Approaches to identify, clone, and express symbiont bioactive metabolite genes
Mark
Hildebrand
,
Laura E.
Waggoner
,
Grace E.
Lim
,
Katherine H.
Sharp
,
Christian P.
Ridley
and
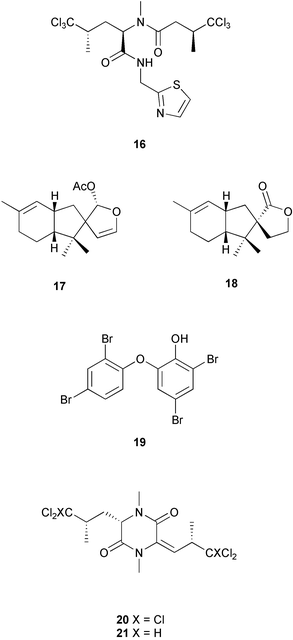
Margo G.
Haygood
*
Scripps Institution of Oceanography, Marine Biology Research Division; Center for Marine Biotechnology and Biomedicine; and UCSD Cancer Center, University of California, San Diego, La Jolla, California 92093, USA
First published on 15th December 2003
Abstract
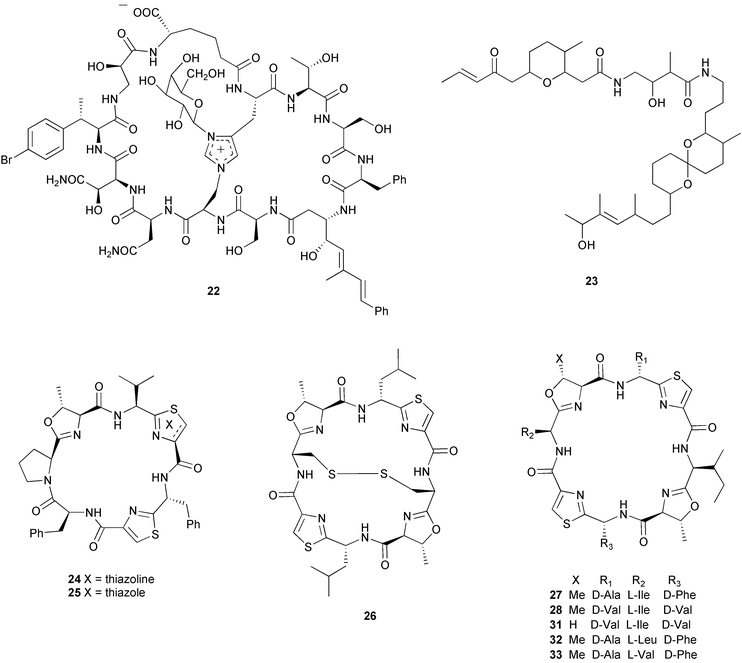
Covering: 1981–2003
This review discusses approaches to identify, clone, and express bioactive metabolite genes from symbionts of marine invertebrates. Criteria for proving symbiotic origin of bioactive metabolites are presented, followed by a comprehensive, practically-oriented overview of techniques to be applied. The Bugula neritina/Endobugula sertula association is used as a primary example, but other symbioses are discussed. Thirty-six compounds are presented and 111 references are cited.
 Mark Hildebrand | Mark Hildebrand received a PhD in Biochemistry, with an emphasis on molecular biology, from the University of Arizona in 1987. He did post-doctoral research with Professor Benjamin Volcani at the Scripps Institution of Oceanography and is currently an Associate Project Scientist at Scripps. His research interests include the molecular and cell biology of silicified cell wall synthesis in diatoms, biological applications in nanotechnology, and cloning and expressing bioactive metabolite genes. |
 Laura E. Waggoner | Laura E. Waggoner received a BS in Biology from Duke University in 1995. She completed her PhD in Biology in 1999 at the University of California, San Diego, where she studied the molecular mechanisms governing regulation of egg-laying behavior in nematodes. Combining her experience in molecular biology with a lifelong interest in marine biology, she then took a post-doctoral position at Scripps Institution of Oceanography, where she is currently investigating marine invertebrate symbioses and the bioactive metabolites they produce. |
 Grace E. Lim | Grace Lim received her BS in Molecular Environmental Biology with an emphasis on Microbiology at the University of California, Berkeley in 1998. She is currently pursuing a PhD degree in Marine Biology with Margo Haygood at the Scripps Institution of Oceanography. Grace's interests include bacterial phylogenetics and genomics as applied to the study of symbiosis and secondary metabolism. |
 Katherine H. Sharp | Katherine Sharp received a BA in Biology and Anthropology from Mount Holyoke College in 1998. She is currently a PhD candidate in Marine Biology with Dr Margo Haygood at the Scripps Institution of Oceanography. During her time at Scripps, she has worked within the field of marine bioactive metabolite symbiosis and focused her research efforts on microbial ecology of sponges, as well as symbiont transmission and recruitment mechanisms in marine invertebrate hosts. |
 Christian P. Ridley | Christian Ridley was born in 1977 in Kinnelon, NJ. He received a BS in Marine Chemistry from Southampton College (Long Island University) in 1999. He is currently working on his PhD in marine natural products research at the Scripps Institution of Oceanography, studying symbioses between marine invertebrates and bacteria. In addition to symbiosis, his research interests include the isolation and structural elucidation of natural products as well as the synthesis of natural product analogs to explore structure–activity relationships. |
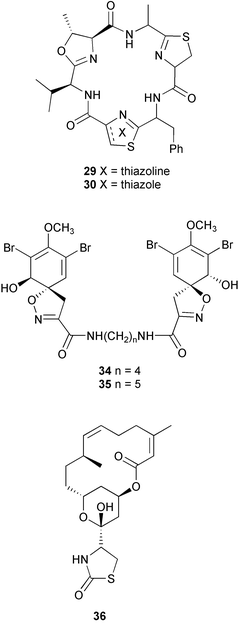
 Margo G. Haygood | Margo Haygood is a Professor of Marine Biology at the Scripps Institution of Oceanography, University of California, San Diego. She studied History of Science at Harvard University, and received her PhD in Marine Biology from Scripps Institution of Oceanography in 1984. She did postdoctoral work in molecular biology with Professor Mary Lidstrom at the University of Washington, and served as a scientific officer for microbiology and molecular biology programs at the US Office of Naval Research. She returned to Scripps as an assistant professor in 1987. Her interests in marine microbiology include iron acquisition and microbial symbioses, especially bioactive metabolite symbioses. |
Introduction
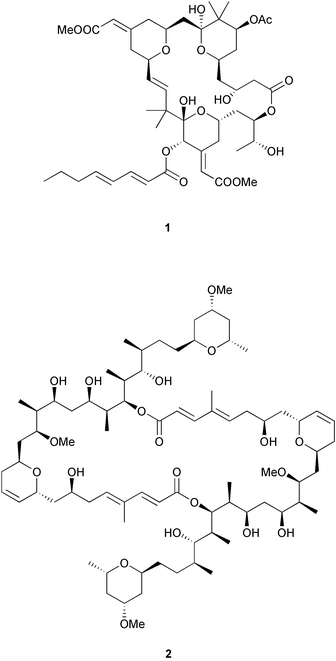
1.1 Marine invertebrate natural products Marine invertebrates have been and continue to be a prolific source of novel and structurally diverse natural products.1,2 Often these compounds display potent and selective bioactivities that trigger biomedical interest.3 Unfortunately, the supply of the bioactive natural product is usually insufficient to meet the demands of pre-clinical and clinical development. A large-scale collection of the source marine invertebrate can be difficult due to scarcity of the organism, and can also have negative environmental consequences. In addition, natural supplies can fluctuate, either seasonally or due to environmental changes. Aquaculture4 or cell culture of an invertebrate could alleviate supply problems, but approaches for these are not yet developed for all organisms. Ideally, an efficient chemical synthesis of the desired natural product could be achieved, however the structural complexity of many natural products such as bryostatin 1 (1) and swinholide A (2) requires inefficient multi-step syntheses that cannot meet the demands of pre-clinical and clinical development.5 Identification of a simpler, more easily synthesized structure which retains the biological activity6 is another option, but the best scenario would be to have a supply of the natural product that can be generated inexpensively and reproducibly in the lab under controlled conditions.1.2 Bioactive metabolite symbioses In most cases, it is likely that marine invertebrates produce their natural products themselves.7 However, on occasion analysis of the structure or localization of a natural product suggests that the molecule is biosynthesized by an associated microbial symbiont. Distinguishing between these possibilities was an area of intensive study in John Faulkner's laboratory. One of Faulkner's most important contributions was to recognize that this topic deserved attention beyond casual speculation, and that rigorous experimental tests were possible and should be pursued.
Symbiotic systems in which there is a strong likelihood of microbial bioactive metabolite synthesis offer attractive alternatives to chemical synthesis or extraction from natural sources. Symbionts that can be cultivated in the laboratory and still produce the bioactive metabolite could be subjected to fermentation technology to produce large amounts of the compound. However, cultivation of tightly integrated microbial symbionts can be difficult because of their dependency on the host,8 and success rates are thus low. In these cases, alternative means of obtaining the compound need to be explored.
Unlike their invertebrate hosts, genomes of bacteria and archaea are small and their biosynthetic pathways tend to be organized in contiguous regions of DNA (operons). These features greatly facilitate cloning of these pathways. Expression technology for bacterial genes is well developed, making cloning and expressing biosynthetic genes of bacterial symbionts entirely feasible. In the case of uncultivable symbionts, this provides the only way to produce bioactive metabolites in a culture system. For both cultivable and non-cultivable symbionts, cloning and expressing bioactive metabolite genes offer the possibility of providing sufficient amounts of compounds for drug development that could not otherwise be obtained, and open an avenue for combinatorial biosynthesis later on.
In this review, we will examine the process of determining whether or not symbionts are in fact likely to be producing a natural product, and outline approaches to identify, clone, characterize, and express bioactive metabolite genes from symbionts that do.
2 Criteria for proving symbiotic origin of bioactive metabolites
The study of symbiosis is evolving and specific criteria to prove hypotheses of the symbiotic origin of bioactive metabolites are emerging from the consensus of scientists in the field. In considering possible criteria, it is profitable to examine those used for a related subject, infectious disease. In classical microbiology, Koch's postulates, described in 1884,9 are the gold standard for determining the causative agent of a disease. They require 1) that the candidate organism always be present in the disease state, and absent in healthy organisms, 2) that it can be isolated in pure culture from diseased tissue, 3) that reintroduction of the agent precipitates the disease in healthy subjects, and 4) that the candidate can be isolated again from the diseased host. Satisfaction of all these principles constitutes a rigorous proof, but there are many cases, usually those in which the disease agent cannot be grown in pure culture, when all of the principles cannot be satisfied. In such situations, modified criteria must be employed to provide supporting evidence for the microbe's involvement in infection.The equivalent to Koch's postulates in bioactive metabolite symbiosis is to 1) correlate presence of the symbiont with a function for the host, 2) remove the symbiont and show loss of function, 3) reintroduce the symbiont and show that function is regained, and 4) isolate the symbiont again. This also is a rigorous approach, but in many symbioses all of these criteria cannot readily be fulfilled. One difficulty lies in obtaining aposymbiotic (symbiont-free) hosts, which are sometimes not viable without their symbiont.10 Also, reintroducing obligate symbionts, which do not maintain populations outside of the host, is far more difficult than reintroducing infectious organisms that have specifically evolved to invade their hosts. Obligate symbionts are often transferred only directly between generations and lack reinfection capability. As with infectious diseases, modified criteria must be employed to substantiate the role of a microbial symbiont in bioactive metabolite synthesis.
An alternative to the microbiological approach described above is to use molecular tools to demonstrate that the biosynthetic machinery for metabolite synthesis resides in the symbiont. Techniques to do so can be applied to purified or partially purified symbionts, or in situ, where a diagnostic signal is localized to the symbiont. The use of nucleotide probes for biosynthetic genes can confirm that these genes reside in the symbiont genome. However, this approach requires an authentic probe, derived from genes that have been independently verified to have the required function. Cloning biosynthetic genes from a symbiont and establishing their function can be a major undertaking in itself; hence initial experiments to generate small probes specific for the genes of interest can be useful. Likewise, specific antibodies could be used to detect the presence of biosynthetic enzymes in a symbiont. Unless one has an antibody that recognizes the same class of enzyme from different species, as in the detection of Rubisco in chemoautotrophic symbionts,11 this requires purifying or expressing the enzyme from the symbiotic association and verifying its function, before it can be used to produce specific antibodies for precise localization. Enzymatic function can also be directly assayed, or visualized in situ, using specific substrates that produce a colored, fluorescent or radioactive signal.
2.1 Criteria for study
Reproductive tissues in gametes and larvae are always important to examine for the presence of microbes. Although symbionts can be recruited from the environment, in many cases, the host has evolved mechanisms to ensure intergenerational (“vertical”) transmission. Microbes persistently associated with eggs and larvae are likely to have important roles in the life of the host, one of which could be synthesis of bioactive metabolites.
Both the criteria for evaluating the role of symbionts in bioactive metabolite production, and the techniques for investigating these symbioses are emerging. Applying these criteria can build substantial support for the involvement of a particular microbe in the synthesis of a bioactive metabolite. The final proof lies in either actually growing the symbiont in culture and subsequently isolating the natural product from the culture, or for non-cultivable symbionts, cloning and expressing target genes in a heterologous, cultivable organism. In this article we will use the research in our laboratory and our collaborations with John Faulkner's group to illustrate issues and describe methods important in investigating marine invertebrate/microbial symbioses and identifying the producer of a bioactive metabolite. Some of the concepts in this review were presented previously,7 however, our goal is to provide a comprehensive, practically oriented overview. We will focus on progress to date in the Bugula neritina–Endobugula sertula association, which has become our model system for developing approaches and methods. In addition, we will discuss examples of research on other invertebrate–microbe symbioses that demonstrate specific techniques and challenges in bioactive metabolite symbiosis research.
3 Evaluation of natural products as bacterially-produced compounds
Three criteria are often used to evaluate whether the structure of a natural product indicates a bacterial origin in a symbiotic association. First is that these compounds share structural similarities with those isolated from cultured microbes. For example, halichondramide (3) from the sponge Halichondria sp. bears a resemblance to scytophycin B (4) from the cyanobacterium Scytonema pseudohofmanni, and therefore is speculated to be microbial in origin.20 Another example is ecteinascidin, ET-743 (5) and related compounds found in the ascidian Ecteinascidia turbinata. They are thought to be biosynthesized by symbiotic bacteria since they share structural similarities with saframycin B (6), which is produced by Streptomyces lavendulae,20 and the safracins from Pseudomonas.21 (It is interesting to note that the production of ET-743 by PharmaMar is by semi-synthesis from safracin-B).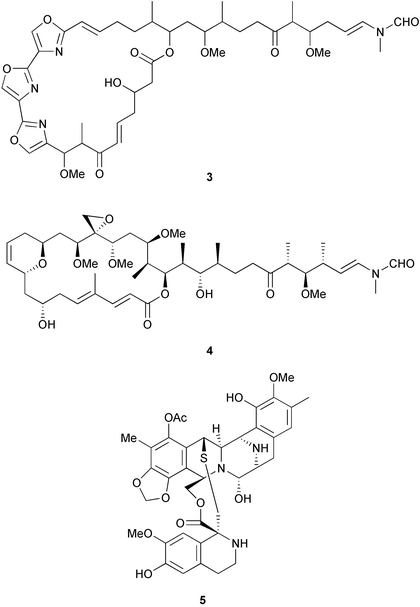
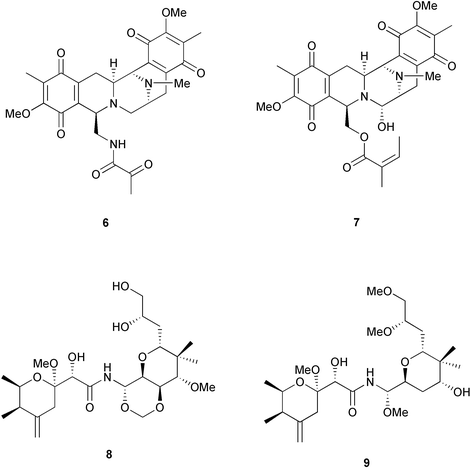
Another criterion used to evaluate whether a compound is microbially-produced is the presence of similar compounds in unrelated host organisms. In this case, it is considered more likely that microbes with a common biosynthetic capacity are found in the different hosts, rather than for the hosts to have undergone convergent evolution to be able to synthesize the same compound. The ecteinascidins also fit this criterion, as they are not only similar to an actinomycete metabolite, but also resemble renieramycin E (7) and its analogs, which were isolated from sponges of the genus Reniera.20 Similarly, mycalamide A (8) from the sponge Mycale sp. shares a striking resemblance to pederin (9), a metabolite isolated from the blister beetle Paederus sp.20
A final criterion is that even if the compounds do not share structural similarity with known metabolites from cultured microbes, or from unrelated organisms, a symbiotic origin is hypothesized if the metabolites appear to be synthesized by known microbial enzymes. For example, while bryostatin (1), isolated from the bryozoan Bugula neritina, does not superficially resemble any microbial product, it is a complex polyketide. Complex polyketides (non-aromatic macrolides) are typically produced by bacteria and fungi,22 and hence, it was suggested that bryostatin is produced by a microbial symbiont of the bryozoan.23 Cyclic peptides, and peptides with non-proteinogenic amino acids, are synthesized by NRPSs, which are enzymes typically found in microbes.18
It is important to note that these three criteria are more of a suggestive rather than a substantive way of targeting a symbiont as the source of a bioactive metabolite, because of the possibility that different hosts have evolved similar biosynthetic capacities. However, these criteria can be valuable in devising experiments to directly test such hypotheses. An example is the hypothesis that the B. neritina symbiont “Candidatus Endobugula sertula” is the synthetic source of bryostatin. By developing a probe to a modular PKS based on sequences from other microbes, Davidson et al.14 were able to demonstrate expression of a PKS in E. sertula, and this probe has enabled the cloning of the putative bryostatin PKS (unpublished data).
4 Localization of bioactive metabolites in marine invertebrates to specific cell types
A number of studies have been carried out on marine invertebrates where a natural product has been localized to a specific cell type. Although this information must be evaluated with the caveats described in section 2.1, namely that the site of synthesis may not be where the metabolite is ultimately localized, or the metabolite could be dietary-derived, cell type localization can still be a useful piece of information in determining if the compound is microbial in origin. A schematic diagram depicting approaches to localizing bioactive metabolites is shown in Fig. 1.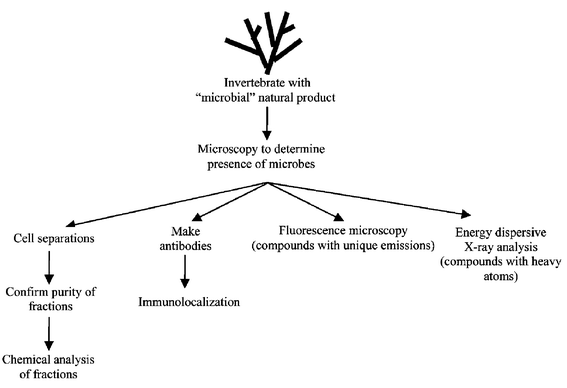 | ||
| Fig. 1 Approaches for localizing bioactive metabolites. | ||
Because marine invertebrates frequently contain large and diverse bacterial populations, exemplified by Aplysina aerophobia, Rhopaloides odorabile, and Theonella swinhoei,24 it is quite common to find potential natural product-producing bacteria in these organisms. However, in spite of the abundance of bacteria, most localization studies have implicated the host sponge (Table 1), or ascidian (Table 2) as the biosynthetic source of their bioactive metabolites. One important consideration of these data is that some host cell types may contain bacteria, either intracellularly or tightly associated with the exterior of the cell, and although this occurs frequently, in some studies it has been overlooked. When cell separation studies are done, it is important to rigorously analyze the bacterial content of the “host cell” fraction to evaluate whether bacteria are present.
| Species | Natural product(s) | Compound class | Ref |
|---|---|---|---|
| a Other sterols and non-brominated long chain fatty acids are found in sponge cells.106–108 | |||
| Amphimedon terpenensis | diisocyanoadociane |
diterpene
sterols |
100 |
| Amphimedon terpenensis | 3 brominated fatty acids | fatty acids | 101 |
| Aplysina fistularis |
aerothionin (34)
homoaerothionin (35) |
brominated tyrosine dev. | 37 |
| Crambe crambe | crambines and/or crambescidins | guanidine alkaloids | 102 |
| Dysidea avara | avarol | sesquiterpene hydroquinone | 103, 104 |
| Dysidea herbacea | spirodysin (17), herbadysidolide (18) | sesquiterpenes | 29 |
| Haliclona sp. | haliclonacyclamines A and B | pyridine alkaloids | 105 |
| Negombata magnifica | latrunculin B (36) | macrolide | 38 |
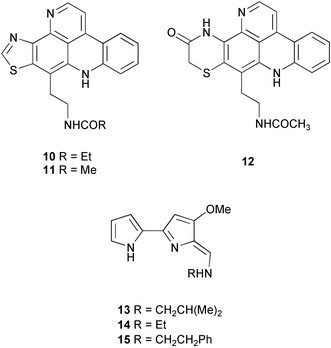
| Oceanapia sagittaria | dercitamide (10) | pyridoacridine alkaloid | 26 |
| Species | Natural product(s) | Compound class | Ref |
|---|---|---|---|
| a The relative stereochemistry of the tetrahydropyranyl and spiroketal moieties has been proposed.110 b Study results conflict with other studies shown in Table 3. c An example of a number of peptides, including tunichromes and larger polypeptides,111 isolated from the blood cells of ascidians. | |||
| Atapozoa sp. | tambjamines C, E, F (13–15) | bipyrrole alkaloids | 28 |
| Cystodytes dellechiajei | kuanoniamine D (11), shermilamine B (12) | pyridoacridine alkaloids | 27 |
| Lissoclinum bistratum | bistramide A (23)a,b (= bistratene A) | macrocyclic ether | 34 |
| Lissoclinum patella | patellamides A–C (31–33)b | cyclic peptides | 36 |
| Styela plicata | plicatamidec | octapeptide | 109 |
These localization studies (Tables 1 and 2) have revealed a few surprises. The pyridoacridines had been proposed to originate in a symbiont since they were isolated from unrelated organisms such as tunicates, sponges, an anemone (Cnidaria), and a prosobranch mollusc.25 However, Salomon and Faulkner utilized the pH-dependent fluorescent properties of dercitamide (10) to localize the metabolite to “inclusional” sponge cells in Oceanapia sagittaria.26 Further examination by transmission electron microscopy (TEM) revealed that no intracellular symbionts were present in these cells, providing further support that these metabolites were synthesized de novo by the sponge. A similar study conducted on the tunic of the ascidian Cystodytes dellechiajei using the pH-dependent properties of kuanoniamine D (11) and shermilamine B (12), indicated that the pyridoacridines were contained in ascidian bladder cells and pigment cells.27 The tambjamines have been isolated from bryozoans, ascidians, and a mutant strain of the bacterium Serretia marcescens, and therefore were also thought to be produced by associated bacteria in the ascidian Atapozoa sp.28 A study of the tissue by microscopy led to the proposal that tambjamine C, E and F (13–15) are found in granular amebocyte blood cells based on the fact that these compounds have a bright yellow coloration and the lack of intense pigmentation in other cells.28 Although this did not rule out the possibility that another pigment was responsible for the coloration of the granular amebocytes, the authors also indicated that there was no significant amount of intra- or extracellular bacteria in the ascidian, which provided further support that these compounds were biosynthesized by the Atapozoa sp. These methods do not exclude the possibility that the compound-containing cells are storage sites for natural products that are produced elsewhere. However, there is no known case of an extracellular bacterium in a marine invertebrate producing a natural product and transferring it to specific host cells. Instead, metabolite production in these organisms is likely due to convergent evolution to produce natural products that possess useful biological activities, or possibly due to gene transfer events.
Other studies have successfully identified a microbial symbiont responsible for the production of certain secondary metabolites (Table 3). Taking advantage of the auto-fluorescence of cyanobacteria, the sponge cells of Dysidea herbacea were separated from associated Oscillatoriaspongeliae filaments using a fluorescence activated cell sorter, and the chlorinated amino acid derivative 13-demethylisodysidenin (16) was shown to exist only in the filamentous cyanobacterial cells, while the sesquiterpenes spirodysin (17) and herbadysidolide (18) were found only in the sponge cells.29 Using the same technique on a different specimen of D. herbacea, Unson et al. demonstrated that a brominated diphenyl ether (19) was located only in the cyanobacterial filaments.17 Host cells and cyanobacterial cells from a sample of D. herbacea that contained the chlorinated diketopiperazines dihydrodysamide C (20) and didechlorodihydrodysamide C (21) were separated on a centrifugation density gradient, and the chlorinated metabolites were shown to exist only in the cyanobacterial fraction.30 Interestingly, one O. spongeliae fraction did not contain the chlorinated amino acid derivatives, leaving open the possibility that there may be closely related strains of cyanobacteria in the sponge. A study of the sponge Theonella swinhoei indicated that swinholide A (2) and theopalauamide (22) were localized to unicellular heterotrophic bacteria and a filamentous heterotrophic bacterium, respectively.31 This was accomplished through the use of differential centrifugation, a technique in which dissociated cells are exposed to increasing speeds of centrifugation to yield different fractions of cells. The filamentous heterotrophic bacterium was later identified as a δ-proteobacterium, “Candidatus Entotheonella palauensis”.32
| Host species | Natural product(s) | Compound class | Bacterium | Ref |
|---|---|---|---|---|
| a Study results conflict with other studies shown in Table 2. | ||||
| Dysidea herbacea | 13-demethylisodysidenin (16) | chlorinated amino acid dev. | Oscillatoria spongeliae | 29 |
| Dysidea herbacea | brominated diphenyl ether (19) | brominated diphenyl ether | Oscillatoria spongeliae | 17 |
| Dysidea herbacea |
dihydrodysamide C (20)
didechlorodihydrodysamide C (21) |
chlorinated diketopiperazines | Oscillatoria spongeliae | 30 |
| Lissoclinum bistratum | bistratamide A (29) and B (30)a | cyclic peptides | Prochloron sp. | 34 |
| Lissoclinum bistratum | bistramide A (23)a | macrocyclic ether | Prochloron sp. | 35 |
| Lissoclinum patella | lissoclinamide 4 (24) and 5 (25), ulithiacyclamide (26), patellamide D (27), ascidiacyclamide (28)a | cyclic peptides | Prochloron sp. | 33 |
| Theonella swinhoei | swinholide A (2) | macrolide | unicellular heterotrophic | 31 |
| Theonella swinhoei | theopalauamide (22) | bicyclic glycopeptide | “Candidatus Entotheonella palauensis” | 32 |
Cellular localization studies do not always definitively identify the source organism. A good example is several studies conducted on the ascidians Lissoclinum bistratum and Lissoclinum patella, where it was attempted to determine whether the cyclic peptides and the macrocyclic ether bistramide A (= bistratene A) (23) were located in ascidian cells or in associated cyanobacterial cells of the genus Prochloron. Initial studies based on separated cyanobacterial cells from L. patella indicated that lissoclinamides 4 (24) and 5 (25), ulithiacyclamide (26), patellamide D (27) and ascidiacyclamide (28) were produced by the symbiont, as they could be isolated from the Prochloron cells in equal or greater amounts on a weight-to-weight basis than could be found in the entire colony.33 From Lissoclinum bistratum, using the same technique, Degnan et al.34 reported that the peptides bistratamide A (29) and B (30) were found in the cyanobacteria, while bistramide A (23) was not. A second study of L. bistratum contradicted these results, concluding that bistramide A (23) was found in Prochloron cells at concentrations 4 to 6 times greater than in the intact ascidian.35 A recent study of L. patella has indicated that the cyclic peptides patellamides A–C (31–33) are not found in separated Prochloron cells, but are distributed throughout the tunic.36 Based on these experiments, the source of the cyclic peptides and bistramide A (23) is unclear and awaits further studies.
Other techniques to localize natural products to specific cell types are available. If the natural product is halogenated, as in the case of aerothionin (34) and homoaerothionin (35) isolated from the sponge Aplysina fistularis, energy dispersive X-ray microanalysis can be used to determine the cellular location of the metabolite in sections of tissue.37 In situations where the natural product is not halogenated and cellular dissociation is not easy, immunolocalization of the compound may be possible. This technique requires the production and isolation of antibodies that specifically bind to the natural product, which can be used as a probe to determine the cellular location of the compound in a tissue section. This was accomplished in the localization of latrunculin B (36) in the sponge Negombata magnifica.38
5 Investigating microbe presence
An important criterion for demonstrating microbial origin of a natural product is to confirm the presence of a persistently associated microbe with the animal host. Following conventional environmental microbiology research, one can employ techniques from both molecular biology and microscopy, as diagrammed in Fig. 2. Polymerase chain reaction (PCR)-based techniques can be used to identify bacterial small subunit (16S) ribosomal RNA (rRNA) gene sequences, revealing phylogenetic affiliations of microbes in or on animal tissues. Probes targeting these sequences must be used to confirm the presence of and localize specific microbes within a sample. When it is feasible to use probes for biosynthetic genes, probing for in situ expression of candidate biosynthetic genes can identify producers of bioactive compounds. Here we summarize approaches using probing methods for the investigation of symbiotic production of bioactive metabolites.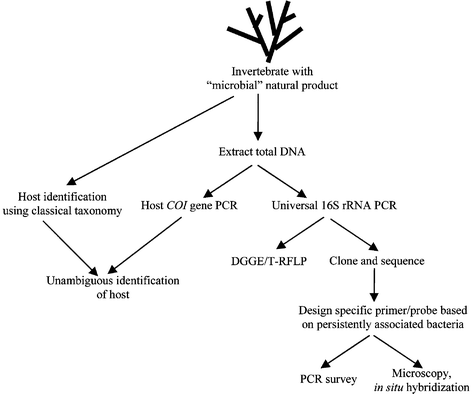 | ||
| Fig. 2 Approaches for investigating microbe presence. | ||
The 16S rRNA gene is typically amplified by PCR from a total DNA preparation of the invertebrate and its associated microbes (Fig. 2). Universal primers are used so that 16S rRNA genes from all associated bacteria are amplified. Plasmid clone libraries are constructed from the mixed pool of PCR products, and clones are sequenced to determine what microbes are associated with the invertebrate (Fig. 2). One concern with PCR-based methods is a phenomenon known as PCR bias in which universal primers may actually favor certain sequences over others.44 PCR can also 1) produce chimeras in which portions of a sequence are derived from different species, 2) produce sequence errors due to misincorporation by the DNA polymerase, and 3) form heteroduplexes consisting of imperfectly matching strands of DNA hybridized to each other. However, these artifacts can be minimized by using high fidelity enzymes, adjusting PCR conditions, and post-PCR purification.45
As with all environmental sampling methods, determining the sampling number (evaluating when enough clones have been sequenced to provide a representative picture of the bacterial community) is an important consideration. Because of nonspecific association of environmental bacteria in an invertebrate, a bacterial species that is most abundant in a sample is not necessarily significant to the host. In addition, the abundance of a sequence in a clone library does not necessarily reflect its abundance in a natural sample due to possible PCR artifacts. Most analyses to date on bacterial biodiversity in sponges have been based on sequencing 50–70 clones of 16S rRNA.8,24 Depending on the number of microbes present and their relative abundance, this may lead to an underestimation of the total diversity present in an organism. Statistical approaches for estimating microbial biodiversity and determining the number of sequences required for accurate representation of the natural sample have been the subject of several reviews.46–48 Although a number of tools are available, none appear superior, and different methods on the same sample can yield biodiversity estimates that differ by an order of magnitude or greater.46,49,50 Despite these problems, it seems likely that improved statistical analyses will become routinely incorporated into studies of microbial diversity of marine invertebrates.
Sequencing clone libraries from several invertebrate samples can be tedious, but other molecular approaches allow surveys of microbial diversity in invertebrates (Fig. 2). Denaturing gradient gel electrophoresis (DGGE) separates DNA according to the temperature required to separate the two DNA strands (the melting temperature), which differs depending on the nucleotide composition of the DNA.51,52 Therefore, a mixed pool of 16S rRNA gene fragments from different organisms generated by PCR can be separated by DGGE (Fig. 3). Ideally, identical sequences migrate to the same position in the gel, so the use of DGGE to profile PCR products from multiple samples can reveal bacterial sequences that are common among different samples (Fig. 3). One problem with DGGE is that heteroduplexes are formed when amplifying from a mixed population of DNA, which will migrate as separate bands but need to be excluded from the analysis. A way to minimize heteroduplex formation is through the use of reconditioning PCR,53 in which a final PCR product is diluted and reamplified with excess primers for a few cycles. Another potential issue is that different sequences may have similar melting temperatures, and can co-migrate. Running additional gels with less steep temperature gradients can provide better resolving power, although only sequencing of bands will confirm that they represent only one species.
 | ||
| Fig. 3 Denaturing gradient gel electrophoresis of 16S rRNA from Bugula neritina. Samples are PCR amplifications of 1) a cloned 16S rRNA from E. sertula, 2) DNA isolated from adult B. neritina, 3) DNA isolated from a bacterially enriched fraction of adult B. neritina, and 4) DNA from B. neritina larvae. Arrow denotes the 16S rRNA band from E. sertula, other bands (lanes 2 and 3) are from other bacteria associated with the host. | ||
Another method for comparing microbial communities among different communities of the host invertebrate is terminal restriction fragment length polymorphism (T-RFLP). This technique involves amplifying community DNA with fluorescently labeled universal 16S rRNA primers and then generating DNA fragments of different lengths depending on their sequence by restriction enzyme digestion.54 These fragments are separated electrophoretically, and their sizes are diagnostic of the individual microbe 16S rRNA gene sequences present. This is a rapid method to profile similarities and differences among many samples; however, because sequencing is not involved, it does not permit direct identification of the microbes.
5.3 Probes for bioactive metabolite genes An alternative and complementary approach to 16S rRNA-based probes is biosynthetic gene probes based on characteristics of the secondary metabolite of interest (Fig. 2). For example, complex polyketides such as bryostatin 1 are synthesized by modular PKSs, enzymes that have distinct functional domains within their larger protein sequence. The amino acid sequence of certain domains is relatively well-conserved across species, as is the case of the type I bacterial PKS β-ketoacyl synthase (KS) domain. By comparing amino acid sequences of this domain in several bacteria, Davidson et al. (2001)14 identified conserved amino acids that were used to design degenerate oligonucleotide primers complementary to the gene sequence encoding those amino acids. Degenerate primers compensate for the redundancy of the genetic code, and will amplify from all sequences that encode the chosen amino acid sequence, enabling the isolation of genes even when the exact DNA sequence is unknown. Using these degenerate primers under specific PCR conditions, a fragment of a KS gene sequence was obtained from a B. neritina DNA extract. These primers and the KS gene fragment were invaluable in other characterizations of the B. neritina/E. sertula symbiosis.14Isolating even a short (ca. 250 base pair) DNA fragment specific to the symbiont of interest is an extremely valuable tool in the characterization and eventual cloning of a bioactive metabolite pathway. Another method, which does not rely on oligonucleotide primers, is the isolation of a symbiont-specific DNA fragment using a DNA fragment probe derived from the same type of gene in another organism. This approach, called “heterologous hybridization”, depends on gene sequences from the two organisms being similar enough. Hybridization refers to the complementary base pairing of two DNA sequences; if one is labeled the other can be identified. For successful heterologous hybridization the two genes usually must be from closely related species. There are other potential complicating issues in this approach; however, heterologous hybridization can be considered as another approach for isolating gene fragments and entire genes from natural product biosynthetic pathways.
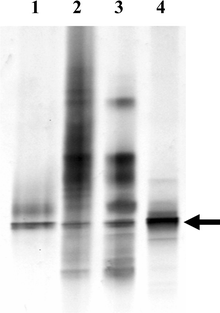
5.4 Testing persistent association of microbes with their hosts by PCR or DGGE Once candidate 16S rRNA or biosynthetic gene fragment sequences are obtained, they can be used to demonstrate the association of a possible symbiont with its host (Fig. 2). PCR or DGGE techniques are especially useful for this purpose. A PCR survey of B. neritina isolated from a variety of locations, and other bryozoans, showed consistent presence of the KS gene fragment described above along with the presence of bryostatin in B. neritina, providing evidence that this KS gene was involved in bryostatin synthesis.14 DGGE can be used to compare the microbial communities of multiple samples or different life cycle stages of the same species (Fig. 3). This enables discrimination between microbes that are only transiently or sporadically associated with the invertebrate and those that are true symbionts.It is important to consider the life cycle stage of the host when attempting to identify candidate symbiotic microbes. As mentioned previously, direct transmission of a microbe from generation to generation is indicative of an important functional interaction; therefore, analyzing gametes or reproductive tissues (e.g. developing embryos or larvae) can be valuable. In addition, levels of non-persistent microbes may be reduced at particular stages in the life cycle. For example, non-feeding B. neritina larvae do not contain microbes from a gut, in contrast to adult B. neritina. DGGE analysis of adult and larval B. neritina DNA extracts indicates a significant enrichment of E. sertula in the larvae relative to adult tissue (Fig. 3).
Once a microbial species is shown to be persistently associated with an animal, experimental manipulation of the bacterial population in the host can help to determine if there is a microbial role in the bioactive metabolite biosynthesis. Antibiotics interacting with bacterial but not eukaryotic ribosomes can be applied in an attempt to reduce the numbers of bacteria. This was done using the antibiotic gentamycin sulfate on developing colonies of B. neritina.14 After subsequent growth, PCR screening with E. sertula-specific primers indicated that levels of E. sertula were reduced, and subsequent analysis showed that bryostatin levels were as well.14 There was not a strict correlation between the amount of reduction in the symbiont and bryostatin; possible reasons for this were discussed in section 2.1, however, the result is consistent with an E. sertula involvement in bryostatin synthesis.
5.5 Investigating microbe presence by microscopy Conventional light microscopy, scanning electron microscopy, and transmission electron microscopy have historically been used for observing bacteria in environmental samples and animal tissues. In addition, development of fluorescent stains for application in epifluorescence microscopy has increased capabilities for observing microbes in complex environmental samples. For example, the fluorescent dye 4′-6-diamidino-2-phenylindole (DAPI), which binds to DNA,55 allows the researcher to distinguish between cells with genetic material and inorganic bacteria-sized particles. These tools allow determination of whether microbes are associated with the animal of interest. However, only labeled specific nucleotide probes enable researchers to localize a specific microbe or the expression of particular microbial genes in a given sample.From PCR and sequencing, a researcher can obtain a 16S rRNA sequence to identify microbes in a host animal. Microscopy then becomes an essential complement to the molecular data (Fig. 2). Persistent microbial associates of invertebrates can be identified by PCR or DGGE, but the source of a given sequence must be confirmed by localizing the sequence to microbial cells in the sample.
5.6 Localization of microbes in animal hosts using in situ hybridization Analyzing the microbial community of filter-feeding animals such as sponges, tunicates, and bryozoans can be a daunting task. In situ hybridization (ISH), a technique in which probes labeled with fluorescent molecules or enzymes that catalyze colorimetric reactions bind to a desired target, is a powerful tool for localizing microbes in complex environmental communities and correlating expression of specific genes to specific microbes. The method involves incubating labeled oligonucleotide or polynucleotide probes, which can be specific to groups of microbes or to individual species, with fixed animal tissue, and then visualizing a probe-specific signal with the microscope. Images of labeled microbes in animal tissue allow confirmation of the presence and abundance of specific microbes, in addition to localization on a microscopic scale. The ability to localize microbes in animal hosts is indispensable for investigating symbioses. Localization of a particular 16S sequence in microbial cells is necessary for confirmation that the bacteria are associated with the host rather than incidentally in the seawater or on the animal surface during sampling. Haygood and Davidson used this approach to localize E. sertula in B. neritina larvae, showing that the larval pallial sinus exclusively contained E. sertula.56 Schmidt et al.32 used cell separations, DGGE, PCR and fluorescent in situ hybridization (FISH) to identify and localize a filamentous δ-proteobacterium, “Candidatus Entotheonella palauensis”, in the sponge T. swinhoei (Fig. 4). Localization of the symbiont by FISH confirmed that the sequence obtained did indeed originate in the filamentous microbe, and that the candidate symbiont was sufficiently abundant for the production of theopalauamide (22), which was previously localized to the filaments.31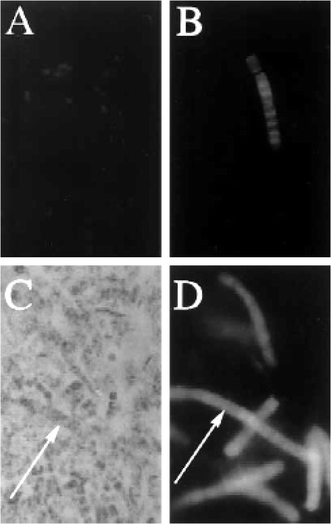 | ||
| Fig. 4 Fluorescent in situ hybridization of E. palauensis in tissue of the sponge Theonella swinhoei.32A, B Universal bacterial 16S rRNA probe; C, DE. palauensis-specific 16S rRNA probe. A Fluorescence micrograph of unicellular bacteria (400 ×); B Fluorescence micrograph of filamentous bacteria (800 ×); C Light micrograph (400 ×); D fluorescence micrograph (400 ×) (arrows indicate identical filaments in C and D). Used with permission of the authors and the journal Marine Biology. | ||
Localizing a particular microbe in situ can also reveal host adaptations to symbiosis, indicative of a tight association between microbe and host. One example is the transmission of symbiotic microbes via reproductive tissues to future offspring, documented in numerous marine invertebrate–microbe associations, including but not limited to bryozoans14,56,57 and bivalves.58–63 In addition, microbes visualized in specialized host structures, such as bacteriocytes,64,65 are likely to be important symbionts. Such adaptations suggest that there has been evolutionary selection for the maintenance of these specific bacteria, and recent research suggests that vertical symbiont transmission may be reflected by highly co-evolved host–symbiont associations.66 Physiological and behavioral adaptations for symbiont transmission have also been noted in many sponge species. The transmission of maternal bacteriocytes into developing embryos has been found in Petrosia ficiformis, Chondrosia reniformis, and at least two species of Oscarella.67–69 Characterizing the complex microbial communities associated with invertebrate tissues remains a significant challenge, but identifying bacteria associated with reproductive structures in invertebrates may offer a targeted approach to identifying those microbes significant to the biology of the host, including those that may be responsible for bioactive metabolite biosynthesis.
5.7 Localization of expression of biosynthetic genes in symbiotic systems If a candidate biosynthetic gene (section 2.1) has been isolated from a sample, nucleotide probes targeting the messenger RNA (mRNA) transcripts of the gene can be designed, labeled, and used to localize its expression. Co-localization of a biosynthetic transcript and a specific 16S rRNA sequence in the same microbial cell can confirm that the biosynthetic gene is expressed in the microbe. Davidson et al. used this approach in B. neritina by constructing a ribonucleotide probe targeting the transcript containing one of the KS domains in the putative bryostatin biosynthetic gene cluster. This ribonucleotide probe hybridized to mRNA within E. sertula cells, providing conclusive evidence that E. sertula expressed this domain.14 5.8 Obstacles with in situ hybridization in symbiotic systems There are technical challenges involved in using in situ hybridization to localize microbes within animals, in contrast to pure cultures or environmental microbial samples. One problem is the autofluorescence of the host tissue, and in some cases, of the microbial biomass as well. Commonly used fluorescent labels, such as fluoroscein, rhodamine, and their derivatives, absorb and fluoresce at similar wavelengths as many endogenous organic compounds. In epifluorescence microscopy, filters for fluorescently tagged probes are designed to detect emission within a range of approximately 50–60 nanometers. With the advent of confocal laser scanning microscopy and improvements in imaging software and technology, it is now possible to detect emission over a narrower range. This allows the researcher to specifically target the emission wavelength of the fluorophore molecule, blocking out most autofluorescence from the unhybridized portion of the sample and increasing the signal to noise ratio. Systems that rely on colorimetric detection have an analogous difficulty. Because these detection schemes employ probe labels such as biotin and rely on activity from enzymes such as phosphatase and peroxidase, any biotin or enzyme activity endogenous to the host tissues will result in a false signal. Reagents and protocol modifications that successfully block endogenous activity have been developed, and are crucial for these types of detection schemes. Because of background issues, extensive controls are required for all in situ hybridization experiments.Another technical challenge is that because symbionts have reduced growth rates relative to free-living microbes,70 this results in lower levels of rRNA, and hence reduced signals in ISH.71–73 Protocols have been developed for signal amplification that allow the visualization of smaller microbes with decreased metabolic activity.74,75 Catalyzed reporter deposition-FISH (CARD-FISH), utilizing tyramide signal amplification (TSA), has been developed and adapted to increase signal above background levels.74 This can also be done with colorimetric detection schemes. In addition to CARD-FISH, ribonucleotide probes, which target 16S rRNA and contain multiple labels, have been used for detection of slow-growing microbial populations.71,75,76 These modifications in detection methods should significantly improve the researcher's ability to detect symbiotic microbes in environmental samples and animal tissues.
6 Identifying and isolating biosynthetic genes
When several of the criteria described above are consistent with a microbial symbiont being the source of a bioactive compound, the stage is set to isolate the biosynthetic genes from the symbiont to enable definitive proof. In this section we will discuss approaches to do this (Fig. 5). | ||
| Fig. 5 Approaches for cloning bioactive metabolite genes. | ||
Another method for enrichment is the separation of bacteria using density gradient centrifugation. This procedure involves isolating an enriched bacterial sample, and centrifuging the material on a Percoll™ (Pharmacia) gradient. This will separate bacteria based on their buoyant density and can also remove host cellular material based on the same principle.30 Fluorescence activated cell sorting has also been used to separate symbiotic bacteria from their hosts.29
6.2 DNA isolation, purification, and enrichment procedures Before isolating DNA to characterize and clone symbiont genes, it is important to consider what treatments and characterization steps will be required after the DNA is isolated. For example, if the particular gene cluster is predicted to be large, then clone libraries with larger inserts are desirable, which requires isolating high molecular weight DNA. In general, isolating high molecular weight DNA is advantageous, however, for some characterization steps, lower molecular weight DNA, which requires less care in preparation, may be sufficient. Another consideration is the presence of inhibitors to DNA manipulations (restriction digests, cloning reactions, PCR), and how these may be removed.An effective method for DNA extraction that works on most tissue types is to freeze the material at −80 °C, and then grind aliquots in a small amount of dry ice with a pre-chilled mortar and pestle. After the material is pulverized into a fine powder, it is added to an extraction buffer (see below). With careful manipulation, DNA isolated by this method is of high enough molecular weight for most cloning approaches. An advantage of this technique is that there is little opportunity for endogenous nucleases to degrade the DNA, provided it remains frozen until added to the extraction buffer. A disadvantage is that there is no opportunity for pre-enriching the symbiont, unless enough symbiont material can be isolated prior to freezing.
DNA can be extracted from pulverized frozen material or live material by a short (5 min) incubation in an extraction buffer followed by phenol : chloroform partition. We use the extraction buffer of Davidson et al.14 which inhibits endogenous nuclease activity, lyses cell membranes, and extracts and denatures proteins. This buffer has proven effective on a variety of organisms. For B. neritina we have tested more benign buffers with a goal of digesting the bacterial cell wall with lysozyme prior to extraction or treating enriched bacterial cells with DNase to remove contaminating DNA, and found that the DNA was substantially degraded by endogenous nuclease activity. Depending on the system, it may be worth attempting these procedures; however, in general the more rapidly the cell material is incubated in extraction buffer and treated with phenol : chloroform, the more intact the DNA. To obtain high molecular weight DNA, during the phenol : chloroform partition it is critical to very gently mix the aqueous and organic layers for an extended period of time. We use a rotator apparatus that inverts the tube with the extraction mixture at 25 rpm for 40 min. After centrifugation, the aqueous layer is gently removed with a large-bore pipet into a new tube, and the extraction repeated for 20 min. The use of large-bore pipets and pipet tips is essential to minimize DNA shearing which will reduce the average size of the DNA. After extraction the DNA can be precipitated using standard procedures.77 Precipitated DNA can either be pelleted by centrifugation or if sufficient quantities are present, removed by spooling on a glass rod. The latter technique has two advantages, 1) if inhibitors are present that co-pellet with the precipitated DNA, then a larger proportion of them can be removed, and 2) spooled DNA is easier to resuspend and disperse in solution than the compact DNA pellet resulting from centrifugation.
Uncharacterized inhibitors can be a significant problem for subsequent manipulations with DNA especially considering that they can co-purify during DNA precipitation; several methods can be tried to remove them. A simple one to remove small inhibitors is to pass the DNA solution through a Sephadex-based spin column normally used to remove oligonucleotides from PCR reactions. Another is to use silica-based DNA-binding columns for cleanup. DNA treated in these ways is likely to be sheared by the manipulations, and so may not be suitable for cloning, but can be used for PCR. The best method to maintain high molecular weight DNA during cleanup is CsCl gradient centrifugation.
6.3 Purification and enrichment of DNA on CsCl gradients After DNA has been extracted from a sample, there are options for further purification and enrichment. A most effective method to remove inhibitors and cellular RNA is to subject the bacterial DNA to ethidium bromide–caesium chloride (EtBr–CsCl) equilibrium density gradient ultracentrifugation. The high ionic strength of the solution facilitates dissociation of pigments and inhibitors from the DNA. In the case of DNA preparation from B. neritina, ultracentrifugation resulted in RNA, pigments, and inhibitory compounds migrating to the bottom of the gradient, while the DNA complexed with EtBr was at a higher level (unpublished results). This method successfully removed an inhibitory pigment associated with DNA from B. neritina.Once inhibitory compounds have been removed, there is the option of further enrichment of the symbiont DNA, especially if several bacterial species are present in the sample. Enrichment can be an important factor because it reduces the number of clones required in a library, and increases signal : noise in other analyses such as Southern hybridizations (section 7.5). One enrichment method is to fractionate the DNA on a CsCl gradient containing Hoechst 33258 dye (Behring Diagnostics). Hoechst dye is a bisbenzimide DNA intercalator that will bind differentially to DNA based on the percentage of adenines and thymines (AT%) in the sequence. Ultracentrifugation will separate the DNA into distinct bands on the CsCl gradient, based on differences in AT content. Bands can be removed from the gradient in small fractions and the fractions then screened by PCR for genes known to be contained within the symbiont genome to identify those fractions most highly enriched in symbiont DNA. In attempting to enrich for E. sertula DNA from B. neritina, 5 bands were observed, which corresponded to various environmental bacterial DNAs, the symbiont DNA, and residual host DNA not removed in the bacterial enrichment protocol.
6.4 Monitoring the extent of enrichment Because of the diversity of bacterial communities in marine invertebrates, enrichment procedures can vary in their effectiveness from organism to organism. It is thus vital to monitor the extent of enrichment in each case. There are several methods to accomplish this. Competitive PCR (Fig. 6) can be used to quantify the amount of symbiont DNA in a sample. In this method, known amounts of a cloned symbiont gene fragment, with a small internal deletion or insertion to alter size and allow resolution on an agarose gel (a “competitor”), are added to samples of the target DNA. Primers specific to the gene are then used for amplification. Because of competition between full and altered-sized copies, the ratio of amplified products reflects the ratio of the amount of target and competitor DNA. Because the initial amount of added competitor is known, the amount of target DNA can then be estimated. By comparing enriched with pre-enriched samples, the extent of enrichment can be determined. In the case of the B. neritina/E. sertula system competitive PCR (Fig. 6) indicated a 5.5 fold enrichment of E. sertula DNA by preparing a bacterial fraction by differential centrifugation, and a 2.9 fold additional enrichment using Hoechst dye–CsCl gradient fractionation, for an overall 16 fold enrichment.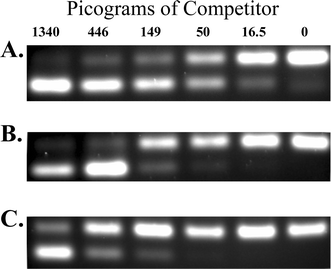 | ||
| Fig. 6 Competitive PCR analysis of DNA preparations from Bugula neritina. Panels depict agarose gel separations of PCR products, amplified from the KSa β-ketoacyl synthase domain of the putative bryostatin gene cluster.14 The upper band in each panel is amplification from the authentic gene copy, and the lower band is from an added amount (in picograms, denoted at the top) of a clone of KSa with a small internal deletion (the competitor). When the amount of competitor DNA is equivalent to the amount of the authentic gene copy, amplification products in the upper and lower bands are of equal intensity (e.g. panel A, 50 pg). By titrating the amount of added competitor, conditions are determined where amplification is equal, and the greater the amount of competitor needed for this (or the transition between lower and upper band predominance), the more enriched the genomic DNA is in the target gene. In this experiment, A) total, B) bacterial enriched, and C) Hoechst dye–CsCl gradient fractionated DNAs (see text for explanation) are compared. The data indicate 5.5-fold enrichment in the bacterial fraction, and 16-fold enrichment in the Hoechst dye–CsCl gradient DNA, relative to the total DNA preparation. | ||
A technique called quantitative real-time (QRT) PCR provides a more accurate method for determining levels of specific DNA sequences in a sample but requires specialized equipment. This method is based on detection of a fluorescent signal that changes proportionally during amplification of a PCR product. There are two general methods for QRT PCR (see78 for a brief review). The first involves adding the fluorescent dye, SYBR Green I, to a PCR reaction.79 SYBR Green I binds to double stranded DNA, and as products from the PCR accumulate, an increasing fluorescent signal is generated. By comparison with standards, one can determine the initial quantity of the gene being amplified. There are variations on the second method, which in general involves fluorescently labeled primers whose fluorescence increases or decreases (due to quenching) in relationship to the progress of the PCR reaction.78 A significant advantage of the SYBR Green method is that standard primers are used rather than more expensive fluorescently labeled primers. QRT PCR has advantages over competitive PCR, the most notable being the precision of detection and the rapidity of data collection,78 but for most cloning procedures only an approximation of the extent of enrichment is necessary.
Another, less quantitative, method to monitor enrichment is to use Southern blot hybridizations. This method (described in section 7.5) involves hybridizing symbiont specific probes to a blot containing enrichments of the symbiont DNA and comparing the intensity of hybridization. A drawback of this approach is that more input DNA is required and a certain level of enrichment may be necessary to even detect a signal.
6.5 DNA preparation for pulsed field gel electrophoresis (PFGE) In pulsed field gel electrophoresis, controlled changes in the direction of an electric field through an agarose gel enable the separation of very large DNA, on the order of entire bacterial genomes, 4–5 megabase pairs (Mbp). Isolating very high molecular weight DNA (on the order of 0.5 Mbp or larger) as one does for PFGE is essential for cloning very large fragments in bacterial artificial chromosomes (BACs), and can provide accurate genome sizing information if only a single bacterial species is present. In theory PFGE could be used to separate chromosomes of different bacteria in a mixture, provided their genome sizes are sufficiently different.There are special procedures to prepare DNA for PFGE. The first step is to determine whether sufficient bacteria of interest can be isolated from the organism. This is critical because unless enough bacteria are present, DNA will not be visible on the pulsed field gels. The amount of bacteria is substantial; for a 4.3 Mbp genome, a concentrated pellet containing on the order of 1 × 1010 cells resuspended in 1 ml is required. This number can be difficult, if not impossible, to achieve with most symbiotic bacteria. In addition to the large number of cells, a complicating factor can be the presence of residual host cell material; if this cannot be adequately removed, then it may not be possible to obtain a sufficiently concentrated bacterial sample. Both difficulties have occurred in our work on the B. neritina/E. sertula association, and PFGE has not been successful. If a sufficient amount of bacteria can be isolated, then a concentrated solution of the bacteria is mixed with molten agarose and poured into a mold to form a plug. Once solidified, plugs are placed in an EDTA solution and then treated with SDS and protease over an extended period to lyse the bacteria and digest their proteins. The semi-solid nature of the agarose prevents the cells from bursting during lysis, which is a primary cause of DNA shearing during isolation. For PFGE, the plugs are then incorporated into a gel and electrophoresed using parameters optimal for the desired size range separation. DNA can be digested for cloning by equilibrating plugs with restriction buffer and adding restriction enzyme in a prolonged incubation.80 DNA can then be isolated from gels for cloning by using agarose digesting enzymes or electroelution, taking care to minimize manipulations that may reduce the size of the DNA.80
7 Cloning of biosynthetic genes
The strategy to choose for cloning a bioactive metabolite pathway depends on the size of the region to be cloned and whether all elements of the pathway are likely to be located in proximity in the genome. An additional factor is the average molecular weight of DNA that can be isolated. We will address these issues while discussing cloning approaches and their advantages and disadvantages. 7.1 DNA requirements and general cloning procedures As a result of the manipulations during isolation procedures, DNA is sheared to a particular average size. In addition, differences in endogenous nuclease activities can also result in size differences. Clone libraries are usually generated by cutting the DNA into pieces by partial digestion with restriction enzymes that cut frequently. The advantage of this is that a given region of a genome is covered by multiple DNA fragments that overlap each other, which after cloning, usually allows complete coverage of the region of interest. Ideally, the average size of DNA prior to digestion should be five times larger than the desired size for cloning to ensure that the ends of most DNA fragments will result from restriction digestion and not shearing. However, representative libraries can be made from DNA three times larger than the desired size for cloning.Even though large DNA is advantageous for cloning, it is inherently viscous, which creates problems in its manipulation, especially prior to digestion. Precipitated high molecular weight DNA is difficult to resuspend, but this is best accomplished by gently shaking the tube containing the DNA for several hours to overnight. Even with this treatment, dispersal of the DNA evenly throughout the solution can be difficult. Evidence for uneven dispersal can be obtained by reading the absorbance of equal volume aliquots from the same solution of DNA in a spectrophotometer. If the DNA is not evenly dispersed, significantly different absorbances will result in subsequent aliquots. It is important to have evenly dispersed DNA to enable reproducibility in restriction digests and other manipulations. To maximize the homogeneity of large DNA in a solution, gently and repeatedly pipeting the solution with a large bore pipet is advisable. This may result in some DNA shearing, but is often necessary.
Partial digests are done by incubating a standard amount of DNA with different concentrations of restriction enzyme for a given time. An enzyme typically used for partial digests of genomic DNA is Sau3AI, which recognizes the frequently represented sequence GATC. Pilot-scale digests are usually done to estimate the amount of enzyme to use in larger scale digests. It is advantageous to use the same concentration of DNA in all digests because it minimizes variability and enables scaling up. For example, we typically use DNA at 100 ng µl−1, and add restriction enzyme based on units of enzyme per µl. After assembling reactions on ice, restriction enzyme is added, thoroughly mixed, and the sample incubated at 37 °C for 1 h. EDTA is then added to 20 mM, and the sample incubated at 70 °C for 15 min to inactivate the enzyme. Digestion products are electrophoresed on an agarose gel and the average molecular weight of digested DNAs is compared with standards to identify the optimal amount of enzyme. A valuable approach is to digest more DNA than needed for the gel and examine a portion of it for size. In samples having optimal size, the remainder of the digest can be used for cloning. When doing partial digests, one should keep in mind that occasionally the distance between cut sites can be significantly larger than the average molecular weight of the desired digestion product. In this case a particular region may be underrepresented in a clone library. We have encountered this in the E. sertula PKS cluster.
Digested DNA should be size-fractionated prior to cloning to minimize cloning undesirably small fragments and to maximize insert size. For small-insert size libraries (< 10 kilobase pairs – kbp), fractionation can be done by separation in an agarose gel, and DNA extracted from the gel for cloning. Because recovery from gels can be low, it is important to digest sufficient DNA to ensure enough material for ligation. For larger insert size libraries (10–40 kbp), sucrose gradient fractionation of DNA is an effective means of size enrichment. A simple means of generating an approximately linear sucrose gradient is to pour a step gradient with equal volumes of 40%, 30%, 20%, and 10% sucrose in buffer and then freeze and thaw the solution in the tube. During the thawing process, the less concentrated sucrose melts more slowly and as it does so it migrates up the tube, linearizing the gradient. We form gradients containing sucrose in a buffer of 10 mM Tris, pH 8.0, 10 mM NaCl, 1 mm Na2EDTA, and using 2.5 ml of each concentration of sucrose in an SW41 ultracentrifuge rotor (Beckman) tube. Samples, in volumes of 1 ml or less, are layered on the gradients and centrifuged in the SW41 rotor for 22 h at 22,000 rpm and 20 °C. After centrifugation, gradients can be fractionated in small aliquots (200–400 µl) from the top using a pipettor. Fractions are analyzed on an agarose gel to determine those with DNA sized suitably for the desired cloning. One possible drawback of the sucrose gradient method is that large amounts of partially digested DNA (50–100 µg) must be loaded in order to visualize individual fractions. For bacterial artificial chromosome library construction, size fractionation in pulsed-field gels is the method of choice (section 6.5).
7.2 Cloning vectors
As a probe for screening libraries, ideally one would like to have a gene fragment from the metabolite pathway to be cloned. Gene fragments can be generated by PCR, as described previously. If a probe cannot be generated from the symbiont of interest, one can try to use a probe derived from a similar gene in another organism. A potential problem with this approach is that because heterologous probes are not likely to match the gene of interest perfectly, hybridizations must be done under lower stringency conditions. This can lead to higher background, making it difficult to isolate truly positive clones. When using heterologous probes, it is a good idea to determine optimal hybridization conditions by Southern blots (section 7.5) prior to screening a library.
7.4 Cloned fragments that rearrange in, or are detrimental to E. coli The problem of cloning DNA fragments that rearrange or are detrimental to propagation in E. coli can be serious, and one that is difficult to track down. The literature contains little discussion on this subject, because only successful cloning attempts are reported. Most cloning and expression vectors are developed with well-characterized genes encoding small soluble proteins that are stable and allow for maximum levels of expression. When cloning a bioactive metabolite pathway, which in many cases encodes large protein complexes, the situation can be very different. Three possible problems can occur, 1) rearrangement or deletion of portions of a clone, 2) “leaky” expression of genes that produce a protein toxic to E. coli, or 3) leaky or induced expression making a protein that removes significant amounts of metabolic pathway intermediates from E. coli, inhibiting growth.Simply cloning large fragments of DNA in a high copy number vector can put a strain on the E. coli DNA replication machinery. For example, if a 35 kbp fragment is cloned in an 8 kbp cosmid vector maintained at 25 copies per cell, the additional DNA represents 25% of the E. coli genome size. A significantly larger genome is selected against, which encourages mechanisms that reduce genome size. This is one of the advantages of maintaining libraries in lambda phage, because they divert the E. coli replication machinery for their own ends and are not subjected to selective pressures generated by constraints on E. coli growth.
Rearrangements or deletions are caused by recombination between repeated sequences within a cloned region. Recombination across inverted repeats will invert the intervening sequence, whereas recombination between tandemly oriented repeats can delete the intervening sequence. Since tandem repeat recombination decreases the size of the cloned fragment, this can be positively selected for. Maintaining the cloned DNA in recombination deficient hosts (e.g. SURE E. coli strain, Stratagene), a low copy number vector, or using a host that reduces copy number (ABLE E. coli strain, Stratagene) can be helpful in minimizing or eliminating these problems.
Most E. coli cloning and expression vectors contain the T7 phage promoter, which is used for high level induction of expression of cloned genes. However, this promoter is “leaky”, in that it is not completely repressed and there is always a baseline level of transcription occurring. This can result in expression of a cloned protein product when it is not desired, which can be detrimental to E. coli and provide a selection against cloning a gene. We have encountered this situation several times, and have expended a considerable amount of effort troubleshooting cloning protocols, when in fact the trouble was not in the cloning, but in the clone. There can be positional determinants involved; for example, we have successfully cloned larger fragments stably, whereas smaller fragments derived from the larger clones were unstable, or vice versa. A useful test to determine if lethality is due to leaky expression is to clone the fragment in both possible orientations. If clones are only obtained when the gene is cloned in the opposite orientation relative to the T7 promoter, then selection against the insert is likely occurring. Specific host strains and T7 based vectors can minimize leaky expression; however, problems can still occur. Other promoter systems for expression can be tested; however some of these (e.g. the arabinose inducible promoter) are also leaky.
The mechanisms of cloned gene toxicity in E. coli can vary, but generally lie in properties of the expressed protein. For example, proteins containing highly hydrophobic regions can be toxic to E. coli, either through self-association or association and disruption of the cell membrane. Another difficulty can arise if a clone produces an enzymatically active complex that removes significant amounts of E. coli metabolic pathway intermediates, inhibiting growth.
7.5 Restriction mapping and Southern blot hybridization In addition to deletions or rearrangements occurring during cloning, it is also possible to clone separate gene fragments from different parts of the genome into the same vector. Either phenomenon results in gene sequences that appear contiguous but in fact are not. One way to evaluate whether cloned DNA has deleted or rearranged is to compare the cloned DNA sequence with genomic DNA by restriction mapping and Southern blotting.A restriction map positions sequences within a region of DNA by cleavage into defined fragments using restriction endonucleases. For Southern blot analysis, this process involves digestion of the DNA of interest with appropriate restriction enzymes, separation of the resulting DNA fragments by gel electrophoresis, transfer of these fragments to a nylon membrane and then hybridization of labeled probes to visualize specific fragments (Fig. 7). If the pattern of fragment sizes comparing cloned and native DNA match, this indicates that the cloned DNA is not rearranged or deleted (Fig. 7). In addition to confirming a restriction map, this type of analysis can indicate the presence or absence of separate but similar genes of a given type in the DNA, indicated by the number of hybridized bands on the blot. This can be useful in providing evidence that one has cloned the correct gene or that only one gene of a given type exists in a symbiont association, contributing to the proof that an identified gene is responsible for making the natural product of interest.
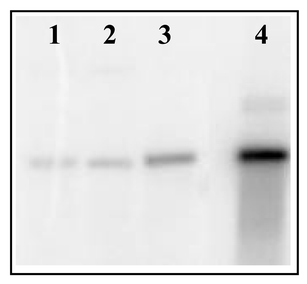 | ||
| Fig. 7 Southern hybridization of a putative bryostatin PKS cluster probe to DNA isolated from B. neritina. Lanes are: 1) total DNA, 2) bacterial-enriched fraction DNA, 3) Hoechst dye–CsCl gradient fractionated DNA, and 4) cosmid clone of the region. | ||
To perform these analyses, it is necessary to have the desired DNA enriched sufficiently from other contaminating DNA. In some instances, a total DNA preparation from B. neritina has not been enriched enough in E. sertula DNA for detection by Southern hybridization. Even when successful, it is clear that enrichment improves the hybridization signal (Fig. 7).
General protocols for Southern blot analysis can be found in Sambrook et al.77 However, several variables specific to the genes of interest should be considered. Restriction enzymes should be chosen to produce DNA fragments of lengths that can be resolved on a gel (size ranges are dependent on gel parameters). The amount of DNA per digest is also important, especially when the DNA is not a pure sample. For E. sertula, 2–3 µg of symbiont enriched DNA per digest was optimal to observe hybridization. For pure bacterial DNA, 0.5–1 µg is sufficient.
7.6 Considerations if the host organism proves to be the synthetic source of the bioactive metabolite Because most natural product synthesis activities in marine invertebrates have been localized to the host and not to their associated bacteria (Tables 1–3), one should consider what to do if one wants to isolate genes encoding such activities from the host. In general, this is a matter of scale up; since the eucaryotic genomes of the hosts are likely to be between one and two orders of magnitude larger than those of their associated microbes, one needs to generate correspondingly larger clone libraries. The techniques of probe generation, localization, DNA purification and enrichment, and cloning of genes will be similar for both microbes and their hosts. One advantage of cloning a host bioactive metabolite gene is that the host DNA is likely to be by far the most abundant in a DNA preparation.8 Strategies for sequence determination
Once clones suspected to contain genes for a bioactive metabolite are obtained, the gene sequence must be determined. There are several approaches to do this, and it is likely that more than one will come into play, especially when sequencing a large region of DNA. An initial approach that is helpful for generating or confirming a restriction map is to digest a larger clone into smaller pieces and subclone these fragments. Sequence determined from the ends of these fragments can then be used to design primers to sequence in the opposite direction on the larger clone from which the subclones were derived. This will result in sequence overlapping the restriction cut sites, allowing determination of adjacent restriction fragments. For complete sequence determination two methods are used, primer walking and shotgun sequencing. With primer walking, after obtaining initial sequence data, one uses those data to design new oligonucleotide primers to extend the sequence in another round of sequencing. This is repeated until the entire sequence is obtained. By determining multiple initial sequences, as in the fragment subcloning approach described above, one can design multiple primers for subsequent rounds of sequencing, speeding the process of obtaining the entire sequence. A drawback of this approach is that there is idle time between designing and obtaining new primers. With shotgun sequencing, a large piece of cloned DNA is randomly sheared by passage of the DNA solution through a narrow orifice (a nebulizer) into fragments of 1–2 kbp average size. These fragments are cloned into a plasmid vector, generating a library of pieces of DNA from the initial large clone. Because the shearing is random, sequencing of the multiple and overlapping clones in this library results in a complete sequence of the region.9 Confirming that cloned genes encode the biosynthetic machinery for a metabolite
The traditional way of determining gene function is to create mutations in the gene to eliminate its function. However, this process relies on the ability to introduce DNA into bacteria by transformation and have the bacteria either integrate and express a mutated gene or insert a foreign piece of DNA into a gene. In situations where the bacterium has not been cultivated this approach cannot be taken, and one must evaluate the likelihood that a gene produces the metabolite in question by analysis of the encoded protein sequence, and if this is promising, express the gene in a heterologous host to synthesize the desired product. 9.1 Analysis of domain content Once a putative bioactive metabolite gene or gene cluster is cloned, analysis of its sequence using comparison programs such as BLAST81 can identify domains with previously identified function. In the case of many biosynthetic pathway genes, such domains are well conserved and can reveal information about gene cluster function as well as the metabolite it produces. For example, in PKS gene clusters, the sequence of the genes often corresponds to the order of formation of the polyketide product, as in the modular PKS that produces erythromycin.82–84 In these situations, one can literally “read” the sequence of domains in the gene and infer the structure of the polyketide it produces. However, this is not always the case. PKSs are continually discovered in which the gene sequence is not co-linear with the formation of the polyketide product or contains too few or excess domains. This is the case in the bryostatin PKS from E. sertula (unpublished data), as well as those PKSs producing stigmatellin and the antibiotic TA.85,86 Some domains can be identified by homology but be nonfunctional due to mutations in their active sites, for example in the pikromycin PKS from Streptomyces venezulae.87 Thus, analysis of domain content and order can not always be relied on to predict the function and product of biosynthetic gene clusters.If the sequence of a gene cluster is not clearly indicative of the product it is responsible for making, one can consider isolating the enzymes responsible for synthesizing the metabolite from the symbiont as a means of confirming that the genes encode these proteins. If a purified protein preparation is shown to synthesize the compound in question, then one could perform amino acid sequencing to determine whether the proteins contain the amino acid sequence predicted by the gene sequence. This would constitute definitive proof that the gene encoded the enzymes that synthesized the compound, but requires a strategy to isolate and purify the synthesizing activity. This approach could also be helpful for identifying accessory proteins that might associate with a core enzyme but might not have been identified through gene sequencing alone. One method to aid in this process is to subclone and express individual domains from the sequenced region and use these domain proteins to make specific antibodies. Antibodies could be used to identify the entire enzyme complex from whole protein preparations of the sample, and to co-localize activity with protein.
9.2 Expressing cloned genes for the bioactive metabolite A definitive method to verify that a gene cluster encodes enzymes responsible for making a bioactive metabolite is to actually synthesize the metabolite by expressing the entire pathway in a culturable host. The size of the gene cluster becomes a factor even in the subcloning manipulations required to make constructs for expression, and if a cluster is large (> 30 kb) it might be advantageous to express smaller portions and combine the proteins later. If the biosynthetic machinery exists as a complex that forms by self-assembly of different subunits, then expressing and purifying the different subunits and then combining them would be a viable way to attempt to reconstitute activity. Using an expression system with an inducible promoter would allow one to control expression, which can be important if a gene product is detrimental to the expression host. A native gene's promoter (RNA polymerase recognition site) may not work nor be well regulated in the expression host.The choice of expression vector and host can vary depending on the sequence of the genes cloned and the metabolite made. The AT% of a gene can be an important factor, because if it is not similar to the AT% of the host, the gene may not be efficiently expressed. The underlying mechanism for this has to do with bias towards particular transfer RNAs (tRNAs) used in a given organism, which may not match well with those found in the cloned gene. Analyzing the AT content of a cloned gene as well as performing homology searches to identify species containing the most closely related genes can aid in making effective decisions regarding the host for expression. E. coli is most commonly used for expression because of a thorough understanding of its genetics, flexible DNA manipulation technologies, well-developed and varied expression vector systems, and rapid growth of cells. Strains of E. coli are available (Invitrogen) that have been engineered to correct for tRNA usage bias, by containing plasmids expressing rarely used tRNAs.
For gene clusters that produce complex natural products, there can be accessory genes or proteins needed for complete function, which might not be provided for in E. coli. One solution is to clone the desired accessory genes from another organism into E. coli, to attempt to provide a functional counterpart. Alternatively, one can explore other hosts. Many species of the genus Streptomyces are naturally “tuned” to produce complex polyketide compounds in significant quantities and contain the required accessory proteins (e.g. phosphopantetheinyl transferase genes necessary for PKS function). Furthermore, Streptomyces contains a network of metabolic pathways that produce many of the starter units necessary for metabolite production,24,88 and there have been many reports on heterologous expression of natural or hybrid PKS genes in this genus. However, the low AT% (30%) of Streptomyces can be a drawback. Bacillus subtilis is another established host for the expression of recombinant proteins, with an AT% closer to 50%.
An alternative to bacterial expression hosts is to use the eucaryotic methylotropic yeast, Pichia pastoris. Expression in P. pastoris is under the control of the alcohol oxidase promoter, which provides highly regulated, high-level expression of the protein of interest.89 This system is particularly well suited for expression of soluble proteins in their native form, which might be essential for functional enzymes. However there are limitations to this system, which include higher protease activity and lower levels of heterologous protein expressed compared with bacterial hosts.
The choice of what heterologous expression system to use, and what constructs to make, has an empirical aspect. With expression of any large, complex pathway, unanticipated problems can arise that are difficult to track down and resolve. Problems that can arise include the possible instability of cloned DNA in a given host and the production of a protein that is detrimental to the host, even when expression is not induced (section 7.4). If difficulties arise in one system, sometimes the solution is just to try another system.
Although there may be difficulties, the benefits of cloning and expressing bioactive metabolite genes are enormous. If successful, expression not only allows definitive proof of function, but allows the synthesis of unlimited amounts of a compound. In addition, once cloned, genetic manipulations can alter the gene sequence to create new and possibly more effective bioactive metabolites. Bioengineering can also help us to understand the interaction between complex enzymes and pathways.
10 Genome-based methods in symbiont bioactive metabolite research
10.1 Why use genome-based methods? Whole genome sequencing can be very useful in the study of symbiont secondary metabolism. By determining the complete sequence of a genome, one can identify all of the biosynthetic clusters in an organism, which will include the pathway of interest, as well as other cryptic pathways. Even well-studied microbes such as Streptomyces coelicolor have yielded several unknown secondary metabolite pathways when sequenced.90 The genome sequence also gives us insight into the regulation of biosynthetic pathways. In addition, if a symbiont cannot be cultivated, genomics can provide clues to aid in cultivation efforts by identifying genes lacking in the symbiont whose function is compensated for by the host. 10.2 Genome sequencing and metagenomes Genome sequencing capabilities have increased tremendously over the past few years. There are at least 145 complete genomes and hundreds more in the pipeline (www.genomesonline.org). Microbial genomes are typically sequenced using a whole genome shotgun method. This entails fragmenting the genome into smaller pieces, cloning these fragments into a vector, and sequencing the clones. The raw sequences from many clones are then compared to one another and pieced together by matching up identical sequences. This process is called assembly and two or more raw sequences that have been matched up form a contiguous segment, also called a contig.Though the majority of the microbes being sequenced are pathogens, microbes that have interesting secondary metabolisms are being sequenced as well. Most of these microbes are readily cultivated, but it is also possible to sequence the entire genome of uncultured obligate symbionts or parasites. There are two completed genomes of Streptomyces,91 and at least three more are being sequenced. Buchnera sp., the symbiont of aphids,92Wigglesworthia glossinidia, the symbiont of tsetse flies,93 and Rickettsia prowazekii, an intracellular parasite in eukaryotic cells94 have been sequenced. The genome sequencing of a microbial symbiont of the marine sponge Axinella sp. is currently underway. In these systems, DNA of the microbe of interest is purified from host DNA and DNA of other associated microbes. This approach may work well if the microbial community of the host is comprised of only one or a few species. Alternatively, one could enrich for the symbiont or symbiont DNA (section 6.1). Symbiont enrichment was used to construct a library of the archaeal symbionts of A. mexicana.95 In the B. neritina/E. sertula system, enrichment of symbiont DNA is likely to yield the purest DNA for library construction and sequencing. However, in some systems such as T. swinhoei and the “zoo” of microbes it harbors, it may prove difficult to purify each symbiont away from the others, and although enrichment for symbiont DNA could be explored, yet another approach should be considered. The complex microbial community in sponges is not unlike soil microbiota; hence, determining the metagenome of symbiotic microbes may be more appropriate. The metagenome refers to the collective genomes of a biological community.96 BAC or cosmid libraries of soil metagenomes have been constructed and screened for biological activity as a result of expression of cloned genes in E. coli, or screened for biosynthetic genes.97 Soil DNA has also been cloned and expressed in S. lividans, and novel natural products were isolated from the transformants.98
10.3 Whole genome sequencing: practical considerations and challenges There are some issues to consider when sequencing the genome of a microbial symbiont. Because obligate symbionts of marine invertebrates have not yet been cultured, DNA used to make libraries for genome sequencing has to originate from a mixed environmental pool. Consequently, more sequencing is required compared to a cultivable microbe with similar genome size to obtain the same coverage. Coverage refers to the extent to which a nucleotide is represented by raw sequences. Table 4 gives an estimate of the amount of sequencing required to assemble a genome. It is apparent that the more enriched the DNA preparation is of your target organism, the less sequencing is required. Assembly of the symbiont genome is also confounded by the presence of other bacteria. Host sequence can be readily weeded out during assembly since its genome size will be two or more orders of magnitude larger than that of the symbiont. Thus, contigs of the host genome will be rare, while the symbiont genome will be easily assembled. Contigs of transiently associated non-symbiotic bacteria can be excluded if their 16S rRNA gene is located on a contig, enabling their identification.| Organism | Genome size/Mb a | % purity b | # Mb for 10X coverage | # of clones (2 kb insert size) |
|---|---|---|---|---|
| a Host genome size estimated from an average of organisms in the same phyla (www.genomesize.com). b Proportion of DNA that belongs to organism of interest. c Genome size of E. sertula estimated by flow cytometry (unpublished data). d Estimate of number of dominant symbionts in T. swinhoei which will have the most representation in a library. e Genomes of higher eukaryotes are usually not shotgun sequenced using small insert libraries. | ||||
| E. sertula | ∼2 c | 100 | 20 | 10 000 |
| E. sertula + B. neritina | ∼2 (host ∼200) | 50 | 40 | 20 000 |
| E. sertula + B. neritina | ∼2 (host ∼200) | 10 | 200 | 100 000 |
| T. swinhoei dominant symbionts (15) d | ∼60 (est. 4 per symbiont) | 100 | 600 | 300 000 |
| T. swinhoei dominant symbionts (15) + sponge | ∼60 (host ∼1500) | 50 | 1200 | 600 000 |
| Homo sapiens | 3000 | 100 | 30000 | n/a e |
There are other alternatives to whole genome sequencing. One promising approach is high throughput genome scanning.99 This method takes advantage of clustering of biosynthetic genes in microbes, and can be used on enriched DNA from one symbiont or a DNA sample from a microbial community. Briefly, two libraries are constructed: one small-insert library that is shotgun sequenced, and one BAC library. The sequences are identified, and those that match biosynthetic genes of interest are used as probes to screen the BAC library. The entire biosynthetic cluster can then be sequenced from the BAC clone. Zazopoulos et al.99 successfully used this method to isolate a class of biosynthetic genes from microbes that were not known to produce those metabolites.
11 Summary
Detailed investigation of bioactive metabolite symbioses is a field that is still in its infancy. Every organism and every symbiotic system requires effort to understand and overcome the unique problems it presents. From apparently simple tasks such as extracting useful DNA, to identifying symbiont candidates and seeking biosynthetic genes, attention to detail in building a strong foundation of basic knowledge ultimately enhances the chances of success. Despite the intrinsic challenges of this field, we believe that skillful application of research approaches borrowed from molecular biology and microbial ecology, combined with natural products chemistry, will yield a greater understanding of these fascinating systems. This understanding will ultimately be used to help realize the biomedical potential of marine natural products.12 Acknowledgements
John Faulkner was our cherished colleague, mentor and unflagging cheerleader in this work. We also thank Carolyn Sheehan and Deeanne Edwards for their assistance in experiments, and Eric Schmidt for permission to use Fig. 4. We appreciate assistance from CalBioMarine Technologies, Inc. (Carlsbad, CA). L. E. W. is supported by a Department of Defense Postdoctoral Traineeship Award (DAMD17-00-1-0181), and G. E. L. is a Howard Hughes Predoctoral Fellow. K. H. S. is a Los Angeles Chapter of Achievement Rewards for Collegiate Scientists Foundation fellow, and C. P. R. was funded by a grant from the National Science Foundation (CHE 98-16169). Work in the lab was supported by the National Institute of Health (5R01CA079678-03), California Sea Grant (R/MP-88), and the Department of Defense (DAMD 17-00-1-0183).13 References
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2003, 20, 1 RSC.
- D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1 RSC.
- A. M. S. Mayer and M. T. Hamann, Comp. Biochem. Phys. C, 2002, 132, 315 Search PubMed.
- D. Mendola, in Drugs from the Sea, ed. N. Fusetani, S. Karger A. G., Basel, 2000, p. 120 Search PubMed.
- D. J. Faulkner, Antonie Van Leeuwenhoek, 2000, 77, 135 CrossRef CAS.
- P. A. Wender, J. L. Baryza, C. E. Bennett, C. Bi, S. E. Brenner, M. O. Clarke, J. C. Horan, C. Kan, E. Lacote, B. Lippa, P. G. Nell and T. M. Turner, J. Am. Chem. Soc., 2002, 124, 13648 CrossRef CAS.
- D. J. Faulkner, M. K. Harper, M. G. Haygood, C. E. Salomon and E. W. Schmidt, in Drugs from the Sea, ed. N. Fusetani, S. Karger A. G., Basel, 2000, p. 107 Search PubMed.
- N. S. Webster, K. J. Wilson, L. L. Blackall and R. T. Hill, Appl. Environ. Microbiol., 2001, 67, 434 CrossRef CAS.
- M. T. Madigan, J. M. Martinko, J. Parker, Brock Biology of Microorganisms, 9th edn, Prentice Hall, New Jersey, 2000 Search PubMed.
- M. Bright, H. Keckeis and C. R. Fisher, Mar. Biol., 2000, 136, 621 CrossRef.
- C. M. Cavanaugh, M. S. Abbott and M. Veenhuis, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7786 CAS.
- N. Kondo, N. Nikoh, N. Ijichi, M. Shimada and T. Fukatsu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14280 CrossRef CAS.
- Q. B. Zhang, M. C. Cone, S. J. Gould and T. M. Zabriskie, Tetrahedron, 2000, 56, 693 CrossRef CAS.
- S. K. Davidson, S. W. Allen, G. E. Lim, C. M. Anderson and M. G. Haygood, Appl. Environ. Microbiol., 2001, 67, 4531 CrossRef CAS.
- J. A. Doino and M. J. McFallNgai, Biol. Bull., 1995, 189, 347 Search PubMed.
- F. Dedeine, F. Vavre, F. Fleury, B. Loppin, M. E. Hochberg and M. Bouletreau, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6247 CrossRef CAS.
- M. D. Unson, N. D. Holland and D. J. Faulkner, Mar. Biol., 1994, 119, 1 CAS.
- D. Schwarzer, R. Finking and M. A. Marahiel, Nat. Prod. Rep., 2003, 20, 275 RSC.
- S. Davidson, S. W. Allen, G. E. Lim, C. M. Anderson and M. G. Haygood, Appl. Environ. Microbiol., 2001, 67, 4531 CrossRef CAS.
- J. Kobayashi and M. Ishibashi, Chem. Rev., 1993, 93, 1753 CrossRef CAS.
- Y. Ikeda, H. Idemoto, F. Hirayama, K. Yamamoto, K. Iwao, T. Asao and T. Munakata, J. Antibiot., 1983, 36, 1279 CAS.
- B. J. Rawlings, Nat. Prod. Rep., 1997, 14, 523 RSC.
- U. Anthoni, P. H. Nielsen, M. Perieira and C. Christophersen, Comp. Biochem. Physiol., 1990, 96B, 431 Search PubMed.
- U. Hentschel, J. Hopke, M. Horn, A. B. Friedrich, M. Wagner, J. Hacker and B. S. Moore, Appl. Environ. Microbiol., 2002, 68, 4431 CrossRef CAS.
- T. F. Molinski, Chem. Rev., 1993, 93, 1825 CrossRef CAS.
- C. E. Salomon, T. Deerink, M. H. Ellisman and D. J. Faulkner, Mar. Biol., 2001, 139, 313 Search PubMed.
- E.-M. Rottmayr, B. Steffan and G. Wanner, Zoomorphology, 2001, 120, 159 Search PubMed.
- N. Lindquist and W. Fenical, Experientia, 1991, 47, 504 Search PubMed.
- M. D. Unson and D. J. Faulkner, Experientia, 1993, 49, 349 Search PubMed.
- A. E. Flowers, M. J. Garson, R. I. Webb, E. J. Dumdei and R. D. Charan, Cell Tissue Res., 1998, 292, 597 CrossRef CAS.
- C. A. Bewley, N. D. Holland and D. J. Faulkner, Experientia, 1996, 52, 716 Search PubMed.
- E. W. Schmidt, A. Y. Obraztsova, S. K. Davidson, D. J. Faulkner and M. G. Haygood, Mar. Biol., 2000, 136, 969 CrossRef CAS.
- B. M. Degnan, C. J. Hawkins, M. F. Lavin, E. J. McCaffrey, D. L. Parry, A. L. van den Brenk and D. J. Watters, J. Med. Chem., 1989, 32, 1349 CrossRef CAS.
- B. M. Degnan, C. J. Hawkins, M. F. Lavin, E. J. McCaffrey, D. L. Parry and D. J. Watters, J. Med. Chem., 1989, 32, 1354 CrossRef CAS.
- J.-F. Biard, C. Grivois, J.-F. Verbist, C. Debitus and J. B. Carre, J. Mar. Biol. Assoc. U. K., 1990, 70, 741 Search PubMed.
- C. E. Salomon and D. J. Faulkner, J. Nat. Prod., 2002, 65, 689 CrossRef CAS.
- J. E. Thompson, K. D. Barrow and D. J. Faulkner, Acta Zool., 1983, 64, 199.
- O. Gillor, S. Carmeli, Y. Rahamim, Z. Fishelson and M. Ilan, Mar. Biotechnol., 2000, 2, 213 CAS.
- N. Knowlton, Annu. Rev. Ecol. Syst., 1993, 24, 189 Search PubMed.
- S. K. Davidson and M. G. Haygood, Biol. Bull., 1999, 196, 273 Search PubMed.
- T. M. McGovern and M. E. Hellberg, Mol. Ecol., 2003, 12, 1207 CAS.
- D. Erpenbeck, J. A. J. Breeuwer, H. C. van der Velde and R. W. M. van Soest, Mar. Biol., 2002, 141, 377 Search PubMed.
- B. L. Maidak, G. J. Olsen, N. Larsen, R. Overbeek, M. J. McCaughey and C. R. Woese, Nucleic Acids Res., 1996, 24, 82 CrossRef CAS.
- M. F. Polz and C. M. Cavanaugh, Appl. Environ. Microbiol., 1998, 64, 3724 CAS.
- X. Y. Qiu, L. Y. Wu, H. S. Huang, P. E. McDonel, A. V. Palumbo, J. M. Tiedje and J. Z. Zhou, Appl. Environ. Microbiol., 2001, 67, 880 CrossRef CAS.
- B. J. M. Bohannan and J. Hughes, Curr. Opin. Microbiol., 2003, 6, 282 CrossRef CAS.
- A. P. Martin, Appl. Environ. Microbiol., 2002, 68, 3673 CrossRef CAS.
- J. B. Hughes, J. J. Hellmann, T. H. Ricketts and B. J. M. Bohannan, Appl. Environ. Microbiol., 2002, 68, 448 CrossRef CAS.
- B. B. Ward, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10234 CrossRef CAS.
- T. P. Curtis, W. T. Sloan and J. W. Scannell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10494 CrossRef CAS.
- S. G. Fischer and L. S. Lerman, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 1579 CAS.
- G. Muyzer and K. Smalla, Antonie Van Leeuwenhoek, 1998, 73, 127 CrossRef CAS.
- J. R. Thompson, L. A. Marcelino and M. F. Polz, Nucleic Acids Res., 2002, 30, 2083 CrossRef CAS.
- C. B. Blackwood, T. Marsh, S. H. Kim and E. A. Paul, Appl. Environ. Microbiol., 2003, 69, 926 CrossRef CAS.
- D. H. Williamson, D. J. Fennell, in Methods in Cell Biology, ed. D. M. Prescott, Academic Press, New York, 1975, p. 335 Search PubMed.
- M. G. Haygood and S. K. Davidson, Appl. Environ. Microbiol., 1997, 63, 4612 CAS.
- R. M. Woollacott, Mar. Biol., 1981, 65, 155 CrossRef.
- A. Fiala-Medioni, Z. P. McKiness, P. Dando, J. Boulegue, A. Mariotti, A. M. Alayse-Danet, J. J. Robinson and C. M. Cavanaugh, Mar. Biol., 2002, 141, 1035 Search PubMed.
- D. L. Distel, D. J. Beaudoin and W. Morrill, Appl. Environ. Microbiol., 2002, 68, 6292 CrossRef CAS.
- O. Gros, L. Frenkiel and H. Felbeck, Symbiosis, 2000, 29, 293 Search PubMed.
- D. M. Krueger, R. G. Gustafson and C. M. Cavanaugh, Biol. Bull., 1996, 190, 195 Search PubMed.
- A. R. Sipe, A. E. Wilbur and S. C. Cary, Appl. Environ. Microbiol., 2000, 66, 1685 CrossRef CAS.
- S. C. Cary, Eos, 1994, 75, 60 Search PubMed.
- N. A. Moran and J. J. Wernegreen, Trends Ecol. Evol., 2000, 15, 321 CrossRef.
- M. A. Johnson and C. Fernandez, J. Mar. Biol. Assoc. U. K., 2001, 81, 251 Search PubMed.
- A. S. Peek, R. A. Feldman, R. A. Lutz and R. C. Vrijenhoek, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9962 CrossRef CAS.
- M. Sciscioli, E. Lepore, M. Gherardi and L. S. Liaci, Cah. Biol. Mar., 1994, 35, 471 Search PubMed.
- G. Muricy, C. Bezac, M. F. Gallissian and N. Boury-Esnault, J. Nat. Hist., 2002, 33, 159 Search PubMed.
- A. V. Ereskovksy and N. Boury-Esnault, J. Nat. Hist., 2002, 36, 1761 Search PubMed.
- M. G. Haygood, B. M. Tebo and K. H. Nealson, Mar. Biol., 1984, 78, 249 CrossRef.
- A. Pernthaler, C. M. Preston, J. Pernthaler, E. F. DeLong and R. Amann, Appl. Environ. Microbiol., 2002, 68, 661 CrossRef CAS.
- H. Eilers, J. Pernthaler, F. O. Gloeckner and R. Amann, Appl. Environ. Microbiol., 2000, 66, 3044 CrossRef CAS.
- Y. Oda, S. J. Slagman, W. G. Meijer, L. J. Forney and J. C. Gottschal, FEMS Microbiol. Ecol., 2000, 32, 205 CrossRef CAS.
- A. Pernthaler, J. Pernthaler and R. Amann, Appl. Environ. Microbiol., 2002, 68, 3094 CrossRef CAS.
- E. F. DeLong, L. T. Taylor, T. L. Marsh and C. M. Preston, Appl. Environ. Microbiol., 1999, 65, 5554 CAS.
- M. Karner, E. F. DeLong and D. M. Karl., Nature, 2001, 409, 507 CrossRef CAS.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: a laboratory manual, 2nd edn, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1989 Search PubMed.
- W. Gloffke, Scientist, 2003, 17, 41 Search PubMed.
- A. K. R. Dhar and K. R. Klimpel, J. Clin. Microbiol., 2001, 39, 2835 CrossRef CAS.
- B. Birren, E. D. Green, S. Klapholz, R. M. Myers, H. Riethman and J. Roskams, Genome analysis: A laboratory manual (Volume 3: Cloning Systems), 1st edn, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1999 Search PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Wyers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403 CrossRef CAS.
- J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, Nature, 1990, 348, 176 CrossRef CAS.
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, Science, 1991, 759, 675.
- D. J. Bevitt, J. Cortes, S. F. Haydock and P. F. Leadlay, Eur. J. Biochem., 1992, 204, 39 CAS.
- N. Gaitatzis, B. Silakowski, B. Kunze, G. Nordsiek, H. Bloecker, G. Hoefle and R. Mueller, J. Biol. Chem., 2002, 277, 13082 CrossRef CAS.
- Y. Paitan, G. Alon, E. Orr, E. Z. Ron and E. Rosenberg, J. Mol. Biol., 1999, 286, 465 CrossRef CAS.
- Y. Xue, D. Wilson and D. H. Sherman, Gene, 2000, 245, 203 CrossRef CAS.
- H. Liu and K. A. Reynolds, J. Bacteriol., 1999, 181, 6806 CAS.
- J. M. Cregg, T. S. Vedvick and W. C. Raschke, Bio-technology, 1993, 11, 905 Search PubMed.
- S. D. Bentley, K. F. Chater, A. M. Cerdeno-Tarraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C. H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M. A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141 CrossRef.
- H. Ikeda, J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori and S. Omura, Nat. Biotechnol., 2003, 21, 526 CrossRef.
- S. Shigenobu, H. Watanabe, M. Hattori, Y. Sakaki and H. Ishikawa, Nature, 2000, 407, 81 CrossRef CAS.
- L. Akman, A. Yamashita, H. Watanabe, K. Oshima, T. Shiba, M. Hattori and S. Aksoy, Nat. Genet., 2002, 32, 402 CrossRef CAS.
- S. G. E. Andersson, A. Zomorodipour, J. O. Andersson, T. Sicheritz-Ponten, U. C. M. Alsmark, R. M. Podowski, A. K. Naslund, A. S. Eriksson, H. H. Winkler and C. G. Kurland, Nature, 1998, 396, 133 CrossRef CAS.
- C. Schleper, E. F. DeLong, C. M. Preston, R. A. Feldman, K. Y. Wu and R. V. Swanson, J. Bacteriol., 1998, 180, 5003 CAS.
- J. Handelsman, M. R. Rondon, S. F. Brady, J. Clardy and R. M. Goodman, Chem. Biol., 1998, 5, R245 CAS.
- S. Courtois, C. M. Cappellano, M. Ball, F. X. Francou, P. Normand, G. Helynck, A. Martinez, S. J. Kolvek, J. Hopke, M. S. Osburne, P. R. August, R. Nalin, M. Guerineau, P. Jeannin, P. Simonet and J. L. Pernodet, Appl. Environ. Microbiol., 2003, 69, 49 CrossRef CAS.
- G. Y. S. Wang, E. Graziani, B. Waters, W. B. Pan, X. Li, J. McDermott, G. Meurer, G. Saxena, R. J. Andersen and J. Davies, Org. Lett., 2000, 2, 2401 CrossRef CAS.
- E. Zazopoulos, K. X. Huang, A. Staffa, W. Liu, B. O. Bachmann, K. Nonaka, J. Ahlert, J. S. Thorson, B. Shen and C. M. Farnet, Nat. Biotechnol., 2003, 21, 187 CrossRef CAS.
- M. J. Garson, J. E. Thompson, R. M. Larsen, C. N. Battershill, P. T. Murphy and P. R. Berquist, Lipids, 1992, 27, 378 Search PubMed.
- M. J. Garson, M. P. Zimmerman, C. N. Battershill, J. L. Holden and P. T. Murphy, Lipids, 1994, 29, 509 Search PubMed.
- M. J. Uriz, M. A. Becerro, J. M. Tur and X. Turon, Mar. Biol., 1996, 124, 583 CAS.
- W. E. G. Muller, B. Diehl-Seifert, C. Sobel, A. Bechtold, Z. Kljajic and A. Dorn, J. Histochem. Cytochem., 1986, 34, 1687 Search PubMed.
- M. J. Uriz, X. Turon, J. Galera and J. M. Tur, Cell Tissue Res., 1996, 285, 519 CrossRef.
- M. J. Garson, A. E. Flowers, R. I. Webb, R. D. Charan and E. J. McCaffrey, Cell Tissue Res., 1998, 293, 365 CrossRef CAS.
- M. P. Lawson, I. L. Stoilov, J. E. Thompson and C. Djerassi, Lipids, 1988, 23, 750 Search PubMed.
- M. P. Lawson, J. E. Thompson and C. Djerassi, Lipids, 1988, 23, 741 Search PubMed.
- M. P. Zimmerman, F. C. Thomas, J. E. Thompson, C. Djerassi, H. Streiner, E. Evans and P. T. Murphy, Lipids, 1989, 24, 210 Search PubMed.
- J. A. Tincu, A. G. Craig and S. W. Taylor, Biochem. Biophys. Res. Commun., 2000, 270, 421 CrossRef CAS.
- P. O. Gallagher, C. S. P. McErlean, M. F. Jacobs, D. J. Watters and W. Kitching, Tetrahedron Lett., 2002, 43, 531 CrossRef CAS.
- S. W. Taylor, B. Kammerer and E. Bayer, Chem. Rev., 1997, 97, 333 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |