemistry of nine chiral centers in amphidinolide A (1) to be 1a on the basis of NOESY correlations and 1H–1H coupling constants.43 The proposed structure (1a) was synthesized by three groups independently. Pattenden and coworkers synthesized 1a and its epimer (1b) at C-20 and C-21, through inter- and intramolecular sp2–sp2 Stille coupling reaction between the bisalkenyl stannane (36) derived from 37 and the iodide acetate (38 or 39) prepared from the aldehyde 40 and 41 through Julia olefination44 (Scheme 1). Maleczka et al. have achieved the total synthesis of 1a together with two analogues (1c and 1d).45 Their convergent synthesis was performed using a Stille coupling with copper thiophenecarboxylate between 42 and 43 and then a ring-closing metathesis, the latter compound being formed by Mitsunobu esterification between 44 and 45 (Scheme 2). On the other hand, Trost and coworkers have succeeded in the total synthesis of 1a from three subunits 46–48 using ruthenium-catalyzed alkene–alkyne coupling46 (Scheme 3). However, the spectroscopic data of synthetic compounds 1a, 1b, 1c, and 1d were not coincident with those of the natural specimen. The stereostructure of amphidinolide A (1) is currently being reinvestigated by our group.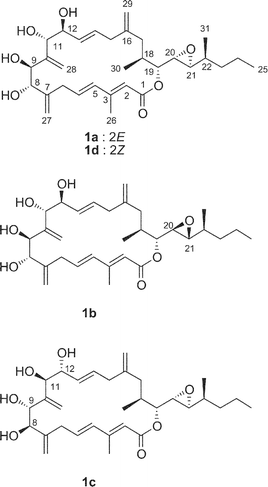
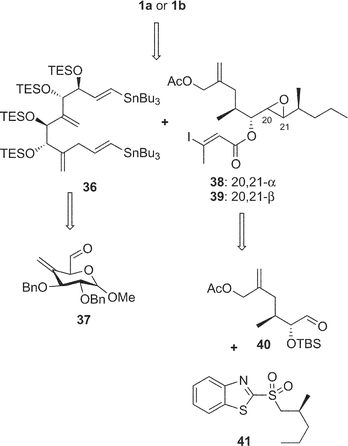 | ||
| Scheme 1 Retrosynthetic analysis of proposed structure (1a) and its 20,21-diastereomer (1b) of amphidinolide A by Pattenden's group. | ||
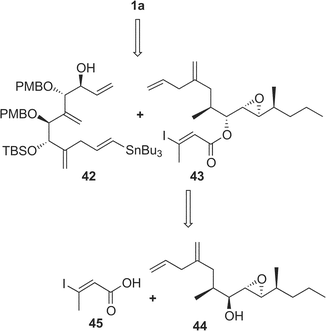 | ||
| Scheme 2 Retrosynthetic analysis of proposed structure (1a) of amphidinolide A by Maleczka's group. | ||
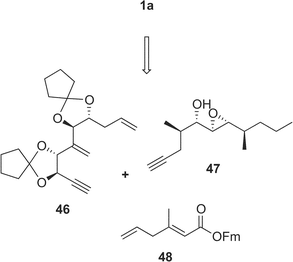 | ||
| Scheme 3 Retrosynthetic analysis of proposed structure (1a) of amphidinolide A by Trost's group. | ||
4 Amphidinolide B- and H-type macrolides
Amphidinolide B (2) is a 26-membered macrolide with an allyl epoxide and an S-cis diene moiety, and shows potent cytotoxicity (IC50 0.00014 and 0.0042 µg mL−1 against L1210 and KB cells, respectively).19,20 Amphidinolide B (2) caused a concentration-dependent increase in the contractile force of skeletal muscle skinned fibers.47 Shimizu and coworkers isolated three amphidinolide B-congeners,48 amphidinolides B1 (2), B2 (49), and B3 (50) from a free-swimming dinoflagellate Amphidinium operculatum ver nov Gibbosum.49 The relative stereochemistry of nine chiral centers in amphidinolide B1 (2) was determined by X-ray analysis. Amphidinolide B1 (2) was shown to be identical with amphidinolide B (2) by comparison of HPLC retention times, 1H NMR data, and optical rotations.48 The absolute stereochemistry of 2 was assigned as 8S, 9S, 11R, 16R, 18S, 21R, 22S, 23R, and 25S on the basis of chiral HPLC analyses of the C-22–C-26 segment (51a), which was obtained from 2 by oxidation with NaIO4, reduction with NaBH4, and acetylation followed by HPLC separation, and the synthetic C-22–C-26 segments (51a and 51b) prepared from (2S,4S)- and (2R,4R)-pentanediol.50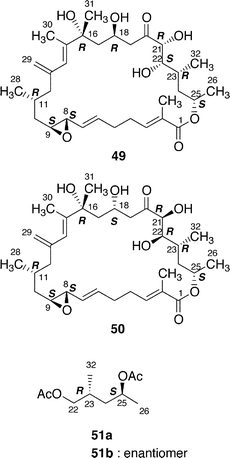
Shimizu and coworkers reported that amphidinolides B2 (49) and B3 (50) were C-18 and C-22 epimers of 2, respectively.48 The 1H NMR spectra of amphidinolide B2 (49) and D (4), the latter of which was assigned as a C-21 epimer of 2 by us, were quite similar to each other, indicating that the two compounds were identical.
Amphidinolides G (7) and H (8), 27- and 26-membered macrolides, respectively, are regioisomers at C-26 and C-25, respectively, and are also different in the position of a hydroxyl group (C-16 and C-26, respectively).32 Recently, amphidinolide H (8) was crystallized from hexane–benzene as colorless needles, mp 131–132 °C.51 The relative stereochemistry of 9 chiral centers in 8 was obtained from a single crystal X-ray diffraction analysis, and the perspective view of the final X-ray model is shown in Fig. 1. Amphidinolide H (8) was revealed to have a rectangular shape, which was bridged in the middle by an intramolecular hydrogen bond (1.99 Å) between O(O6)H and the epoxide O(O3). The bond length of one C–O bond [O3–C8; 1.470(3) Å] at the epoxide ring was greater than that of the other C–O bond [O3–C9; 1.448(3) Å], probably due to the effect of the intramolecular hydrogen bond. Another intramolecular hydrogen bond (1.92 Å) between O(O7)H-22 and O(O4)-18 assists the formation of an envelope-boat-shaped eight-membered ring in the C18–C19–C20–C21–C22–O7–O(O7)H–O4 portion. In this crystal structure the S-cis diene portion at C15–C14–C13–C29 was revealed to be twisted [torsion angle – 35.6(5)°]. The X-ray structures of amphidinolides H (8) and B (2) were close to each other, as the overlay of backbone structures shows in Fig. 2. Both conformations had an intramolecular hydrogen bond (2; 2.02 Å) between the hydroxyl group at C-21 and the epoxide oxygen atom, and their macrocyclic skeletons overlapped well with each other.
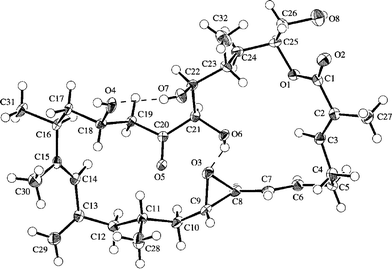 | ||
| Fig. 1 X-ray structure of amphidinolide H (8a). Intramolecular hydrogen bonds are shown by dashed lines. | ||
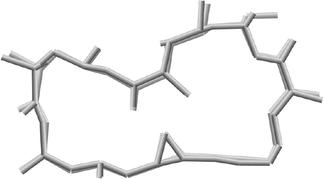 | ||
| Fig. 2 Overlay of X-ray structures of amphidinolides H (8a) and B (2). | ||
The absolute stereochemistry of amphidinolide H (8) was concluded to be 8S, 9S, 11R, 16S, 18S, 21R, 22S, 23R, and 25R on the basis of comparison of the 1H NMR data of the tris-(S)-MTPA ester (52a) of the C-22–C-26 segment derived from natural 8 by four-step conversion (reduction with NaBH4, oxidation with NaIO4, reduction with NaBH4, and MTPA-esterification followed by HPLC separation) with those of tris-(S)- and -(R)-MTPA esters (52a and 52b) of the C-22–C-26 segment synthesized from methyl (2S)-3-hydroxy-2-methylpropionate. On the other hand, treatment of amphidinolide H (8) with K2CO3 in EtOH at 4 °C for 18 h yielded a 1 : 1 mixture of 7 and 8. All spectral data of amphidinolide G (7) isolated from this mixture were identical to those of natural product (7). Thus, the absolute configurations of amphidinolide G (7) were concluded to be the same as those of amphidinolide H (8).
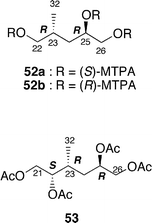
Amphidinolide L (11) is a 26-membered macrolide with a tetrahydropyran moiety, which corresponds to a 20-dihydro-21-dehydro derivative of amphidinolide G (7).33 The absolute configurations at C-21, C-22, C-23, and C-25 in 11 were assigned on the basis of comparison of the 1H NMR data of the C-21–C-26 segment (53) derived from a natural specimen of 11 by a four-step degradation (reduction with NaBH4, oxidation with NaIO4, reduction with NaBH4, and acetylation followed by HPLC separation) with those of the C-21–C-26 segment synthesized from methyl (2S)-3-hydroxy-2-methylpropionate.
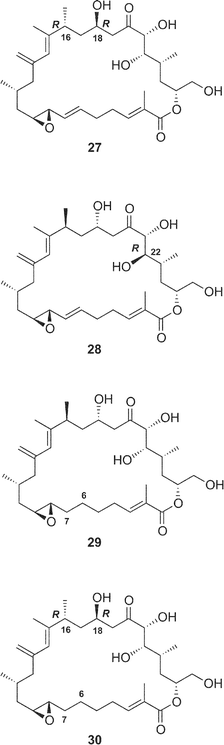
The structures of four amphidinolide H-congeners, amphidinolides H2–H5 (27–30) and two amphidinolide G-congeners, amphidinolides G2 (25) and G3 (26), were deduced from detailed analyses of spectroscopic data including J-based configuration analysis as well as distance-geometry calculation based on NOESY data.39 Amphidinolides H2 (27) and H3 (28) were assigned as 16 and 18-epimer and 22-epimer of amphidinolide H (8), respectively. The structures of amphidinolides H4 (29) and H5 (30) were assigned as the 6,7-dihydro form of amphidinolide H (8) and H2 (27), respectively. Amphidinolides G2 (25) and G3 (26) were assigned as 16 and 18-epimer and 6,7-dihydro form of amphidinolide G (7), respectively.
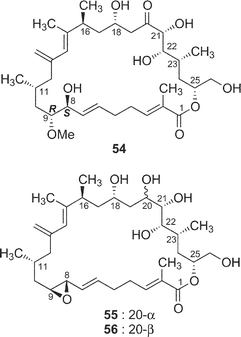
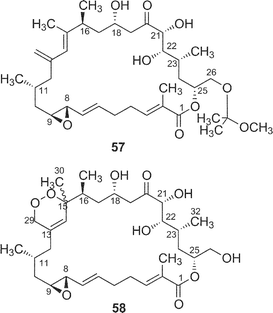
The cytotoxicity of five derivatives (54–58) of amphidinolide H (8) and ten amphidinolides B- and H-related macrolides (2, 4, 7, 8, and 25–30) was examined,37 and the results are summarized in Table 2. Amphidinolide H4 (29), the 6,7-dihydro form of 8, (0.18 and 0.23 µg mL−1 against L1210 and KB cells, respectively) was 300 and 400 times less potent than 8. Compound 54, a ring-opened form of 8, showed no cytotoxicity at 3 µg mL−1. Reduction of the ketone group at C-20 resulted in a remarkable reduction of the activity. Oxidation of the S-cis-diene moiety of 8 into peroxide 58 led to a 400 fold decrease of cytotoxicity (IC50 against L1210 and KB cells, 0.2 and 0.26 µg mL−1, respectively). These results indicate that the presence of an allyl epoxide, an S-cis-diene moiety, and the ketone at C-20 is important for the cytotoxicity of amphidinolide H (8)-type macrolides. Mean-panel studies of amphidinolides G (7) and H (8) using a 39 human tumor cell line panel revealed these macrolides to have a low correlation coefficient against known anticancer drugs, indicating that 7 and 8 are expected to have a different mechanism of action from those of known anticancer drugs. Amphidinolide H (8) exhibits antitumor activity against murine leukemia P388 mice (T/C: 140% at a dose of 0.2 mg kg−1).
| Compounds | IC50a/µg mL−1 | |
|---|---|---|
| L1210b | KBc | |
| a 50% inhibition concentration. b Murine lymphoma cell. c Human epidermoid carcinoma cells. | ||
| 2 | 0.00014 | 0.0042 |
| 4 | 0.019 | 0.08 |
| 7 | 0.0054 | 0.0046 |
| 8 | 0.00048 | 0.00052 |
| 25 | 0.3 | 0.8 |
| 26 | 0.72 | 1.3 |
| 27 | 0.06 | 0.06 |
| 28 | 0.002 | 0.022 |
| 29 | 0.18 | 0.23 |
| 30 | 0.2 | 0.6 |
| 54 | > 3 | > 3 |
| 55 | 0.3 | 0.2 |
| 56 | 0.2 | 0.2 |
| 57 | 0.0021 | 0.0064 |
| 58 | 0.2 | 0.26 |
Synthetic studies of amphidinolides B (2) and H (8) were carried out by some groups including our group. We synthesized two segments of amphidinolide B (2), the C-1–C-13 segment (59) and a 15-desmethyl form of the C-14–C-26 segment (60), esterification of which using Keck reaction afforded a key intermediate (61) (Scheme 4),52–54 but palladium-catalyzed coupling reaction between C-13 and C-14 in 61 did not succeed. Pattenden and coworkers also prepared the C-1–C-13 (62) and C-14–C-26 segments (63), which were coupled through an esterification by the Yamaguchi procedure to give an intermediate 64 (Scheme 5).55 The intramolecular Stille coupling in 64 using copper thiophene-2-carboxylate did not yield any macrocyclic compound. Nishiyama et al. synthesized the C-1–C-19 segment (65) of 2 through a coupling reaction between acetylene 66 and aldehyde 67, oxidation at C-13, construction of the exo-methylene by Tebbe's procedure, and then C2-elongation (Scheme 6).56
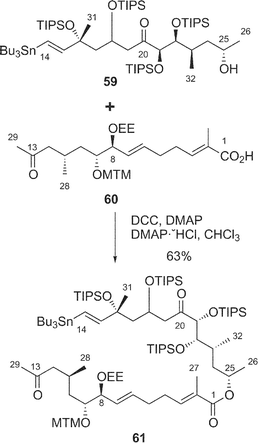 | ||
| Scheme 4 Our synthetic approach to amphidinolide B (2). | ||
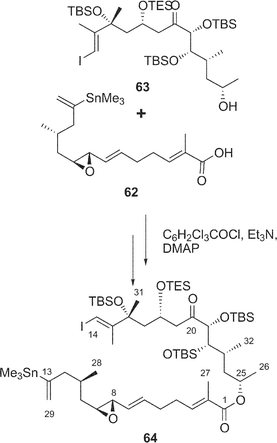 | ||
| Scheme 5 Pattenden's synthetic approach to amphidinolide B (2). | ||
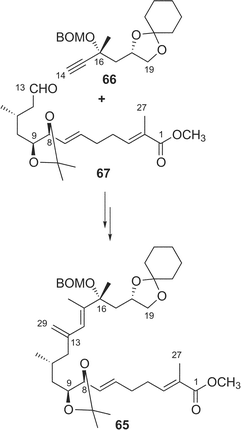 | ||
| Scheme 6 Nishiyama's synthetic approach to amphidinolide B (2). | ||
5 Amphidinolides C, F, and U
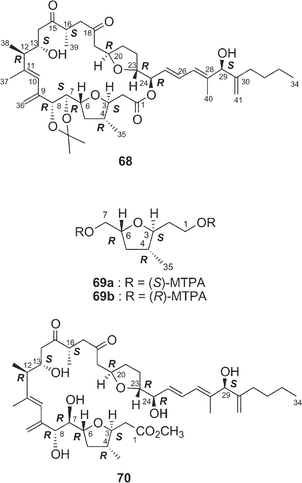
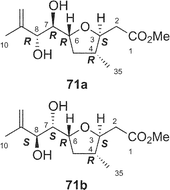
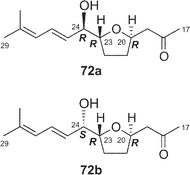
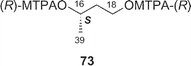
Amphidinolides C21 (3) and F33 (6) are 25-membered macrolides having two tetrahydrofuran rings and vicinally-located one-carbon branches. Amphidinolide C (3) exhibited potent cytotoxicity against tumor cells. Recently, relatively large amounts of amphidinolide C (3) have been isolated from three strains (Y-56, Y-62, and Y-71) of the genus Amphidinium, which were separated from the internal cells of the marine acoel flatworm Amphiscolops sp. This sample was used to reinvestigate the relative stereochemistry and to determine the absolute configurations at the twelve chiral centers in 3.57The relative stereochemistry of the C-1–C-8 and C-20–C-23 portions has been previously assigned tentatively by NOESY correlations of 3 and its 7,8-O-isopropylidene derivative (68).1,58 Application of the J-based configuration analysis59 revealed the erythro-relation for the C-12–C-13 bond and threo-relation for the C-23–C-24 bond. The absolute configurations of two oxymethine carbons at C-13 and C-29 were determined by the modified Mosher's method.60 To investigate the absolute stereochemistry at C-3, C-4, and C-5, reduction of 3 with DIBAL, oxidative cleavage of the 7,8-diol unit with NaIO4, reduction with NaBH4, esterification with (R)-(−)-MTPACl, followed by HPLC separation furnished the bis-(S)-MTPA ester (69a) of the C-1–C-7 segment. Both bis-(S)- and -(R)-MTPA esters (69a and 69b, respectively) of the C-1–C-7 segment were prepared from D-glutamic acid. 1H NMR data of the bis-(S)-MTPA ester (69a) derived from a natural specimen were identical with those of the synthetic bis-(S)-MTPA ester (69a). Therefore, the absolute configurations at C-3, C-4, and C-6 were established to be S, R, and R, respectively. The absolute configurations at C-7, C-8, and C-24 were elucidated by application of modified Mosher's method for linear methyl ester (70) of 3. Furthermore, from comparison of the 1H NMR chemical shifts of MTPA esters of each diastereomer of the C-1–C-10 (71a and 71b) and C-17–C-29 segments (72a and 72b) with those of linear methyl ester 70, the absolute configurations at C-7, C-8, C-20, C-23, and C-24 in 3 were confirmed to be all R. To determine the absolute configuration at C-16 of 3, a three-step degradation reaction [Baeyer–Villiger oxidation using trifluoroperacetic acid (TFPA), reduction with LiAlH4, and MTPA-esterification followed by HPLC separation] was applied to 3 to afford a bis-(R)-MTPA ester (73) of 1,3-butanediol corresponding to the C-16–C-18 segment of 3. Authentic bis-(R)-MTPA esters of (S)-(+)- and (R)-(−)-1,3-butanediols were also prepared. 1H NMR data of compound 73 derived from a natural specimen were identical with those of authentic bis-(R)-MTPA ester of (S)-(+)-1,3-butanediol, indicating that the absolute configuration at C-16 of 3 was S. Therefore, the absolute configurations at twelve chiral centers in amphidinolide C (3) were assigned as 3S, 4R, 6R, 7R, 8R, 12R, 13S, 16S, 20R, 23R, 24R, and 29S.
Amphidinolide F34 (6) is a congener of amphidinolide C (3) with a shorter side chain by a C6 unit than that of 3. Since 1H and 13C chemical shifts of 6 were close to those of 3, the relative stereochemistry of eleven chiral centers in 6 was suggested to be the same as that of amphidinolide C (3). Amphidinolide U38 (20) is a novel 20-membered macrolide possessing a tetrahydrofuran ring, two exo-methylenes, three branched methyls, two ketones, two hydroxyl groups, and a C10 linear side-chain. The gross structure of the C-9–C-29 unit in amphidinolide U (20) corresponds to that of C-14–C-34 of amphidinolide C19 (3), while the carbon skeleton of the C-1–C-8 unit in 20 is very close to that of C-1–C-8 in amphidinolide A18 (1). This observation suggests that amphidinolide U (20) may be biogenetically related to amphidinolides C (3) and A (1).
Amphidinolide C (3) exhibited potent cytotoxic activity against L1210 and KB cells in vitro with IC50 values of 0.0058 and 0.0046 µg mL−1, respectively. Amphidinolides F (7) and U (20) showed only weak cytotoxicity against L1210 (IC50: 1.5 and 12 µg mL−1, respectively) and KB cells (IC50: 3.2 and 20 µg mL−1, respectively). The 25-membered macrolactone ring may be essential for cytotoxicity, and the length of side chain is considered to affect the potency of cytotoxic activity significantly.
6 Amphidinolide E
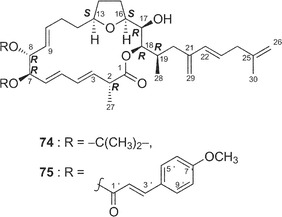
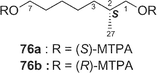
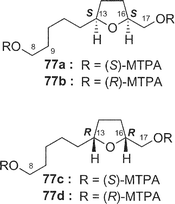
Amphidinolide E22 (5) is a 19-membered macrolide possessing a tetrahydrofuran ring, four C1 branches, and three hydroxyl groups, and exhibits cytotoxicity against L1210 cells (IC50: 2.0 µg mL−1). In the previous studies the stereochemistry of eight chiral centers in 5 remained unresolved due to lack of sample. Recently, we established the absolute stereochemistry of 5 using a substantial amount of sample (2 mg) obtained through repeated cultivation.61The relative stereochemistry of C-7 and C-8 was elucidated to be threo on the basis of NOESY data of the 7,8-isopropylidene derivative (74) of 5, while the relative stereochemistry of C-13, C-16, C-17, C-18, and C-19 was assigned as H-13/H-16-syn, C-16/C-17-threo, C-17/C-18-threo, and C-18/C-19-erythro by a combination of J-based configuration analysis4 and detailed analyses of NOESY data. The absolute stereochemistry was determined to be 7R and 8R by application of the exciton chirality method using 7,8-bis-O-p-methoxycinnamate (75). The 17R-configuration was assigned by modified Mosher's method for a hydroxyl group at C-17 in 5. To elucidate the absolute configuration at C-2, a five-step oxidative degradation of 5 was performed as follows: 1) catalytic hydrogenation using rhodium–alumina, 2) reduction with LiAlH4, 3) oxidation with NaIO4, 4) reduction with NaBH4, and 5) MTPA-esterification. Separation of reaction products furnished the 1,7-bis-(S)- and -(R)-MTPA esters of the C-1–C-7 (76a and 76b, respectively) and the 8,17-bis-(S)- and -(R)-MTPA esters of the C-8–C-17 segments (77a and 77b, respectively). The absolute configuration at C-2 with a methyl group was elucidated to be R on the basis of chemical shift differences and signal patterns of the two geminal protons at C-1 of 76a and 76b. The absolute stereochemistry at C-13 and C-16 of the C-8–C-17 segment was established by comparison of NMR data of 77a and 77b with those of the corresponding synthetic segments (77c and 77d), which were enantiomers of 77a and 77b, respectively. Therefore, the absolute configurations at eight chiral centers in amphidinolide E (5) were elucidated to be 2R, 7R, 8R, 13S, 16S, 17R, 18R, and 19R.
7 Amphidinolide J-type macrolides
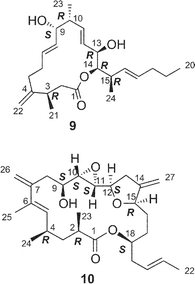
Amphidinolide J23 (9) is a 15-membered macrolide with two hydroxyl groups and four C1 branches, two of which are adjacent to each other. The absolute stereochemistry of 9 was determined on the basis of synthesis of three segments, C-1–C-7 (78), C-8–C-11 (79), and C-12–C-16 (80) obtained by ozonolysis. Recently, Williams and Kissel have succeeded in the total synthesis of amphidinolide J (9) through organozinc-mediated coupling between C-1–C-12 and C-13–C-20 subunits (81 and 82, respectively) followed by macrocyclization by the Yamaguchi procedure62 (Scheme 7).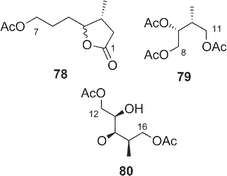
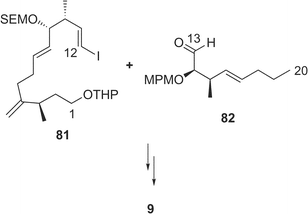 | ||
| Scheme 7 Synthesis of amphidinolide J (9) by Williams' group. | ||
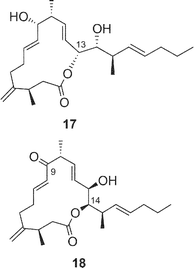
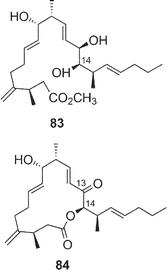
Amphidinolides R (17) and S (18) are minor congeners of amphidinolide J (9).29 The structure of 17 was assigned as a regioisomer of 9 having a 14-membered macrolactone ring, since treatment of 9 and 17 with sodium methoxide yielded an identical linear methyl ester (83). On the other hand, amphidinolide S (18) was concluded to be the 9-dehydro form of 9 by spectroscopic data. Interestingly, treatment of amphidinolide J (9) with MnO2 in benzene afforded the 13-keto derivative (84) but not 18. When oxidation of 9 with MnO2 was carried out in DMF solution, the 9-keto form (18) was produced. No 9,13-didehydro form was detected under either of the oxidative conditions. Since comparison of the 1H NMR spectra of 9 in DMF-d7 with those in benzene-d6 disclosed a small difference in J(H-8,H-9) values (6.0 Hz and 8.8 Hz, respectively), the selective oxidation of the hydroxyl group at C-9 or C-13 seems to depend on these slight conformational differences around C-9 in both solvents.
8 Amphidinolide K
The structure of amphidinolide K24 (10), a 19-membered macrolide containing a tetrahydrofuran ring, an epoxide, and an S-trans diene, was deduced from 2D NMR data using a submilligram of sample (0.3 mg). The relative stereochemistry of the epoxide–tetrahydrofuran portion was proposed as shown in 85 on the basis of NOESY data and coupling constants. Recently, Williams and Meyer achieved total synthesis of a stereoisomer (86) of the proposed structure (85) (Scheme 8).63 Their convergent strategy was initialized by construction of the C-7–C-22 subunit (87) via a coupling reaction between C-7–C-12 and C-13–C-22 segments (88 and 89, respectively) with a chiral bromoborane 90. The coupling reaction between C-1–C-6 (91) and C-7–C-22 (87) with copper thiophene-2-carboxylate as a cocatalyst followed by macrocyclization with C-18 inversion under Mitsunobu conditions furnished 86. The spectral data of 86 were identical with those of a natural specimen of amphidinolide K, but the sign of the [α] value of 76 was the opposite of that measured for amphidinolide K. Therefore the structure of amphidinolide K was concluded to be 10.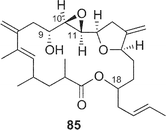
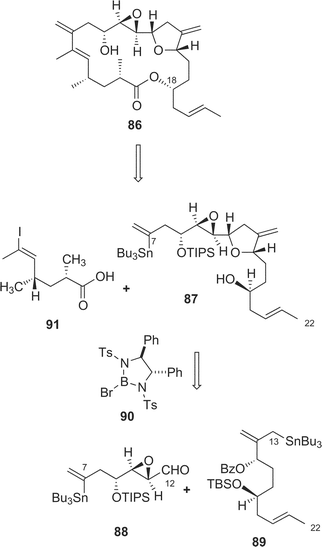 | ||
| Scheme 8 Retrosynthetic analysis of enantiomer (86) of amphidinolide K (10) by Williams' group. | ||
9 Amphidinolides M and N
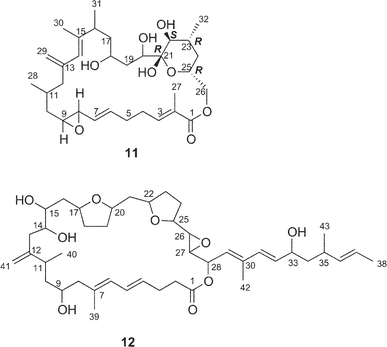
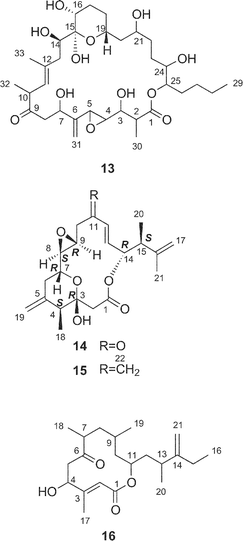
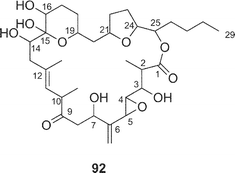
Amphidinolide M25 (12) has a 29-membered macrocyclic lactone ring with two tetrahydrofuran rings, an epoxide, two diene moieties, and two vicinally located methyl or exo-methylene groups. The stereochemistry of 12 remains undetermined, though the angular hydrogens of two tetrahydrofuran portions were both implied as trans-relations. Amphidinolide M (12) exhibited cytotoxicity against L1210 (IC50: 1.1 µg mL−1) and KB cells (IC50: 0.44 µg mL−1).The structure of amphidinolide N26 (13) was interpreted to be a 26-membered macrolide containing a 6-membered hemiacetal ring, an epoxide, a ketone carbonyl, four C1 branches, and seven hydroxyl groups. This compound was extremely cytotoxic against L1210 and KB cells (IC50: 0.00005 and 0.00006 µg mL−1, respectively). Although the relative stereochemistry of C-14, C-15, C-16, and C-19 was indicated as shown, the absolute stereochemistry of 13 was not determined. Shimizu and coworkers isolated an amphidinolide N-type macrolide, named caribenolide I (92), from a free-swimming dinoflagellate Amphidinium operculatum ver nov Gibbosum.64 Caribenolide I (92) was reported to show potent cytotoxicity against human colon tumor cells HCT116 and its drug-resistant strain HCT116/VM46 (IC50: both 0.001 µg mL−1), of which the IC50 value was about 100 times higher than that of amphidinolide B (IC50: 0.122 µg mL−1). Caribenolide I (92) showed antitumor activity against murine leukemia P388 (T/C: 150% at a dose of 0.03 mg kg−1) in vivo.
10 Amphidinolides O–Q and V and amphidinin A
Amphidinolides O (14) and P (15) are 15-membered macrolides possessing a tetrahydropyran ring, an epoxide, and two vicinally located methyl or exo-methylene groups.27 The structural difference between 14 and 15 is at the C-11 position, a ketone group for 14 and an exo-methylene group for 15. Natural product analogs with this type of structural difference are quite rare. Williams et al. achieved the total synthesis of (−)-amphidinolide P (93) in a convergent and highly enantio-controlled manner.65 The strategy was as follows; Sakurai reaction between the C-3–C-6 and the C-7–C-11 subunits (94 and 95 respectively) and then a C2-elongation to prepare the C-1–C-11 segment (96), Stille coupling between 96 and C-12–C-17 segment (97) using tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3), followed by intramolecular esterification (Scheme 9). The optical rotation of 15 was opposite to that of 93, thus indicating that the absolute stereochemistry of amphidinolide P was enantiomeric to the synthetic (−)-amphidinolide P (93). Amphidinolides O (14) and P (15) were cytotoxic against L1210 (IC50: 1.7 and 1.6 µg mL−1, respectively) and KB cells (IC50: 3.6 and 5.8 µg mL−1, respectively).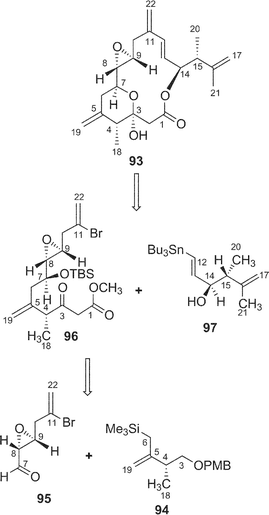 | ||
| Scheme 9 Retrosynthetic analysis of amphidinolide P (15) by Williams' group. | ||
Amphidinolide Q28 (16) is a 12-membered macrolide with four branched methyls, an exo-methylene, a ketone carbonyl, and a hydroxyl group. Convincing evidence for stereochemical assignment of 16 has not been provided. The 14-membered macrolide, amphidinolide V (21), possessed five exo-methylenes and one epoxide.30 The relative configurations at four chiral centers (C-8, C-9, C-10, and C-13) in 21 were deduced from the 1H–1H coupling constants and NOESY data. Amphidinolides Q (16) and V (21) showed cytotoxicity against L1210 (IC50: 6.4 and 3.2 µg mL−1, respectively) and KB cells (IC50: > 10 and 7 µg mL−1, respectively).
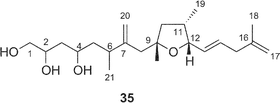
A cytotoxic linear polyketide, amphidinin A31 (35), was isolated together with amphidinolides O (14), P (15), Q (16). The structural features of 35 include vicinally located one-carbon branches, which are one of the unique structural features of the amphidinolides (vide infra).
11 Amphidinolide T-type macrolides
Amphidinolide T135 (19) is a 19-membered macrolide possessing a tetrahydrofuran ring, one exo-methylene, three branched methyls, one ketone, and one hydroxyl group. The absolute configurations at four (C-2, C-13, C-14, and C-18) of the seven chiral centers were determined to be S, S, R, and R, respectively, by modified Mosher's method for 19 and C-1–C-12 (98a and 98b) and C-13–C-21 segments (99a and 99b) (Scheme 10). The absolute configurations at C-7, C-8, and C-10 were elucidated to be S, S, and R, respectively, by comparison of 1H NMR data of C-1–C-12 segments (98a and 98b) with those of synthetic model compounds (100a and 100b) for the tetrahydrofuran portion.36 Furthermore, the structure of 19 was confirmed by a single crystal X-ray analysis37 (Fig. 3). Amphidinolide T1 (19) possesses an odd-numbered macrocyclic lactone ring but has no vicinally located one-carbon branches. The C21 backbone with four C1 branches (C-2, C-8, C-14, and C-16) of 19 is different from the carbon frameworks of two other 19-membered macrolides, amphidinolides E20 and K,22 which have a C26 backbone with four C1 branches (C-2, C-19, C-21, and C-25) and a C22 backbone with five C1 branches (C-2, C-4, C-6, C-7, and C-14), respectively.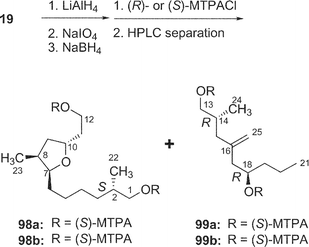 | ||
| Scheme 10 Oxidative degradation of amphidinolide T1 (19). | ||
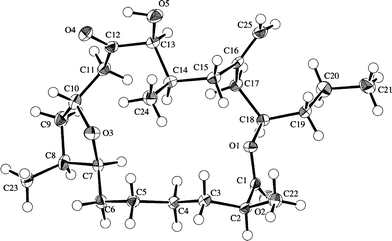 | ||
| Fig. 3 X-ray structure of amphidinolide T1 (19a). | ||
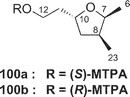
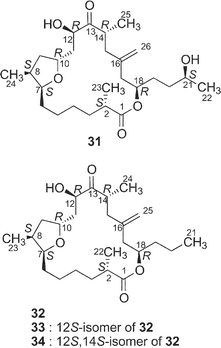
Amphidinolide T236 (31) is a congener of amphidinolide T1 (19) with one-carbon elongation at C-21, while amphidinolides T336 (32), T436 (33), and T536 (34) are 12-dihydro-13-dehydro isomers of 19. The absolute stereochemistry of 31–33 was elucidated by chemical methods similar to those applied for the determination of 19. The structure of amphidinolide T5 (34) was assigned by interconversion of 33 to 34 with K2CO3.
Recently, Fürstner and coworkers achieved the total synthesis of amphidinolide T4 (33),66 and the Ghosh group also reported the total synthesis of amphidinolide T1 (19).67 Key reactions in the Fürstner synthesis of 33 were 1) stannane-mediated cross coupling between anomeric sulfone 101 (C-4–C-10) and tert-butyldimethylsilyl enol ether 102 (C-11–C-15), 2) Pd2dba3 and tris(2-furyl)phosphane-catalyzed acylation reaction between 103 and 104, 3) ring-closing metathesis at the C-4/C-5 bond of 105 using a “second-generation” ruthenium–carbene complex, and construction of exo-methylene using Nysted's reagent (Scheme 11). Ghosh's strategy for the synthesis of 19 was the assembly of C-1–C-10 and C-11–C-21 segments (106 and 107, respectively) by oxocarbenium ion-mediated alkylation with AlCl3, macrolactonization of 108 under Yamaguchi conditions, and final reductive unmasking of bromoether (protecting group of exo-methylene) in 109 with Zn and NH4Cl (Scheme 12).
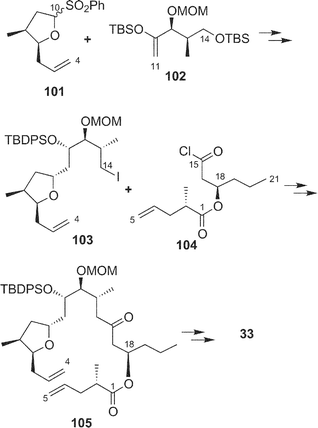 | ||
| Scheme 11 Strategy for total synthesis of amphidinolide T4 (33) by Fürstner's group. | ||
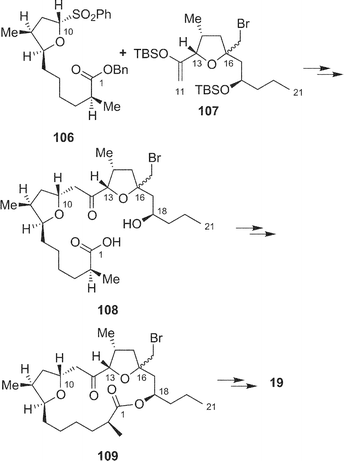 | ||
| Scheme 12 Strategy for total synthesis of amphidinolide T1 (19) by Ghosh's group. | ||
Amphidinolides T1–T5 (19 and 31–34) exhibit modest cytotoxicity against L1210 cells in vitro with IC50 values of 18, 10, 7.0, 11, and 12 µg mL−1, respectively.
12 Amphidinolide W
Further investigation of the less-polar fractions of Y-42 strain resulted in the isolation of a cytotoxic 12-membered macrolide, amphidinolide W40 (22). The gross structure was elucidated on the basis of the spectroscopic data including 13C–13C NMR correlations obtained from an INADEQUATE spectrum. The relative stereochemistry of C-11, C-12, and C-14 was elucidated to be 11,12-threo and 12,13-syn by J-based configuration analysis, and the absolute configuration at C-12 was assigned as S by modified Mosher's method. A 2S-configuration was deduced from 1H NMR data of 1,5,11,12-tetrakis-MTPA esters of a reductive product of 22, while the absolute configuration at C-6 was elucidated to be S on the basis of 1H NMR data of 6,11,12-tris-MTPA esters of C-6–C-20 segment (110a and 110b) obtained by the Baeyer–Villiger reaction of 22. Amphidinolide W (22) is the first macrolide without an exo-methylene unit among all of the amphidinolides isolated so far. The gross structure of the C-9–C-16 moiety of 22 corresponds to that of C-6–C-15 of amphidinolide H (8), which was contained in strain Y-42, suggesting that amphidinolide W (22) may be biogenetically related to amphidinolide H (8). Amphidinolide W (22) exhibits cytotoxicity against L1210 cells in vitro with an IC50 value of 3.9 µg mL−1.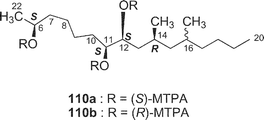
13 Amphidinolides X and Y
A cytotoxic 16-membered macrodiolide, amphidinolide X41 (23), has been isolated from a marine dinoflagellate Amphidinium sp. (strain Y-42). The gross structure of 23 was elucidated on the basis of spectroscopic data including one-bond and long-range 13C–13C correlations obtained from 2D DEPT C–C Relay and 2D DEPT C–C Long-Range Relay experiments The relative stereochemistry for C-10/C-11 was elucidated to be erythro by J-based configuration analysis, while that of the tetrahydrofuran portion was assigned on the basis of NOESY data. The absolute configurations at C-10 and C-17 were elucidated to be S and R, respectively, by application of modified Mosher's method for the C-8–C-22 segments (111a and 111b), which were produced together with the C-1–C-6 segments (112a and 112b) by reduction of 23 with LiAlH4. A 4S-configuration was deduced from comparison of 1H NMR data of MTPA esters (112a and 112b) of the C-1–C-6 segments with those of the synthetic 1,6-bis-(R)-MTPA ester (112b). Amphidinolide X (23) is the first macrodiolide consisting of polyketide-derived diacid and diol units from natural sources.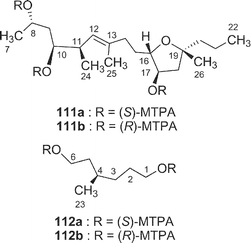
More recently, a 17-membered macrolide, amphidinolide Y42 (24), was obtained from the same strain, and it was elucidated to exist as a 9 : 1 equilibrium mixture of 6-keto and 6(9)-hemiacetal forms (24a and 24b, respectively) on the basis of 2D NMR data. The structure and absolute stereochemistry of the 6-keto form (24a) were assigned on the basis of spectroscopic data and chemical conversion of 24 into amphidinolide X (23) by Pb(OAc)4 oxidation. The 6-keto form (24a) of amphidinolide Y is a 17-membered macrolide possessing a tetrahydrofuran ring, five branched methyls, a ketone, and two hydroxyl groups.
Amphidinolides X (23) and Y (24) show cytotoxicity against L1210 (IC50: 0.6 and 0.8 µg mL−1, respectively) and KB cells (IC50: 7.5 and 8.0 µg mL−1, respectively).
14 Bioactivity of amphidinolides B, H, and N
Among all amphidinolides isolated so far, amphidinolides B (2), H (8), and N (13) show potent cytotoxicity against L1210 and KB cells in vitro (Table 1). The inhibitory pattern of amphidinolide N (13) against a 38 human tumor cell line panel was similar to that of rhodamin-123 with a high affinity for the mitochondria of malignant cells (prostate cancer, etc.), while that of amphidinolide H (8) was different from those of other known anticancer drugs with different mechanisms of action. It was found that amphidinolide B (2), belonging to the same group as amphidinolide H (8), activates actomyosin ATPase.45 The molecular target of amphidinolide H (8) is currently being investigated.15 Biosynthetic studies of amphidinolides B, C, H, J, T1, W, X, and Y
Naturally occurring macrolides generally possess even-numbered macrocyclic lactone rings. However, more than half of the amphidinolides have odd-numbered lactone rings. Amphidinolides have other unique structural features: (a) most of them contain at least one exo-methylene unit, and (b) vicinally located one-carbon branches are present in some of them. In our studies of the biosynthesis of amphidinolides, incorporation patterns of 13C-labeled acetates were investigated for the most abundant macrolide, amphidinolide J (9), in the Y-5 strain of Amphidinium sp.68 (Fig. 4). The incorporation patterns revealed that it is generated through non-successive mixed polyketides including two tetraketides (C-4–C-11 and C-13–C-20), one acetate unit (C-1–C-2), two isolated C1 units (m) from C-2 of acetates and four “m”-labeled C1 branches. This may explain the generation of the odd-numbered macrolactone ring.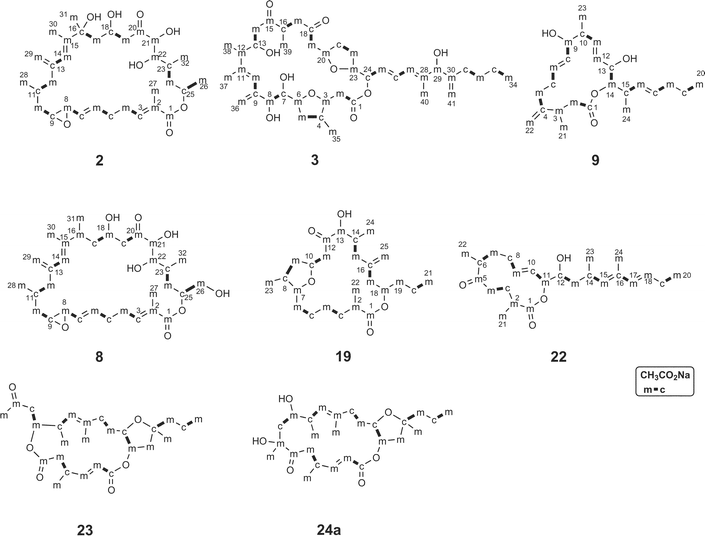 | ||
| Fig. 4 Acetate-incorporation patterns of amphidinolides B (2), C (3), H (8), J (9), T1 (19), W (22), X (23), and Y (24). | ||
Biosynthetic studies of amphidinolides B69 (2), C70 (3), and T136 (19) were performed on the basis of 13C NMR data of 13C enriched samples obtained by feeding experiments with [1-13C], [2-13C], and [1,2-13C2] sodium acetate in cultures of a dinoflagellate Amphidinium sp. (strain Y-71), while those of amphidinolides H71 (8), W72 (22), X41 (23), and Y42 (24) were carried out using the Y-42 and/or Y-72 strains. The incorporation patterns of amphidinolide H (8) suggested that 8 was generated from three unusual C2 units “m–m” derived only from C-2 of acetates in addition to three successive polyketide chains.71 It is noted that the six oxygenated carbons C-1, C-18, C-20, C-21, C-22, and C-26 in 8 were not derived from the C-1 carbonyl but from the C-2 methyl of acetates. The incorporation patterns of amphidinolide B (2) suggested it was generated from three successive polyketide chains, an isolated C1 unit from C-2 of acetate, six branched C1 units from C-2 of acetate, and an “m–m” and an “m–m–m” unit derived only from C-2 of acetates.69 The labeling patterns of amphidinolide B (2) were different from those of amphidinolide H (8), although the structures including absolute configurations were quite similar to each other. Amphidinolide W (22) might be generated from a hexaketide chain, two acetate units, four isolated C1 units from C-2 of acetates, and four branched C1 units from C-2 of acetates.72 The acetate-incorporation patterns for C-1–C-2–(C-21) and C-8–C-18–(C-23, C-24) of 22 corresponded well to those for C-1–C-2–(C-27) and C-5–C-15–(C-28, C-29) of 8.
The incorporation patterns suggested that amphidinolide C70 (3) was generated from four diketide units, four acetate units, five isolated C1 units from C-2 of acetates, seven branched C1 units from C-2 of acetates, and an “m–m” and an “m–m–m” unit derived only from C-2 of acetates. The C-9–C-12 portion including the vicinally located one-carbon branches in 3 disclosed the same labeling pattern, “c(m)–m–m(m)–m(m)”, as those of 8. Amphidinolide T136 (19) also showed a unique labeling pattern consisting of four successive polyketide chains, an isolated C1 unit formed from C-2 of acetate, and three unusual C2 units derived only from C-2 of acetate. It is unique that five oxygenated carbons of C-1, C-7, C-12, C-13, and C-18 were not derived from the C-1 carbonyl, but from the C-2 methyl of acetate. Two tetrahydrofuran portions in amphidinolide C (3) showed the “m–c–m–m” labeling pattern, while the tetrahydrofuran portion of amphidinolide T1 (19) was revealed to be “m–c–m–c”.
The incorporation patterns for the 9-keto form (24a) of amphidinolide Y42 suggested that 24a was generated from three diketide units, two acetate units, three isolated “m” units from C-2 of acetates, an “m–m” unit, and five branched C1 units from C-2 of acetates. The labeling patterns at C-1–C-6 and C-23–C-7–C-21 parts in 24a corresponded to those for the diacid and diol units in amphidinolide X (23),41 indicating that 23 might be generated from 24a through oxidative cleavage at the C-6–C-7 position of the minor 6(9)-hemiacetal form (24b).
16 Long-chain polyketides from dinoflagellates Amphidinium sp.
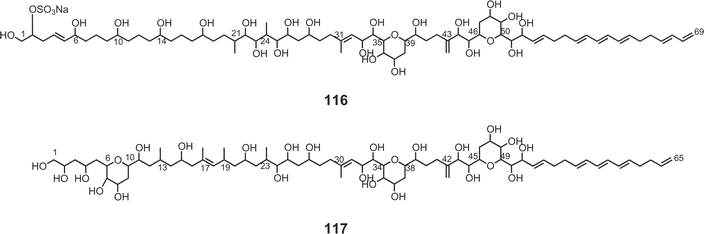
Luteophanols A–C73,74 (113–115) have been isolated from the Y-52 strain of the dinoflagellate Amphidinium sp., which was a symbiont of acoel flatworm Pseudaphanostoma luteocoloris. The gross structure of luteophanol A (113) was elucidated to be a C57-aliphatic chain with a sulfate ester, two tetrahydropyrans, nineteen hydroxyl groups, one exo-methylene, and two branched methyls by NMR data as well as FABMS/MS data. Both luteophanols B (114) and C (115) possess two tetrahydropyrans and twenty-three hydroxyl groups on a C64-linear aliphatic chain with three C1-branches. The central portions of these compounds as well as the C-15–C-42 moiety of 113 are structurally common to those of the amphidinols75–78 such as amphidinol-175 (116), -276 (117), -377 (118), and -477 (119), potent antifungal metabolites from A. klebii NIES 613 and A. carterae. Recently, Murata and co-workers reported the absolute stereostructures of amphidinol-377 (118) and the acetate labeling patterns of amphidinols78 (Fig. 5).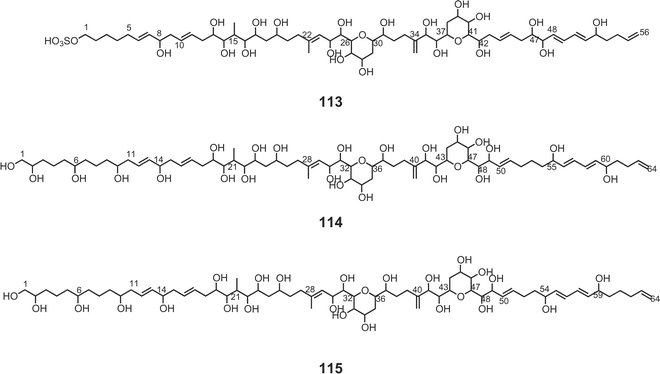
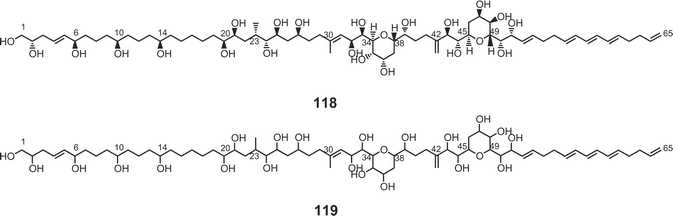
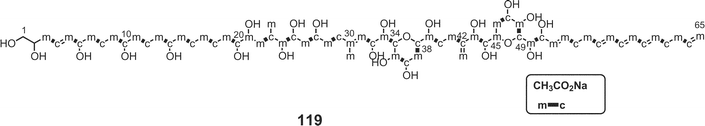 | ||
| Fig. 5 Acetate-incorporation patterns of amphidinol-4 (119). | ||
Colopsinols A–E79–81 (120–124) are the first members of a new class of polyketide natural products possessing a glucoside moiety and a sulfate ester. They were isolated from more polar fractions than the macrolide-containing fraction of the Y-5 strain of the dinoflagellate Amphidinium sp. The gross structures were elucidated on the basis of extensive spectroscopic analyses including CH2-selected editing HSQC NMR as well as FABMS/MS experiments and chemical means. The polyketide aglycone of colopsinol A81 (120) consists of a C56-linear aliphatic chain with three C1 branches (one exo-methylene and two methyls) as well as two ketones, five hydroxyl groups, and a tetrahydropyran and two epoxide rings. Colopsinol A (120) exhibited potent inhibitory activity against DNA polymerase α and β with IC50 values of 13 and 7 µM, respectively. On the other hand, colopsinols B (121) and C (122) are new polyhydroxyl compounds possessing a C53-linear carbon chain including three C1 branches as well as a tetrahydropyran, a tetrahydrofuran, and an epoxide ring, six hydroxyl groups, a glucoside moiety, and a sulfate ester.82 Colopsinol C (122) exhibited cytotoxicity against L1210 cells in vitro with the IC50 value of 7.8 µg mL−1, while colopsinol B (121) did not show such activity (IC50 > 10 µg mL−1). Colopsinols D (123) and E (124) are congeners of 120; 123 has a tetrahydrofuran ring at C-9–C-12, while 124 is the mono-deglucosyl form of 120.83 Colopsinol E (124) exhibited cytotoxicity against L1210 cells (IC50 value: 7 µg mL−1), while colopsinol D (123) did not show such activity (IC50 > 20 µg mL−1). Biosynthetically it is interesting that quite different types of polyketides such as colopsinols and amphidinolides are produced from the same dinoflagellate strain.
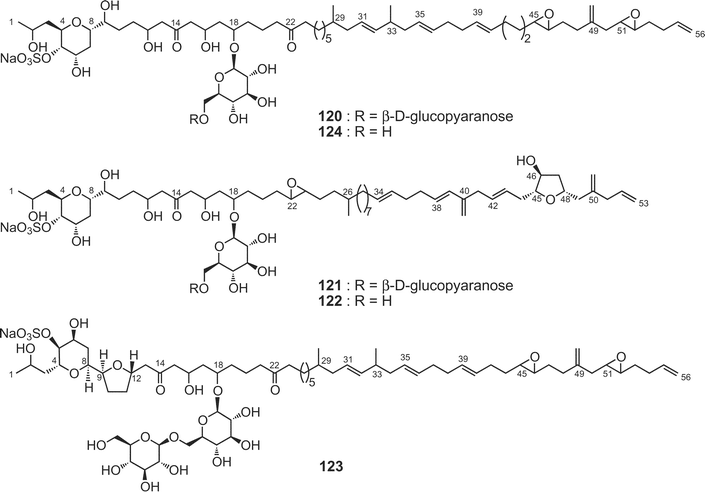
17 18S rDNA Phylogeny of dinoflagellates Amphidinium sp.
For the dinoflagellate Amphidinium sp. (strain number Y-42), from which amphidinolides G (7), H (8), G239 (25), G339 (26), H2–H539 (27–30), W40 (22), X41 (23), and Y42 (24) were isolated, total genomic DNA was extracted from the cultured cells. The small subunit ribosomal RNA gene (SSU rDNA) was amplified, and both the coding and non-coding strands were sequenced. The DNA sequence was compared with those of the SSU rDNA in the databases using BLAST SEARCH and the SSU rDNA of Amphidinium belauense (accession no. L13719), which was originally described as a symbiont of a flatworm Haplodiscus sp.,84 was found to be the closest relative (> 99% identity). Amphidinolides B1 (2), B2 (49) and B3 (50) and carbenolide I (92) were obtained from a free-swimming dinoflagellate Amphidinium operculatum ver nov Gibbosum,49 while amphidinols75–78 were isolated from Amphidinium klebii NIES 613 and Amphidinium carterae.18 Prospects
Marine dinoflagellates of the genus Amphidinium produce a variety of cytotoxic macrolides (amphidinolides, carbenolide I, etc.) and/or long-chain polyketides (colopsinols, luteophanols, amphidinols, etc.). Among all the amphidinolides, amphidinolides H (8) and N (13) exhibit remarkable potent cytotoxicity against human tumor cell lines and are expected to be lead compounds for new anticancer drugs. For further biological testing, the poor productivity of these macrolides needs to be considerably improved. One of the approaches is to identify the polyketide synthase in the dinoflagellate Amphidinium sp., although it will be quite difficult to find it in the huge genome of the dinoflagellate.85 Such an approach is also important for understanding the mechanisms of the unique biosynthesis of polyketides such as amphidinolides, amphidinols, and so on. Another approach is to clarify mechanisms of multiplication of this unicellular alga. For example, it is known that other toxic dinoflagellates (Prorocentrum lima,86Gymnodium breve,87Hetereocapsa circularisquama,88etc.) are responsible for algal blooms (red tides) through extraordinary multiplication, while in the case of laboratory cultivation of the dinoflagellate Amphidinium sp. multiplication of the microalga automatically ceases after limited cell division. Further development of the cell biology and molecular biology of dinoflagellates is required for biomedical and pharmaceutical application of dinoflagellate metabolites such as the amphidinolides.19 Acknowledgements
We thank Professor T. Yamasu, University of the Ryukyus, and Professor M. Ishibashi, Chiba University, for help with dinoflagellate collection, Dr T. Horiguchi and Mr K. Katsumata, Hokkaido University, for 18S rDNA analysis, Dr E. Fukushi and Professor J. Kawabata, Graduate School of Agriculture, Hokkaido University, for NMR measurements, Ms. S. Oka and Miss M. Kiuchi, Center for Instrumental Analysis, Hokkaido University, for ESIMS and FABMS measurements, and Ms. Y. Kariya for her assistance.20 References
- J. Kobayashi and M. Ishibashi, Chem. Rev., 1993, 93, 1753 CrossRef CAS.
- M. Ishibashi and J. Kobayashi, Heterocycles, 1997, 44, 543 CAS.
- J. Kobayashi, M. Ishibashi, in Comprehensive Natural Products Chemistry, Vol. 8. Miscellaneous Natural Products Including Marine Products, Pheromones, Plant Hormones, and Aspects of Ecology, ed. K. Mori, Elsevier Science, Oxford, 1999, pp. 415–650 Search PubMed.
- J. Kobayashi, M. Tsuda and M. Ishibashi, Pure Appl. Chem., 1999, 71, 1123 CAS.
- J. Kobayashi, K. Shimbo, T. Kubota and M. Tsuda, Pure Appl. Chem., 2003, 75, 337 Search PubMed.
- T. K. Chakraborty and S. Das, Curr. Med. Chem., Anti-cancer Agents, 2001, 1, 131 Search PubMed.
- Y. Shimizu, Curr. Opin. Microbiol., 2003, 6, 236 CrossRef CAS.
- A. H. Daranas, M. Norte and J. J. Fernández, Toxicon, 2001, 31, 1101 CrossRef CAS.
- B. S. Moore, Nat. Prod. Rep., 1999, 16, 653 RSC.
- K. S. Rein and J. Borrone, Comp. Biotechnol. Physiol., B: Biochem. Mol. Biol., 1999, 124, 117 Search PubMed.
- T. Yasumoto and M. Satake, Chimia, 1998, 52, 63 CAS.
- T. Yasumoto and M. Satake, J. Toxicol., Toxin Rev., 1996, 15, 91 Search PubMed.
- J. Kobayashi, M. Ishibashi, H. Nakamura, Y. Hirata, T. Yamasu, T. Sasaki and Y. Ohizumi, Experientia, 1988, 44, 800 Search PubMed.
- J. Kobayashi, M. Ishibashi, H. Nakamura, Y. Hirata, T. Ohizumi and Y. Ohizumi, J. Chem. Soc., Perkin Trans. 1, 1989, 101–103 RSC.
- T. Yamasu and A. Okazaki, Galaxea, 1987, 6, 61 Search PubMed.
- L. Provasoli, in Culture and Collection of Algae, ed. A. Watanabe and A. Hattori, Japanese Society of Plant Physiology, Tokyo, 1968, pp. 63–75 Search PubMed.
- H. Iwasaki, in Sourui Kenkyuhou, ed. K. Nishizawa and M. Chihara, Kyouritsu-Shuppan, Tokyo, 1979, pp. 281–293 Search PubMed.
- J. Kobayashi, M. Ishibashi, H. Nakamura, Y. Ohizumi, T. Yamasu, T. Sasaki and Y. Hirata, Tetrahedron Lett., 1986, 27, 5755 CrossRef CAS.
- M. Ishibashi, Y. Ohizumi, M. Hamashima, H. Nakamura, Y. Hirata, T. Sasaki and J. Kobayashi, J. Chem. Soc., Chem. Commun., 1987, 1127 RSC.
- J. Kobayashi, M. Ishibashi, H. Nakamura, Y. Ohizumi, T. Yamasu, Y. Hirata, T. Sasaki, T. Ohta and S. Nozoe, J. Nat. Prod., 1989, 52, 1036 CAS.
- J. Kobayashi, M. Ishibashi, M. R. Walchli, H. Nakamura, Y. Hirata, T. Sasaki and Y. Ohizumi, J. Am. Chem. Soc., 1988, 110, 490 CrossRef CAS.
- J. Kobayashi, M. Ishibashi, T. Murayama, M. Takamatsu, M. Iwamura, Y. Ohizumi and T. Sasaki, J. Org. Chem., 1990, 55, 3421 CrossRef CAS.
- J. Kobayashi, M. Sato and M. Ishibashi, J. Org. Chem., 1993, 58, 2645 CrossRef CAS.
- M. Ishibashi, M. Sato and J. Kobayashi, J. Org. Chem., 1993, 58, 6928 CrossRef CAS.
- J. Kobayashi, N. Yamaguchi and M. Ishibashi, J. Org. Chem., 1994, 59, 4698 CrossRef CAS.
- M. Ishibashi, N. Yamaguchi, T. Sasaki and J. Kobayashi, J. Chem. Soc., Chem. Commun., 1994, 1445 RSC.
- M. Ishibashi, M. Takahashi and J. Kobayashi, J. Org. Chem., 1995, 60, 6062 CrossRef CAS.
- J. Kobayashi, M. Takahashi and M. Ishibashi, Tetrahedron Lett., 1996, 37, 1449 CrossRef CAS.
- M. Ishibashi, M. Takahashi and J. Kobayashi, Tetrahedron, 1997, 53, 7827 CrossRef CAS.
- T. Kubota, M. Tsuda and J. Kobayashi, Tetrahedron Lett., 2000, 41, 713 CrossRef.
- J. Kobayashi, N. Yamaguchi and M. Ishibashi, Tetrahedron Lett., 1994, 35, 7049 CrossRef CAS.
- J. Kobayashi, H. Shigemori, M. Ishibashi, T. Yamasu, H. Hirota and T. Sasaki, J. Org. Chem., 1991, 56, 5221 CrossRef CAS.
- M. Tsuda, T. Sasaki and J. Kobayashi, J. Org. Chem., 1994, 59, 3734 CrossRef CAS.
- J. Kobayashi, M. Tsuda, M. Ishibashi, H. Shigemori, T. Yamasu, H. Hirota and T. Sasaki, J. Antibiot., 1991, 44, 1259 CAS.
- M. Tsuda, T. Endo and J. Kobayashi, J. Org. Chem., 2000, 65, 1349 CrossRef CAS.
- J. Kobayashi, T. Kubota, T. Endo and M. Tsuda, J. Org. Chem., 2001, 66, 134 CrossRef CAS.
- T. Kubota, T. Endo. M. Tsuda, M. Shiro and J. Kobayashi, Tetrahedron, 2001, 57, 6175 CrossRef CAS.
- M. Tsuda, T. Endo and J. Kobayashi, Tetrahedron, 1999, 55, 14565 CrossRef CAS.
- J. Kobayashi, J. K. Simbo, K. Sato and M. Tsuda, J. Org. Chem., 2002, 67, 6585 CrossRef CAS.
- K. Shimbo, M. Tsuda, N. Izui and J. Kobayashi, J. Org. Chem., 2002, 67, 1020 CrossRef CAS.
- M. Tsuda, N. Izui, K. Shimbo, M. Sato, E. Fukushi, J. Kawabata, K. Katsumata, T. Horiguchi and J. Kobayashi, J. Org. Chem., 2003, 68, 5339 CrossRef CAS.
- M. Tsuda, N. Izui, K. Shimbo, M. Sato, E. Fukushi, J. Kawabata and J. Kobayashi, J. Org. Chem., 2003, 68, 9109 CrossRef CAS.
- J. Kobayashi, M. Ishibashi and H. Hirota, J. Nat. Prod., 1991, 54, 1435 CrossRef CAS.
- H. W. Lam and G. Pattenden, Angew. Chem., Int. Ed., 2002, 41, 508 CrossRef CAS.
- R. E. Maleczka, Jr., L. R. Terrell, F. Geng and J. S. Ward, III, Org. Chem., 2002, 4, 2841 Search PubMed.
- B. M. Trost, J. D. Chisholm, S. T. Wrobleski and M. Jung, J. Am. Chem. Soc., 2002, 124, 12421.
- K. Matsunaga, K. Nakatani, M. Ishibashi, J. Kobayashi and Y. Ohizumi, Biochim. Biophys. Acta, 1999, 1427, 24 CrossRef CAS.
- I. Bauer, L. Maranda, Y. Shimizu, R. W. Peterson and L. Cornell, J. Am. Chem. Soc., 1994, 116, 2657 CrossRef CAS.
- L. Maranda and Y. Shimizu, J. Phycol., 1996, 32, 873 Search PubMed.
- M. Ishibashi, H. Ishiyama and J. Kobayashi, Tetrahedron Lett., 1994, 35, 8241 CrossRef CAS.
- J. Kobayashi, K. Shimbo, M. Sato, M. Shiro and M. Tsuda, Org. Lett., 2000, 2, 2805 CrossRef CAS.
- H. Ishiyama, T. Takemura, M. Tsuda and J. Kobayashi, Tetrahedron, 1999, 55, 4583 CrossRef CAS.
- H. Ishiyama, T. Takemura, M. Tsuda and J. Kobayashi, J. Chem. Soc., Perkin Trans 1, 1999, 1163 RSC.
- H. Ishiyama, Ph.D. thesis, 2000, Hokkaido University, Sapporo.
- M. B. Cid and G. Pattenden, Tetrahedron Lett., 2000, 41, 7373 CrossRef CAS.
- K. Ohi, K. Shima, K. Hamada and S. Nishiyama, in Symposium Papers of 41st Symposium on The Chemistry of Natural Products, in Nagoya, Minoru Isobe, Nagoya, 1999, pp. 31–36 Search PubMed.
- T. Kubota, M. Tsuda and J. Kobayashi, Org. Lett., 2001, 3, 1363 CrossRef CAS.
- H. Ishiyama, M. Ishibashi and J. Kobayashi, Chem. Pharm. Bull., 1996, 44, 1819 CAS.
- N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, J. Org. Chem., 1999, 64, 866 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- T. Kubota, M. Tsuda and J. Kobayashi, J. Org. Chem., 2002, 67, 1651 CrossRef CAS.
- D. R. Williams and W. S. Kissel, J. Am. Chem. Soc., 1998, 120, 11198 CrossRef CAS.
- D. R. Williams and K. G. Meyer, J. Am. Chem. Soc., 2001, 123, 765 CrossRef CAS.
- I. Bauer, L. Maranda, K. A. Young, Y. Shimizu, C. Fairchild, L. Cornell, J. MacBeth and S. Huang, J. Org. Chem., 1995, 60, 1084 CrossRef CAS.
- D. R. Williams, K. G. Meyer and L. Mi, Org. Lett., 2000, 2, 945 CrossRef CAS.
- A. Fürstner, C. Aïssa, R. Riveiros and J. Ragot, Angew. Chem., Int. Ed., 2002, 41, 4763 CrossRef CAS.
- A. K. Ghosh and C. Liu, J. Am. Chem. Soc., 2003, 125, 2374 CrossRef CAS.
- J. Kobayashi, M. Takahashi and M. Ishibashi, J. Chem. Soc., Chem. Commun., 1995, 1639 RSC.
- M. Tsuda, T. Kubota, Y. Sakuma and J. Kobayashi, Chem. Pharm. Bull., 2001, 49, 1366 CrossRef CAS.
- T. Kubota, M. Tsuda and J. Kobayashi, Tetrahedron, 2001, 57, 5975 CrossRef CAS.
- M. Sato, K. Shimbo, M. Tsuda and J. Kobayashi, Tetrahedron Lett., 2000, 41, 503 CrossRef CAS.
- M. Tsuda, N. Izui, M. Sato and J. Kobayashi, Chem. Pharm. Bull., 2002, 50, 976 CrossRef CAS.
- Y. Doi, M. Ishibashi, H. Nakamichi, T. Kosaka, T. Ishikawa and J. Kobayashi, J. Org. Chem., 1997, 62, 3820 CrossRef CAS.
- T. Kubota, M. Tsuda, Y. Doi, A. Takahashi, H. Nakamichi, M. Ishibashi, E. Fukushi, J. Kawabata and J. Kobayashi, Tetrahedron, 1998, 54, 14455 CrossRef CAS.
- M. Satake, M. Murata, T. Yasumoto, T. Fujita and H. Naoki, J. Am. Chem. Soc., 1991, 113, 9859 CrossRef CAS.
- G. K. Paul, N. Matusmori, M. Murata and K. Tachibana, Tetrahedron Lett., 1995, 36, 6279 CrossRef CAS.
- G. K. Paul, N. Matusmori, K. Konoki, M. Sasaki, M. Murata and K. Tachibana, in Harmful and Toxic Algal Blooms, ed. T. Yasumoto, Y. Oshima and Y. Fukuyo, UNESCO, Paris, 1996, pp. 503–506 Search PubMed.
- G. K. Paul, N. Matusmori, K. Konoki, M. Murata and K. Tachibana, J. Mar. Biotechnol., 1997, 5, 124 Search PubMed.
- M. Murata, S. Matsuoka, N. Matsumori, G. K. Paul and K. Tachibana, J. Am. Chem. Soc., 1999, 121, 870 CrossRef CAS.
- T. Houdai, S. Matsuoka, M. Murata, M. Satake, S. Ota, Y. Oshima and L. L. Rhodes, Tetrahedron, 2001, 57, 5551 CrossRef CAS.
- J. Kobayashi, T. Kubota, M. Takahashi, M. Ishibashi, M. Tsuda and H. Naoki, J. Org. Chem., 1999, 64, 1478 CrossRef CAS.
- T. Kubota, M. Tsuda, M. Takahashi, M. Ishibashi, N. Naoki and J. Kobayashi, J. Chem. Soc., Perkin Trans. 1, 1999, 3483 RSC.
- T. Kubota, M. Tsuda, M. Takahashi, M. Ishibashi, S. Oka and J. Kobayashi, Chem. Pharm. Bull., 2000, 48, 1447 CAS.
- R. K. Trench and H. Winsor, Symbiosis, 1987, 3, 1 Search PubMed.
- R. V. Snyder, P. D. L. Gibbs, A. Palacios, L. Abiy, R. Dickey, J. V. Lopez and K. S. Rein, Mar. Biotechnol., 2003, 5, 1 CrossRef CAS.
- T. Yasumoto, Y. Ohshima, W. Sugawara, Y. Fukuyo, H. Oguri, T. Igarashi and N. Fujita, Bull. Jpn. Soc. Sci. Fish., 1980, 46, 1397 Search PubMed.
- Y. Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James and K. Nakanishi, J. Am. Chem. Soc., 1981, 103, 6773 CrossRef CAS.
- T. Horiguchi, Phycol. Res., 1995, 43, 129.
Footnote |
| † The authors dedicate this review to the late Professor D. John Faulkner. |
| This journal is © The Royal Society of Chemistry 2004 |