The role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products
Alison
Butler
* and
Jayme N.
Carter-Franklin
Department of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106-9510, USA
Received
(in Cambridge, UK)
11th November 2003
First published on 21st January 2004
Abstract
Covering: 1998–2003
Halogenated natural products are frequently reported metabolites in marine seaweeds. These compounds span a range from halogenated indoles, terpenes, acetogenins, phenols, etc., to volatile halogenated hydrocarbons that are produced on a very large scale. In many cases these halogenated marine metabolites possess biological activities of pharmacological interest. Given the abundance of halogenated marine natural products found in marine organisms and their potentially important biological activities, the biogenesis of these compounds has intrigued marine natural product chemists for decades. Over a quarter of a century ago, a possible role for haloperoxidase enzymes was first suggested in the biogenesis of certain halogenated marine natural products, although this was long before haloperoxidases were discovered in marine organisms. Since that time, FeHeme- and Vanadium-haloperoxidases (V-HPO) have been discovered in many marine organisms. The structure and catalytic activity of vanadium haloperoxidases is reviewed herein, including the importance of V-HPO-catalyzed bromination and cyclization of terpene substrates.
Alison Butler
Alison Butler is a professor of Chemistry and Biochemistry at UC Santa Barbara. Her research interests focus on metallobiochemistry, including the bioinorganic chemistry of the marine environment.
Jayme N. Carter-Franklin
Jayme N. Carter-Franklin is a postdoctoral fellow in the Department of Chemistry and Biochemistry at UC Santa Barbara. She obtained her PhD degree with Alison Butler working on the role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products.
1 Introduction
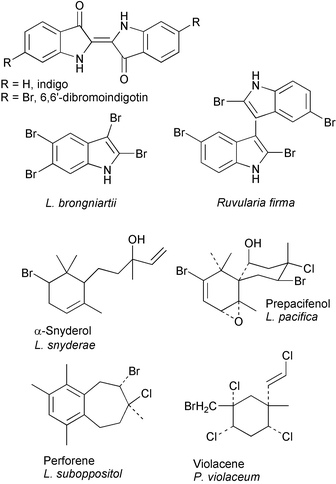
Halogenated natural products are frequently reported metabolites in marine seaweeds, particularly in red macroalgae (Rhodophyceae).1–4 These compounds span a range from halogenated indoles, terpenes, acetogenins, phenolsetc., to volatile halogenated hydrocarbons (e.g., bromoform, chloroform, dibromomethane, etc.) that are produced on a very large scale [Fig. 1].3,5,6 In many cases these halogenated marine metabolites possess biological activities of pharmacological interest, including antifungal, antibacterial, antiviral, and anti-inflammatory activities. Given the abundance of halogenated marine natural products found in marine organisms and their potentially important biological activities, the biogenesis of these compounds has intrigued marine natural product chemists for decades. Over a quarter of a century ago, a possible role for haloperoxidase enzymes was first suggested in the biogenesis of certain halogenated marine natural products,4,7 although this was long before haloperoxidases were discovered in marine organisms. Since that time, haloperoxidases have been discovered in many marine organisms, including FeHeme-vanadium haloperoxidases,8–10 and vanadium(V)-bromoperoxidases,11–13 however the Vanadium-dependent haloperoxidases (V-HPOs) appear to be the most prevalent.14 V-HPOs have now been isolated, purified and cloned from marine algae that produce halogenated secondary metabolites such as the chiral halogenated sesquiterpenes, acetogenins, and indole derivatives. The scope of this review includes the reactivity of vanadium bromoperoxidase (V-BrPO)15 and the applications of V-BrPO in the biosynthesis of halogenated marine natural products.14,16
Fig. 1 Examples of halogenated marine natural products.
2 Vanadium haloperoxidases
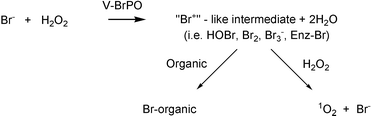
Vanadium haloperoxidases catalyze the oxidation of halides (iodide, bromide, chloride) by hydrogen peroxide. Vanadium haloperoxidases are classified according to the most electronegative halogen oxidized. Thus vanadium chloroperoxidases (V-ClPOs) can oxidize chloride, bromide and iodide,while vanadium bromoperoxidases (V-BrPOs) can oxidize bromide and iodide. Hydrogen peroxide does not have the driving force to oxidize fluoride, however a fluorinating enzyme, fluorinase, has recently been isolated and is proposed to act by an SN2 mechanism.17 Vanadium chloroperoxidases have been isolated primarily from dematiaceous hyphomycete fungi, and have yet to be isolated from marine organisms, whereas V-BrPO has been isolated and characterized from all the different classes of marine algae, including chlorophyta (green algae), phaeophyta (brown algae), and rhodophyta (red algae).16 The overall reaction that vanadium haloperoxidases catalyze is:
H2O2
+ X−
+ R–H + H+
→ R–X + 2H2O
(1)
In the first step V-HPOs catalyze the oxidation of halides (X−) by hydrogen peroxide producing a two electron oxidized halogen intermediate (i.e., “Br+” or biological equivalent).18 In the second step, the oxidized intermediate can halogenate an appropriate organic substrate or react with another equivalent of hydrogen peroxide, forming dioxygen in the singlet-excited state (1O2, 1Δg)
[Scheme 1].19,20
Scheme 1 Overall reaction scheme of vanadium haloperoxidases.19,65
2.1 Characteristics of vanadium bromoperoxidase
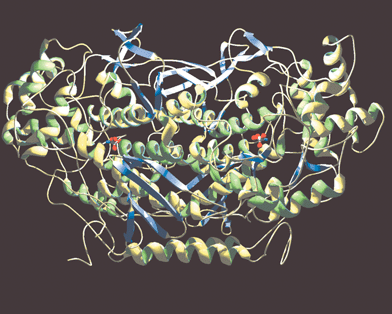
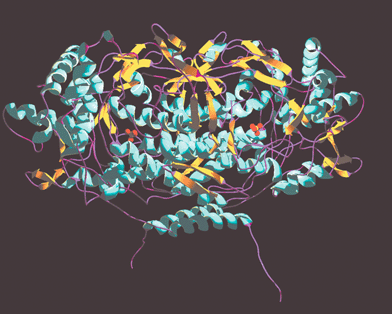
The X-ray crystal structures of native V-BrPO from the brown alga Ascophyllum nodosum and the red alga Corallina officinalis have been reported at 2.0 Å and 2.3 Å resolution, respectively.21,22 The structure of V-BrPO from A. nodosum is a homodimeric protein with approximate dimensions of 90 Å
× 77 Å
× 75 Å, and an estimated molecular weight of 120,400 Da [Fig. 2]. The most recently reported structure of V-BrPO from C. officinalis is a homo-dodecameric protein (MW 740,000), measuring ∼150 Å in diameter with a single subunit dimension of 85 Å
× 56 Å
× 55 Å
[Fig. 3]. Both V-BrPOs are dominated by α-helical secondary structure with a few short β-strands. The main tertiary structural motif in both enzymes is two four-helix bundles similar to the reported X-ray crystal structure of V-ClPO from Curvularia inaequalis.23 Each V-BrPO buries a high percentage (e.g., 33%
C. officinalis, 46%
A. nodosum) of its subunit surface in contacts with neighboring subunits giving rise to the high chemical and thermal stability reported for V-BrPOs.21,22,24–27
Fig. 2 Structure of the dimer of V-BrPO from Ascophyllum nodosum
(PDB identification number, 1QI9). Figure was drawn in Swiss-PDB viewer and rendered with gl_render and POV-ray software. Vanadium cofactors are represented as white/red stick and ball models.
Fig. 3 Structure of the dimer of V-BrPO from Corallina officinalis
(PDB identification number, 1QHB). Figure was drawn in Swiss-PDB viewer and rendered with gl_render and POV-ray software. Phosphate anions are represented as gold/red stick and ball models.
2.2 The vanadium site
The vanadium binding sites of V-BrPO (A. nodosum and C. officinalis) and V-ClPO (C. inaequalis) are remarkably similar, but with certain notable differences. All of the structures have a conserved protein scaffold for the vanadium-binding site. The amino acid residues that participate in vanadium binding are conserved between V-BrPO and V-ClPO, and provide a rigid oxyanion-binding site made up of an intricate hydrogen-bond network between the conserved residues and vanadate.16 The vanadium-binding site in V-BrPO and V-ClPO is located at the core of the four-helix bundle structural motif and is positioned at the bottom of a deep funnel-shaped substrate channel, approximately 15–20 Å deep.21–23
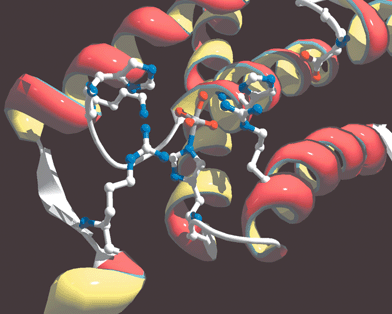
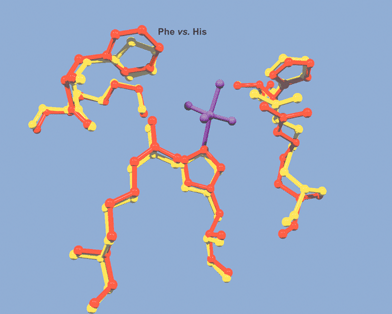
The conserved vanadium binding site motif for V-BrPO and V-ClPO is P[S/A]YPSGHAT, where hydrogen vanadate is bound axially to a single histidine ligand in an overall trigonal bipyramidal coordination geometry.21–23 The three equatorial oxygens of vanadate are hydrogen bonded to two conserved arginines, a lysine, a serine and a glycine amide backbone. The proposed apical hydroxide ligand is hydrogen bonded to a histidine [Fig. 4, shown for A. nodosum V-BrPO].21,23 Overlay of the X-ray coordinates for V-BrPO (A. nodosum) and V-ClPO (C. inaequalis), in addition to sequence alignments, shows the presence of an additional histidine residue in proximity to the vanadate-binding site in V-BrPO. The additional histidine residue in V-BrPO aligns with a phenylalanine in the V-ClPO structure [Fig. 5]. The X-ray crystal structures of V-BrPO indicate that the extra histidine residue does not interact directly with the vanadium cofactor, although it has been proposed that the histidine side chain participates as a proton donor and acceptor during the enzymatic reaction.21,28,29
Fig. 4 The vanadium site of V-BrPO (A. nodosum). Figure was drawn in Swiss-PDB viewer and rendered with gl_render and POV-ray software. Vanadium cofactor is represented as a gray/red stick and ball model.
Fig. 5 Overlay of the X-ray coordinates of the vanadium site of wild-type V-ClPO (Curvularia inaequalis) shown in red and V-BrPO (A. nodosum) shown in yellow. Figure was drawn in Swiss-PDB viewer and rendered with gl_render and POV-ray software. Vanadium cofactor is represented as a purple stick and ball model. Overlap of V-ClPO phenylalanine and V-BrPO histidine is indicated.
Interestingly, comparison of the substrate channels leading to the vanadium active-site in V-BrPO from A. nodosum and C. officinalis reveals only five residues (not including those hydrogen bonded to the bound vanadate ion) out of the 17 lining the substrate cavity that are conserved between the two structures, and none of these five residues are conserved in the V-ClPO structure.21–23 The substrate channels leading to the vanadium binding sites are lined predominantly with hydrophobic residues and a few hydrophilic residues.21,22 The hydrophobic patches and few charged amino acids are proposed to influence substrate specificity as well as provide binding sites for organic substrates.
2.3 Proposed catalytic cycle for vanadium bromoperoxidase
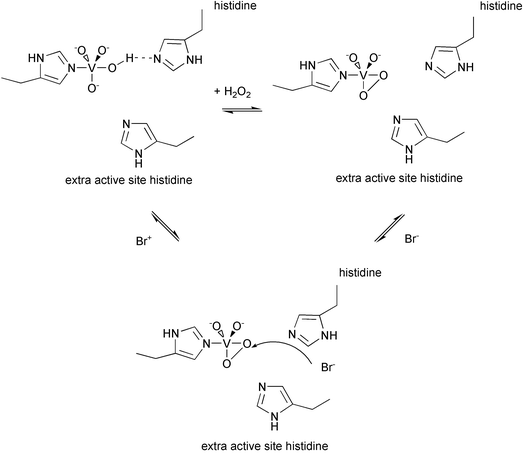
Extensive steady-state kinetic analyses of V-BrPO from A. nodosum and C. officinalis fit a substrate-inhibited bi-bi ping-pong kinetic mechanism, in which hydrogen peroxide binds first followed by bromide oxidation.20,25,26,30,31 Based on these steady state kinetic analyses and consistent with the X-ray crystal structures of V-BrPO from A. nodosum and C. officinalis, and the X-ray crystal structures of the monoperoxo-derivative of V-ClPO, a minimum catalytic mechanism for V-BrPO has been proposed [Fig. 6]. The bound vanadate first coordinates hydrogen peroxide in a side-on bound fashion, producing a square-based pyramidal oxoperoxovanadium(V) intermediate.32 The oxoperoxovanadium(V) intermediate is proposed to oxidize the second substrate in the cycle, bromide, by two electrons to generate a bromonium ion-type intermediate.18 Detection or isolation of the oxidized bromine intermediate is hampered due to its reaction with organic substrates (RH, Scheme 1) or with excess hydrogen peroxide to produce O2, preventing its build up in solution.19,20 As described below, the nature of the oxidized bromine intermediate as either enzyme-trapped, or enzyme-bound, or a freely diffusible species seems to depend on the nature of the organic substrate.33–35
Fig. 6 Proposed reaction scheme for V-BrPO catalysis.
3 Selectivity of vanadium bromoperoxidase
Initial investigations into the reactivity of V-BrPOs with various substrates such as anisole or prochiral aromatic compounds failed to demonstrate any regio- or stereoselectivity upon bromination.36–38 The apparent lack of selectivity at that time was interpreted to mean that V-BrPO produces a diffusible oxidized bromine intermediate such as hypobromite, bromine or tribromide that would then carry out a molecular bromination reaction.30 More recently, however, competitive kinetics studies comparing the bromination of indole substrates by the V-BrPO/H2O2/Br− system to bromination by aqueous bromine (i.e., the equilibrium mixture of HOBr ⇌ Br2
⇌ Br3−) under identical reaction conditions, showed that V-BrPO-catalyzed reactions were not consistent with a freely diffusible brominating intermediate.33,39,40 Vanadium bromoperoxidase catalyzed the preferential bromination and oxidation of indole derivatives when competed against equimolar mixtures of phenol red or monochlorodimedone (MCD), whereas the same reactions performed with aqueous bromine showed the simultaneous bromination of all substrates present in solution.33,34,41
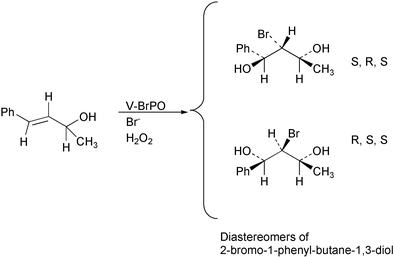
Diastereospecific bromohydrin formation by V-BrPO (C. officinalis) from (E)-4-phenyl-3-buten-2-ol has also been briefly reported.42Bromination of (E)-4-phenyl-3-buten-2-ol by V-BrPO, FeHeme ClPO (C. fumago) and N-bromoacetamide (NBA) produced diastereomers of 2-bromo-1-phenylbutane-1,3-diol [Fig. 7]. The ratios of diastereomers produced, i.e.S, R, S- (or R, S, R-) and R, S, S- (or S, R, R-), differed between the chemical and enzymatic reactions. The corresponding ratio for V-BrPO was 66%
S, R, S- (or R, S, R-) and 34%
R, S, S- (or S, R, R-), while NBA effected bromohydrin formation in a 75% to 25% ratio of the stereoisomers. The difference in diastereospecific product formation of the V-BrPO versus NBA is consistent with an influence from the protein, such as substrate binding to V-BrPO and bromination taking place at or near the active site.
3.1 Vanadium bromoperoxidase reactivity with indole substrates
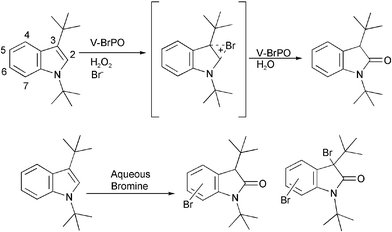
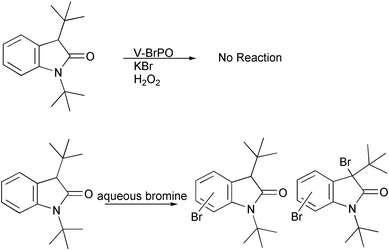
Reactions with the indole derivative 1,3-di-tert-butylindole and V-BrPO resulted in regiospecific brominative oxidation to produce 1,3-di-tert-butyl-2-indolinone as the sole product [Fig. 8].34 The formation of 1,3-di-tert-butyl-2-indolinone likely occurs by an electrophilic attack of the enzyme-generated brominating species on the electron rich C2–C3 double bond of the pyrrole ring producing first a bromoindolinium species, followed by hydration at the C2 position. On the other hand, reactions with aqueous bromine and 1,3-di-tert-butylindole were not regioselective and produced many products including multiply brominated species. Experiments with V-BrPO and 1,3-di-tert-butyl-2-indolinone as the substrate failed to produce any additional brominated products, and did not compete with phenol red in competitive kinetic experiments. In contrast, bromination of 1,3-di-tert-butyl-2-indolinone with aqueous bromine produced multiply brominated species, with bromination occurring on the benzene ring of the indole derivative [Scheme 2]. The regioselective reaction of 1,3-di-tert-butylindole with V-BrPO is consistent with organic substrate in the active site cavity of V-BrPO. The selectivity of V-BrPO can be attributed to 1,3-di-tert-butylindole orienting specifically within the active site cleft of V-BrPO, possibly along a hydrophobic patch of residues. V-BrPO-catalyzed formation of 1,3-di-tert-butyl-2-indolinone is the first example of regiospecific bromination by V-BrPO and suggests the presence of an enzyme-trapped or enzyme-bound brominating intermediate during catalysis.
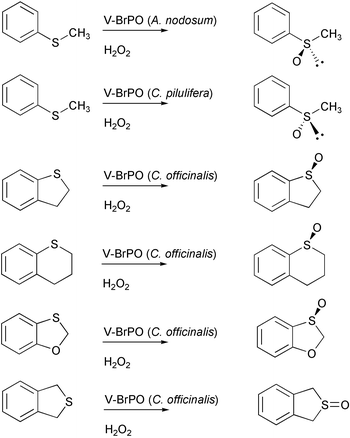
Given the tremendous reactivity of oxoperoxovanadium(V) compounds in organic oxidation reactions, it has been puzzling why V-HPO enzymes do not carry out epoxidation, or hydroxylation reactions, in the absence of halides. Relatively recently the enantioselective oxidation of sulfides to sulfoxides in the absence of halides has been reported for V-BrPO.43–45 The concentration of hydrogen peroxide and its rate of addition to the reaction solutions were found to affect both the yield and enantioselectivity of sulfoxidation. For example, V-BrPO from A. nodosum converts methyl phenyl sulfide to the (R)-enantiomer (91% ee), whereas V-BrPO from C. pilulifera produces the (S)-enantiomer (55% ee)
[Fig. 9].43,44,46 Kinetic resolution experiments using racemic non-aromatic cyclic thioethers with V-BrPO from A. nodosum show that V-BrPO has a kinetic preference for conversion of the (R)-enantiomer to the corresponding sulfoxide.46 Also, V-BrPO isolated from C. officinalis catalyzes the selective sulfoxidation of aromatic bicyclic sulfides to the (S)-sulfoxides in high (up to 91%) ee [Fig. 9].45 However, the selectivity of sulfoxidation is exclusive to the marine algal V-BrPOs, since V-ClPO catalyzes the formation of a racemic mixture of the sulfoxide. The reactivity of V-BrPO (A. nodosum and C. pilulifera) to produce differing enantiomers suggests subtle differences exist in the substrate channel near the vanadium-binding site of these enzymes.
Mechanistic investigations of the source of oxygen in the sulfoxide products were established using 18O-labeled hydrogen peroxide. Experiments with V-BrPO from A. nodosum and methyl phenyl sulfide with H218O2 established only 18O-labeled (R)-methyl phenyl sulfoxide produced in 63% ee.47 These sulfoxidation reactions demonstrated that V-BrPO catalyzes the direct transfer of oxygen from the oxoperoxovanadium(V) intermediate to the sulfide, suggestive of aromatic sulfide binding at or near the vanadium site.
Given the observed regioselective reactions in addition to the stereoselective sulfoxidation reactions catalyzed by V-BrPOs, a picture is emerging in which subtle differences in the active site substrate channel influence the outcome of regioselective and stereoselective products. The ability of V-BrPO to catalyze stereoselective reactions suggests a role for the enzyme in the production of halogenated marine natural products.
4 Biogenesis of halogenated marine natural products
Marine red macroalgae produce an abundance of halogenated secondary metabolites. Halogenated cyclic sesquiterpenes isolated from the red algal genus Laurencia and from herbivorous mollusks, such as Aplysia, that feed on Laurencia are one of the most prevalent structural classes of natural products isolated from the algae.48,49 While their biosyntheses are still poorly understood, biomimetic syntheses and enzyme-catalyzed halogenation reactions have provided useful insight into the biogenesis of halogenated metabolites isolated from marine algae.
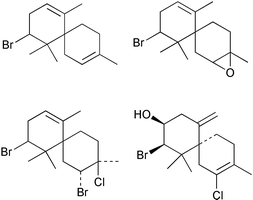
Among the halogenated natural products isolated from marine algae, one of the largest and most varied groups are the halogenated sesquiterpenes, which include halogenated chamigrenes isolated from the red algal genus Laurencia
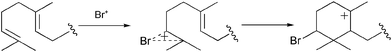
[Fig. 10].50 Biogenesis studies suggest that most brominated cyclic sesquiterpenes are biosynthesized by a bromonium ion induced (or biological equivalent) cyclization of an acyclic terpene precursor [Scheme 3].49,51 Initially biomimetic syntheses of halogenated sesquiterpenes, carried out at a time when marine haloperoxidases had only been hypothesized, utilized direct bromine–carbon bond formation with successive ring closure of the terpene precursor, or alternatively the indirect incorporation of bromine to the cyclized terpene intermediate. Van Tamelen and Hessler reported the first example of a bromonium ion induced cyclization of methyl farnesate in 1966.52 In the reaction methyl farnesate was reacted with NBS in a mixture of water and tetrahydrofuran, to produce small quantities of a brominated bicyclic ester despite the presence of competing nucleophiles.
Fig. 10 Examples of selected halogenated chamigrene natural products.
Scheme 3 Hypothetical bromonium ion initiated cyclization of a terpene precursor.51
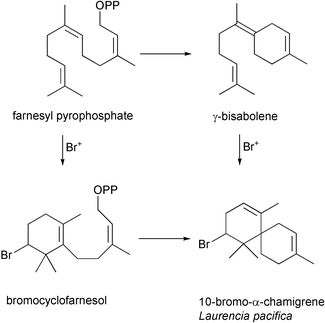
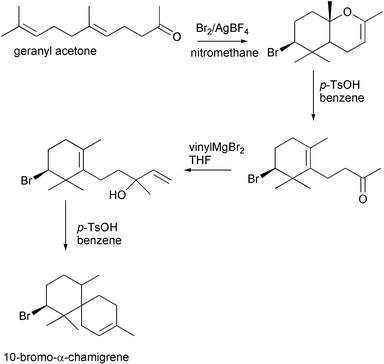
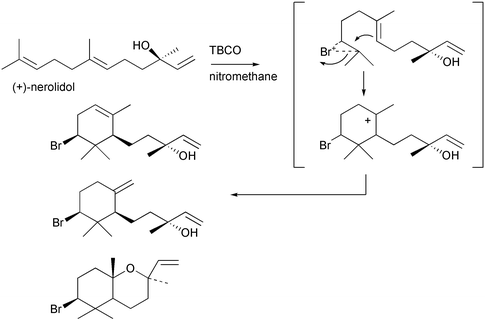
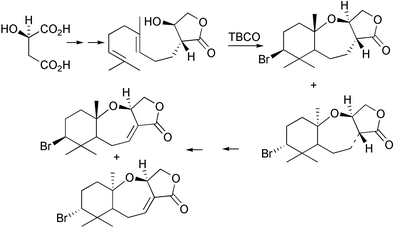
A quarter of a century ago, Faulkner, Fenical, and Gonzalez proposed two biosynthetic routes to the basic halogenated chamigrene skeleton from the sesquiterpene farnesyl pyrophosphate by way of either a γ-bisabolene or a brominated monocyclofarnesyl intermediate [Scheme 4].4,51,53 Faulkner applied the proposed biosynthetic scheme involving a brominated monocyclofarnesol intermediate when he reported the bromonium ion induced cyclization of geranyl acetone in the biomimetic synthesis of 10-bromo-α-chamigrene [Scheme 5].7 Similary, Kato and coworkers used the brominating agent 2,4,4,6-tetrabromocyclohexa-2,5-dienone (TBCO) in nitromethane to direct the cyclization of nerolidol to produce the brominated marine natural products α- and β-snyderol, and 3β-bromo-8-epicaparrapi oxide [Scheme 6].54,55 In addition, the first enantiospecific synthesis of the brominated marine natural product (−)-aplysistatin derived from (R)-(+)-malic acid involved using a bromonium ion induced cyclization reaction [Scheme 7].56 An important feature of the biomimetic reactions is the lack of competing nucleophiles, such as water to quench the bromonium ion intermediate. While biomimetic syntheses produced halogenated natural products in one or two step transformations, the biomimetic reactions suffered from low product yields and lack of specificity in bromine–carbon bond formation.
Scheme 4 Proposed biosynthetic pathway from farnesyl pyrophosphate to 10-bromo-α-chamigrene via a bromonium ion initiated cyclization reaction.4,50,51
Scheme 5 Biomimetic synthesis of 10-bromo-α-chamigrene.7
Scheme 6 Biomimetic synthesis of marine natural products α-, β-snyderol, and 3β-bromo-8-epicaparrapi oxide.54,55
Scheme 7 Biomimetic synthesis of the marine natural product (−)-aplysistatin.56
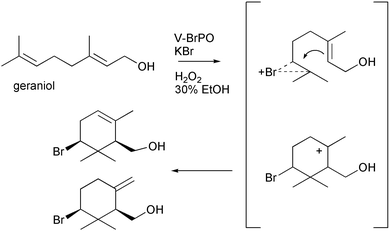
Based on biomimetic approaches, the bromination and cyclization of terpenes to produce cyclic halogenated natural products appear to require a source of electrophilic bromine in a rather non-nucleophilic environment. The active site cavity of V-BrPO possesses both of these qualities. We have begun to explore the utility of V-BrPO in the proposed biosynthetic schemes with terpenes. The reactivity of V-BrPO was evaluated with geraniol, a monoterpene, and geraniol derivatives such as geranyl acetate and geranyl acetone in mixtures of buffer and organic cosolvents (i.e., ethanol, isopropanol, acetonitrile).35 V-BrPO reactivity with geraniol produced bromohydrins as the major products, in addition to epoxide, dibrominated species, and two singly brominated monocyclic alcohols [Scheme 8]. The brominated cyclized alcohols were produced in 1–2% yield and were easily separated from other products. NMR spectroscopy of the cyclized geraniol products showed them to be analogous to α- and β-snyderol marine natural products.57 The mechanism of formation likely involves a bromonium ion attack at the C6–C7 terminal alkene of geraniol, generating a bromonium–geraniol intermediate. The proposed bromonium ion intermediate is subsequently attacked by the electron rich internal olefinic bond, followed by elimination reactions leading to products [Scheme 8].35 The brominated monocyclic products were isolated as single diastereomers without enantioselectivity: the NOE observed between H2 and H6 (geraniol numbering) indicated the bromine substituent in the equatorial position consistent with biomimetic Br+-initiated cyclization reactions.7,55,56,58
Scheme 8 V-BrPO-catalyzed bromination and cyclization of geraniol to the singly brominated α- and β-cyclized geraniol species.35
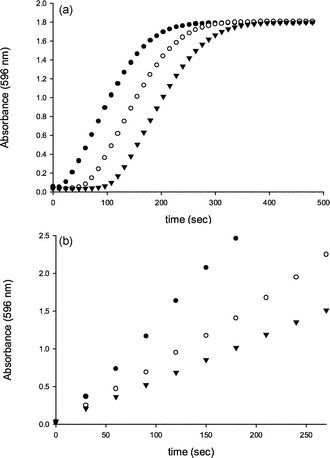
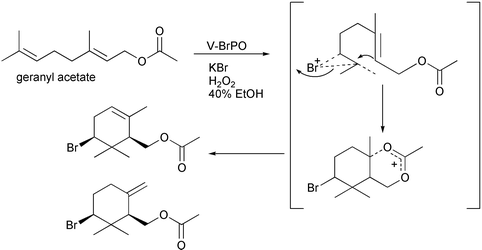
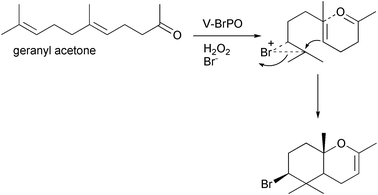
Control reactions with aqueous bromine and geraniol, under identical reaction conditions as V-BrPO, did not produce the brominated monocyclic geraniol species, suggesting bromination and cyclization of geraniol occurred at or near the active site of V-BrPO.35 An enzyme–geraniol interaction to produce the brominated cyclized geraniol species was supported by the V-BrPO-catalyzed sequential bromination of first geraniol followed by phenol red in competitive kinetics studies [Fig. 11]. In contrast, bromination of geraniol and phenol red by aqueous bromine occurred simultaneously, suggesting that the brominating intermediate in V-BrPO reaction with geraniol is enzyme-trapped or enzyme-bound and not a freely-diffusible brominating species. V-BrPO reactivity with derivatives of geraniol such as geranyl acetate [Scheme 9] and geranyl acetone [Scheme 10] also resulted in brominated and cyclized products,35 similar to reactions performed by Faulkner in the biomimetic synthesis of 10-bromo-α-chamigrene.7
Fig. 11 Time course for the bromination of phenol red by (a) V-BrPO/Br−/H2O2 system and (b) aqueous bromine as a function of the geraniol concentration. The reactions were carried out at 25 °C in the presence of 50 µM phenol red and 40 mM KBr in 0.1 M phosphate buffer (pH 5.7) with 30% v/v ethanol. Enzymatic reactions were initiated by addition of 0.5 mM H2O2 and 5 nM V-BrPO. Nonenzymatic reactions were initiated by addition of NaOBr (stock solution of 5 mM) in 10 µL aliquots at 30 s intervals. Production of bromophenol blue was monitored at 596 nm. Concentrations of geraniol: ● 0 µM; ○ 50 µM; ▼ 100 µM.
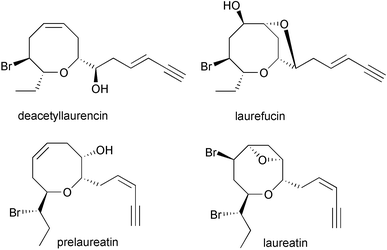
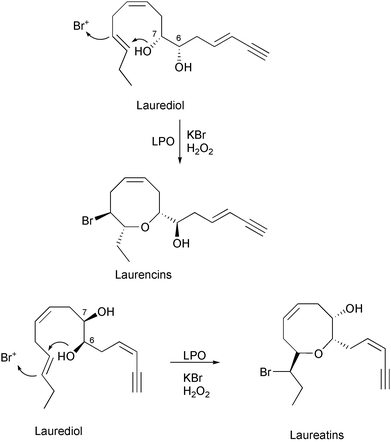
In addition to the biosynthetic schemes for the production of halogenated sesquiterpenes, biomimetic syntheses of halogenated acetylenes or C15 acetogenins have also been explored. The halogenated cyclic acetogenins display features such as varied oxane ring sizes, enyne or allenic side chains, and at least one bromine atom.48 The lauroxocanes are the largest class of Laurencia acetogenins and consist of the laurencin and laureatin structural types [Fig. 12]. The bromoether functionality in the halogenated acetogenins has fascinated organic chemists with respect to construction of the entropically unfavored medium ring ethers. Highly labile cis- and trans-laurediols isolated from Laurencia are assumed to be the common biogenetic precursor of the brominated eight-membered cyclic ethers in Laurencia.59,60 Murai and colleagues in the last ten years have examined the cyclization of (3E)- and (3Z)-laurediol to (E)-deacetyllaurencin and (Z)-prelaureatin using a commercially available lactoperoxidase (LPO) and more recently with a crude preparation of a bromoperoxidase (BPO) isolated from Laurencia nipponica
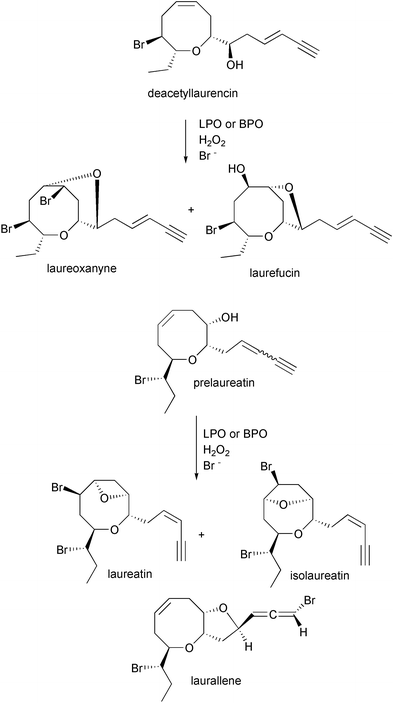
[Scheme 11].61–63 Further reaction of (3E)-deacetyllaurencin with LPO or BPO produced marine natural products laureoxanyne, laurefucin, and bromohydrins as products, and further treatment of (Z)-prelaureatin with LPO or BPO produced laureatin, and isolaureatin [Scheme 12].64 The yields of brominated cyclic products were extremely low, but established a mechanism of bromonium ion initiated cyclization of an acyclic precursor to the eight-membered bromoether functionality.
Fig. 12 Representative structures of halogenated C15 acetogenins from the red algal genus Laurencia.
Scheme 11 Proposed biosynthetic scheme for the LPO- and BPO-catalyzed bromination and cyclization of laurediol to laurencin and laureatin marine natural products.61–63
Scheme 12 Proposed biosynthetic scheme for the LPO- and BPO-catalyzed bromination of deacetyllaurencin and prelaureatin towards the production of marine natural products.64
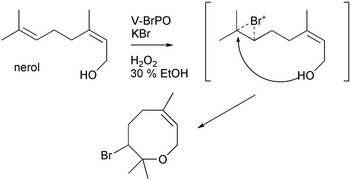
Biosynthesis of the bromoether functionality utilizing V-BrPO was examined using the monoterpene nerol. V-BrPO reaction with nerol in buffered-cosolvent mixtures afforded a novel 8-membered cyclic bromoether structure, reminiscent of the oxane ring present in the halogenated acetogenins.35 Bromohydrins and epoxide products were also isolated from the reaction. The proposed mechanism for formation of the eight-membered bromoether likely involves a bromonium ion attack at on the C6–C7 terminal alkene generating a bromonium–nerol intermediate [Scheme 13]. Internal nucleophilic trapping of the bromonium-intermediate by the pendant alcohol of nerol resulted in the eight-membered bromoether structure. Analogous to V-BrPO reaction with geraniol, the eight-membered bromoether was exclusive to the V-BrPO reaction and was not readily detected in control reactions with aqueous bromine under identical reaction conditions.
Vanadium bromoperoxidase is an enzyme present in marine algae that catalyzes electrophilic halogenation reactions using hydrogen peroxide to oxidize and thereby activate bromide ion. The X-ray structures of V-BrPO and V-ClPO, along with investigations into the overall reactivity of haloperoxidases, are beginning to provide a molecular level understanding of the catalytic cycle of these enzymes. We have shown that V-BrPO which has either been cloned or isolated from marine red algae can catalyze the bromination and cyclization of terpene substrates that are likely biological precursors to halogenated marine natural products. We have barely scratched the surface of the potential reactivity of V-BrPO with these terpene substrates, as well as the role of V-BrPO in the biogenesis of other halogenated natural products. Thus we are further investigating the features of vanadium haloperoxidase that confer halide selectivity and organic substrate selectivity, as well as their reactivity with different terpenes and other organic substrates, under a variety of different conditions.
6 Acknowledgements
John Faulkner's interest in our work on vanadium haloperoxidase and his perceptive questions helped to focus our investigations on vanadium bromoperoxidase. We have missed John over this past year, especially as new results emerge on our V-BrPO-catalyzed reactions that we would wish to report to him. We would love his feedback and especially the challenge to answer every question he would surely ask about the product characterization, the reaction conditions and the mechanism of the reaction.
We are also grateful for funding from the NSF (CHE-0213523) and California Sea Grant (NA06RG0142, project R/MP-94) which supports our research on the marine haloperoxidase enzymes.
7 References
D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1–48 RSC.
D. J. Faulkner, Nat. Prod. Rep., 2001, 18, 1–49 RSC.
D. J. Faulkner, Nat. Prod. Rep., 2000, 17, 7–55 RSC.
A. Butler, J. N. Carter and M. T. Simpson, in Handbook on Metalloproteins; I. Bertini, A. Sigel, H. Sigel, Eds.; M. Dekker: New York, 2001, pp. 153–179 Search PubMed.
D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRefCAS.
H. S. Soedjak, J. V. Walker and A. Butler, Biochemistry, 1995, 34, 12689–12696 CrossRefCAS.
R. Everett and A. Butler, Inorg. Chem., 1989, 28, 393–395 CrossRefCAS.
R. R. Everett, H. S. Soedjak and A. Butler, J. Biol. Chem., 1990, 265, 15671–15679 CAS.
M. Weyand, H. Hecht, M. Kiesz, M. F. Liaud, H. Vitler and D. Schomburg, J. Mol. Biol., 1999, 293, 595–611 CrossRefCAS.
M. N. Isupov, A. R. Dalby, A. A. Brindley, Y. Izumi, T. Tanabe, G. N. Murshudov and J. A. Littlechild, J. Mol. Biol., 2000, 299, 1035–1049 CrossRefCAS.
A. Messerschmidt and R. Wever, Proc. Natl. Acad. Sci. USA, 1996, 93, 392–396 CrossRefCAS.
E. de Boer, H. Plat, M. Tromp, R. Wever, M. Franssen, H. van der Plas, E. Meijer and H. Schoemaker, Biotechnol. Bioeng., 1987, 30, 607–610 CAS.
D. J. Sheffield, T. Harry, A. J. Smith and L. J. Rogers, Phytochemistry, 1993, 32, 21–26 CrossRef.
J. Van Schijndel, P. Barnett, J. Roelse, E. Vollenbroek and R. Wever, Eur. J. Biochem., 1994, 225, 151–157 CrossRefCAS.
J. V. Walker, University of California, Santa Barbara, 1998, PhD Thesis.
G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 3469–3478 CrossRefCAS.
R. Renirie, W. Hemrika and R. Wever, J. Biol. Chem., 2000, 275, 11650–11657 CrossRefCAS.
E. de Boer and R. Wever, J. Biol. Chem., 1988, 236, 12326–12332.
H. S. Soedjak and A. Butler, Biochim. Biophys. Acta, 1991, 1079, 1–7 CrossRefCAS.
A. Messerschmidt, L. Prade and R. Wever, Biol. Chem., 1997, 378, 309–315 CAS.
R. A. Tschirret-Guth and A. Butler, J. Am. Chem. Soc., 1994, 116, 411–412 CrossRefCAS.
J. S. Martinez, G. L. Carroll, R. A. Tschirret-Guth, G. Altenhoff, R. D. Little and A. Butler, J. Am. Chem. Soc., 2001, 123, 3289–3294 CrossRefCAS.
J. Carter-Franklin, J. D. Parrish, R. A. Tschirret-Guth, R. D. Little and A. Butler, J. Am. Chem. Soc., 2003, 125, 3688–3689 CrossRefCAS.
N. Itoh, Y. Izumi and H. Yamada, J. Biol. Chem., 1987, 262, 11982–11987 CAS.
N. Itoh, A. Hasan, Y. Izumi and H. Yamada, Eur. J. Biochem., 1988, 172, 477–484 CAS.
B. E. Krenn, Y. Izumi, H. Yamada and R. Wever, Biochem. Biophys. Acta, 1989, 998, 63–68 Search PubMed.
A. Butler and R. A. Tschirret-Guth, in Mechanisms of Biohalogenation and Dehalogenation; D. B. Janssen, K. Soda, R. Wever, Eds.; Royal Netherlands Academy of Arts and Sciences, Amsterdam: 1996, pp. 55–68 Search PubMed.
R. A. Tschirret-Guth, PhD thesis, University of California, Santa Barbara, 1996.
A. Butler, R. A. Tschirret-Guth and M. T. Simpson, in Vanadium Compounds: Chemistry, Biochemistry and Therapeutic Applications; A. S. Tracey, D. C. Crans, Eds.; American Chemical Society, Washington DC, ACS Symposium Series: 1998; Vol. 711, pp. 202–215 Search PubMed.
P. Coughlin, S. Roberts, C. Rush and A. Willetts, Biotechnol. Lett., 1993, 15, 907–912 CAS.
M. Andersson, A. Willetts and S. Allenmark, J. Org. Chem., 1997, 62, 8455–8458 CrossRefCAS.
H. B. ten Brink, A. Tuynman, H. Dekker, W. Hemrika, Y. Izumi, T. Oshiro, H. E. Schoemaker and R. Wever, Inorg. Chem., 1998, 37, 6780–6784 CrossRefCAS.
M. Andersson and S. Allenmark, Tetrahedron, 1998, 54, 15293–15304 CrossRefCAS.
H. B. ten Brink, H. L. Holland, H. E. Schoemaker, H. van Lingen and R. Wever, Tetrahedron: Asymmetry, 1999, 10, 4563–4572 CrossRefCAS.
H. B. ten Brink, H. E. Schoemaker and R. Wever, Eur. J. Biochem., 2001, 268, 132–138 CrossRefCAS.
K. L. Erickson, in Marine Natural Products; P. J. Scheuer, Ed.; Academic Press Inc., New York: 1983; Vol. V, pp. 131–257 Search PubMed.
J. D. Martin, J. M. Palazon, C. Perez and J. L. Ravelo, Pure Appl. Chem., 1986, 58, 395–406 CAS.
J. D. Martin and J. Darias, in Marine Natural Products: Chemical and Biological Perspectives; Academic Press, New York, 1978; Vol. 1, pp. 125–174 Search PubMed.
D. J. Faulkner, Pure Appl. Chem., 1976, 48, 25–28 CAS.
E. van Tamelen and E. J. Hessler, Chem. Commun., 1966, 411–413 RSC.
A. G. Gonzalez, J. M. Aguiar, J. D. Martin and M. Norte, Tetrahedron Lett., 1975, 2499 CrossRefCAS.
T. Kato, I. Ichinose, A. Kamoshida and Y. Kitahara, J. Chem. Soc., Chem. Commun., 1976, 518–519 RSC.
T. Kato, K. Ishii, I. Ichinose, Y. Nakai and T. Kumagai, J. Chem. Soc., Chem. Commun., 1980, 1106–1108 RSC.
H. Shieh and G. D. Prestwich, Tetrahedron Lett., 1982, 23, 4643–4646 CrossRefCAS.
B. M. Howard and W. Fenical, Tetrahedron Lett., 1976, 1, 41–44 CrossRef.
T. R. Hoye and M. J. Kurth, J. Org. Chem., 1978, 43, 3693–3697 CrossRefCAS.
A. Fukuzawa, M. Aye and A. Murai, Chem. Lett., 1990, 1579–1580 CAS.
A. Fukuzawa, M. Aye, M. Nakamura, M. Tamura and A. Murai, Chem. Lett., 1990, 1287–1290 CAS.
A. Fukuzawa, Y. Takasugi, A. Murai, M. Nakamura and M. Tamura, Tetrahedron Lett., 1992, 33, 2017–2018 CrossRefCAS.
A. Fukuzawa, M. Aye, Y. Takasugi, M. Nakamura, M. Tamura and A. Murai, Chem. Lett., 1994, 2307–2310 CAS.
J. Ishihara, N. Kanoh and A. Murai, Tetrahedron Lett., 1995, 36, 737–740 CrossRefCAS.
J. Ishihara, Y. Shimada, N. Kanoh, Y. Takasugi, A. Fukuzawa and A. Murai, Tetrahedron, 1997, 53, 8371–8382 CrossRefCAS.
A. Butler, Coord. Chem. Rev., 1999, 187, 17–35 CrossRefCAS.