DOI:
10.1039/B310662B
(Perspective)
New J. Chem., 2004,
28, 1-10
Received
(in Montpellier, France)
1st September 2003
, Accepted 3rd November 2003
First published on 19th December 2003
Abstract
Within the context of sustainable technology, catalysis is a means to more efficient processes (lower energy demands, highly selective, waste-free), thereby providing a better use of raw materials. Industry relies heavily on heterogeneous catalysis to perform chemical transformations, but the development of these systems can be slowed down by the difficulty in understanding them, since they are often ill-defined and may contain several types of active sites (which can also be detrimental to selectivity). More recently, homogeneous catalysis has emerged, and it is probably because of a molecular understanding of chemical phenomena with well-defined systems that new processes have been rapidly set up. Our approach, called Surface Organometallic Chemistry (SOMC), has been to bring these two fields together: the result is a molecular approach to the design of heterogeneous catalysts. Within this short review, our strategy is delineated and the case of olefin metathesis is used to exemplify this approach. The design, the preparation and characterisation of a well-defined rhenacarbene are described. Based on a combined use of mass balance analysis, IR spectroscopy, advanced NMR techniques and EXAFS, it has been shown that [Re(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHtBu)(
CHtBu)(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)(CH2tBu)2] reacts with a partially dehydroxylated silica at 700
CtBu)(CH2tBu)2] reacts with a partially dehydroxylated silica at 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C to give a single surface complex: syn-[(SiO)Re(CHtBu)(CtBu)(CH2tBu)(SiOSi)]
(1). This surface complex 1 catalyses olefin metathesis at low temperatures and without co-catalyst. The structure and the activity of this type of catalyst are discussed and compared with other existing homogeneous and heterogeneous catalysis systems.
°C to give a single surface complex: syn-[(SiO)Re(CHtBu)(CtBu)(CH2tBu)(SiOSi)]
(1). This surface complex 1 catalyses olefin metathesis at low temperatures and without co-catalyst. The structure and the activity of this type of catalyst are discussed and compared with other existing homogeneous and heterogeneous catalysis systems.

Christophe Copéret
| Christophe Copéret received his degree of ‘Ingénieur’ in 1992 from Chimie Lyon, France (officially called CPE Lyon). A year before, he joined the PhD program of Purdue University, Indiana (USA) where he worked on the development of synthetic methodology through organometallic chemistry and catalysis under the direction of Prof. E.-i Negishi. In 1996, after obtaining his PhD degree, he joined the group of Prof. Sharpless at the Scripps Research Institute to work on selective oxidation reactions. He then accepted a full-time position at the CNRS (1998), where he is currently developing the surface organometallic chemistry of transition metals on metal oxides directed towards the generation of single-site heterogeneous catalysts. |
Introduction
Catalysis deals with the acceleration and the control of reaction rate and, in turn, allows the understanding and the development of efficient chemical processes that are low-energy, highly selective chemical transformations giving no by-products. This field of chemistry has been traditionally fragmented into three distinctive areas: enzymatic, homogeneous and heterogeneous catalysis. In term of industrial developments, it is often better—when possible—to develop heterogeneous catalysts, because they can be more readily transposed to continuous processes and provide an easy way to separate the products from the catalytic phase. Yet in the past 40 years, homogeneous catalysis has undergone a rapid development and led to several high tonnage chemical processes.1,2 One reason stems from the level of understanding of molecular chemistry, which can readily help to identify the key requirements for better catalytic systems (design of active sites through structure-activity relationship), hence their rapid development. In contrast, heterogeneous catalysts are often more ill-defined (unknown and numerous active sites), which can impede their selectivity and secondly hinder a rational improvement of such systems. Thus, with this in mind, the proposed strategy is to design the active sites of heterogeneous catalysts with a similar approach to that implemented in homogeneous catalysis in order to generate well-defined systems.
The first step is to start by analysing the desired chemical transformation, that is by looking at thermodynamic data and by postulating the chemical events (elementary steps) necessary to perform the desired reaction. Such an approach defines, ab initio, the postulated intermediates and will therefore condition what type of active site (or its precursor) should be generated. The second step consists in constructing the active sites through a careful understanding of the interaction of molecular complexes with a surface. This approach, termed Surface Organometallic Chemistry (SOMC), is targeted at obtaining heterogeneous catalysts with well-defined active sites.3–6 The third step entails the testing of these catalysts in order to gather data on their activities, selectivities and lifetimes, which will be used to decide upon a strategy to improve the catalytic system (new generation of catalysts in the development loop).
In the next section, each step of the process will be detailed and the following section will illustrate this approach through the development of catalysts for olefin metathesis.
General strategy to design and test a heterogeneous catalyst prepared via SOMC
Design of the targeted catalytic system
Chemical reactions rely on successive bond-breaking and bond-making processes, and over the years molecular chemistry has established most, if not all, the possible elementary steps. In homogeneous catalysis, the strategy is to prepare a well-defined stable intermediate, which often consists of a transition metal decorated with the proper ligands: one propagating ligand—the reactive bond (or in some cases just a vacant site)—and ancillary ligands, which will control the stability of the complex (life time) and confer the stereoelectronic properties of the reactive centre (control the activity and the selectivity). In surface organometallic chemistry, the surface will also play a role and must be therefore considered as a ligand (Scheme 1). Depending on the reaction and its corresponding catalytic cycle, it will be necessary to propose an adequate system based on the fundamental rules of organometallic chemistry. For instance, homogeneous catalysis has taught us that olefin polymerisation catalysts will require electrophilic metal-alkyl complexes,7 while olefin metathesis necessitates metal-alkylidene systems (vide infra). Conversely, oxidation catalysts will probably include oxo, peroxo, or alkylperoxy ligands8 while hydrido complexes can be useful in alkane chemistry.9–11
 |
| | Scheme 1 Molecular approach to the construction of active sites of heterogeneous catalysts. | |
Consequently, the design of a heterogeneous catalyst will be based on using the concepts and the level of understanding of molecular chemistry (identification of potential intermediates) and transferring them to the chemistry of surfaces in order to generate well-defined active sites dispersed on a support.
Preparation and characterisation of the catalyst via SOMC.6
After identifying the targeted metal and the desired coordination sphere, it is necessary to devise a strategy to implement them.
Firstly, it is essential to understand the surface structure of the support and how it will interact with a metal complex. Therefore, the solid has to be understood via the measurement of its surface area and morphology (non-porous, porous, mesoporous), but also by understanding its surface structure. The surface of oxide supports (MxOy) are typically composed of various types of (M–OH) and (M–O–M) units, which should be identified and quantified via spectroscopic or chemical methods (number of Lewis and Brønsted acidic sites).
Secondly, the interaction of a molecular complex with a surface needs to be investigated to understand the factors that influence the coordination sphere of the surface complex in order to generate a well-defined system. Therefore, it is necessary to have a “palette” of characterisation tools to probe the structure of the surface complex,12 its exact coordination sphere and the surroundings of the active centre (Scheme 2). These tools can include (1) chemical reactivity studies (including the grafting step) in combination with in situ IR spectroscopy to obtain information on the grafting stage and the reactivity of the surface complex towards further treatments, (2) solid state NMR spectroscopy to determine the nature of the ligands around the metal centre (and their dynamics), (3) EXAFS to further investigate the coordination sphere of the metal centre (distances and average coordination numbers), (4) ESR, UV and XANES to probe the oxidation state, the electronic state and the geometry of the surface metal complex. These techniques, together with the use of molecular models,13–16 molecular modelling and surface science techniques (BET, XPS, …) will provide a molecular understanding of the reaction of an organometallic precursor with a surface and help to establish the molecular structure of the surface complex.
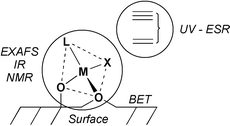 |
| | Scheme 2 General strategy to investigate the coordination sphere of a surface complex. | |
Catalyst testing and improvement of catalytic systems
After ascertaining the structural identity of the surface complex, its reactivity towards the targeted substrate is then investigated. It is typically performed by looking at the reaction conversion and selectivity in batch reactors at various temperatures. However, the use of continuous flow reactors is often highly desirable (especially in gas–solid reactions) to obtain better kinetic data (activation energies, selectivity through contact time studies, identification of primary products, deactivation kinetics, …). Catalytic testing in combination with an understanding of the fate of the catalyst through its characterisation at various stages of the catalytic test are very important to assess structure-activity relationships, which will allow a strategy for improvement/development of the catalysts to be proposed. Based on this approach, a new generation of catalysts can be proposed. This is how the loop of improvement proceeds, as in homogeneous catalysis, the ultimate goal being a viable chemical process (Scheme 3).
 |
| | Scheme 3 Strategy of development of heterogeneous catalysts via Surface Organometallic Chemistry. | |
Preparation of well-defined heterogeneous catalysts for olefin metathesis via SOMC
Background, strategy and design
The olefin metathesis reaction was incidentally discovered using heterogeneous catalysts while studying the reactivity of propene–alkane mixtures over Mo(CO)6 supported on alumina.17 Under these conditions, propene was transformed into a mixture of ethylene and butenes, a disproportionation reaction later named olefin metathesis. The current industrial catalysts typically used are MoO3 or WO3 supported on silica or alumina, which require high reaction temperatures (Scheme 4).18 Despite the discovery by BP a few years later of a catalyst, Re2O7/Al2O3, that works at much lower temperatures (typically 35°C),19–21 it is still not used today in a large scale production process.22 It is noteworthy that this latter system can also be used to transform functionalised olefins when activated by organotin agents.23–25 Despite years of research, the exact nature of the active sites or the initiation steps in these types of heterogeneous catalysts are still unknown.20,21 This had, in fact, led to important discussions in the seventies to decide what was the most likely reaction mechanism to explain olefin metathesis. Today, it is widely accepted that metallocarbenes (and metallacyclobutanes) are the key intermediates [eqn. (1)], as proposed by Herisson and Chauvin of the IFP in 1971.26| |  | (1) |
This proposition directed the search for stable metallocarbene complexes, which could be used in homogeneous catalysis.27,28 After about 30 years of investigation, two main classes of well-defined homogeneous catalysts have emerged: the d0 early transition metals (groups 6 and 7)29–35 and the d4 Ru systems (Scheme 4).36–39 The latter system has changed the field of olefin metathesis by providing easily handled catalysts compatible with most functional groups used in organic synthesis. Noteworthy are the requirements for basic ligands to obtain a good activity, which is exactly the contrary to what has been found for d0 systems, which are all based on generating highly electrophilic systems.30 For well-defined homogeneous catalysts, the initiation step has been established (cross-metathesis), and their stability allows TON of more than ½ million to be reached in some cases.40 Moreover, while the de-activation pathways have been understood to a good extent,41 they have not yet been solved and regeneration is a problem.
 |
| | Scheme 4 State of the art in heterogeneous and homogeneous catalysis applied to olefin metathesis. | |
In contrast, heterogeneous catalysts are not as well understood at the molecular level, but they are readily regenerated under calcination and constitute the main systems used by the chemical industry. Re2O7/Al2O3 is still a promising system, but the low concentration of active sites of unknown nature has slowed down the search for improved catalysts. Several homogeneous models have been prepared (Scheme 5) and showed very good activity, but are equally difficult to understand (initiation-deactivation).42
 |
| | Scheme 5 Homogeneous model and its heterogeneous catalyst equivalent. | |
One question arises: is it therefore possible to generate a well-defined rhenacarbene at the surface of a support? This is a question that falls into the area of surface organometallic chemistry.4,43–45
Choice of support
In order to have a better understanding of the system, the first step is to select a support. The first criterion is to work with a support that does not generate parallel reactions (neutral) and the second one is to have a reasonable knowledge of its surface structure in order to understand its reactivity with an organometallic complex. Silica is a good candidate, since it is available as regular 50 nm spheres, free of metal and carbon traces and with a high surface area (200 m2 g−1). It is a neutral support having a surface covered with siloxane bridges (of various sizes) and silanols (of various types: isolated, vicinal and gem), of which the concentrations can be controlled by thermal pre-treatment.46–48 Silanols are orders of magnitude more reactive than siloxane bridges, and will be used to graft the metal via a (Si–O–M) bond. If a thermal treatment at high temperatures (above 600–700°C) under vacuum is applied, the surface is mainly composed of isolated silanols (0.7±10% OH nm−2) besides siloxane bridges, while at lower temperatures the concentration in silanols is higher (1.4±10% OH nm−2 at 500°C and 3.5±10% OH nm−2 at 200°C) and favours the presence of vicinal and gem silanols. The use of higher temperatures will lower the concentration of surface silanols, but also favours the formation of strained siloxane bridges, which can become reactive. Based on these observations, it is possible to generate selectively mono-grafted complexes by using a silica treated at 500°C and higher (typically 700°C), while the use of dehydroxylation temperatures in the 200–300°C range provides an access to bis-grafted complexes (Scheme 6).49–51
 |
| | Scheme 6 Effect of the temperature of partial dehydroxylation of silica (SiO2-(T); T=temperature of dehydroxylation) on the resulting surface complex structure: the tuneable hapticity of silica. | |
Choice of molecular precursor
The goal is to generate a rhenacarbene on the surface of silica, and one possible route is to use a molecular complex containing both a carbene ligand and a reactive bond, a metal-alkyl for example, which will react with the surface silanols and thus generate a covalent bond between the support and the metal centre. [Re(CtBu)(CHtBu)(CH2tBu)2],52,53 a well-defined molecular complex, has been reported and fulfills the aforementioned requirements. Its reactivity with silica and the methodology applied for the characterisation of the corresponding surface complex will be detailed hereafter.
Preparation and characterisation (Surface Organometallic Chemistry)
Firstly, in situ IR spectroscopy experiments show that there is a chemical grafting of the complex since the surface silanols are consumed during the sublimation step. Secondly, mass balance analysis shows that about 1.06±0.13 equiv. of CH3tBu evolved during grafting and that the corresponding solid contains 15.5±0.8 carbons per grafted Re. Furthermore, 3.9–4.7%wt of Re is typically grafted on silica partially dehydroxylated at 700°C, which corresponds to the consumption of 0.21–0.26 mmol g−1 of silanol groups (0.63–0.78 OH nm−2) and thus to the amount of silanols accessible for bulky complexes on a silica partially dehydroxylated at 700°C (vide supra).54 Therefore, the data are consistent with the loss of approximately one equivalent of neopentane during the grafting, leaving on average three “neopentyl-like” ligands around the metal centre (Scheme 7).
 |
| | Scheme 7 Initial proposed structure from mass balance analysis (elemental analysis and gas evolution during grafting) and IR spectroscopic data. | |
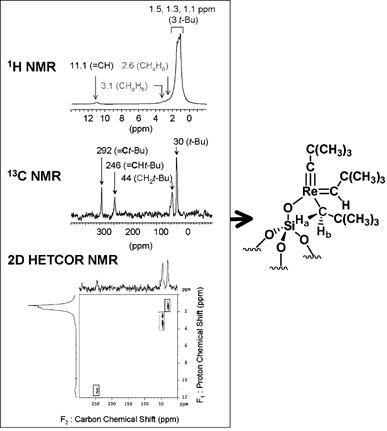
Further investigation of the structure was undertaken through the use of 1D and 2D solid state NMR spectroscopy.54–56 While the 1H solid state NMR spectrum of 1 displays signals at 11.0, 3.0, 2.6, 1.5, 1.3 and 1.1 ppm (Scheme 8), which are assigned to the carbene proton (CH), the two diastereotopic protons (CHaHb) and the methyl groups of the three different tBu groups, the 13C CP/MAS spectrum of the natural abundance sample gives little information. On the other hand, applying H-decoupled 13C solid state NMR on a 10%
13C-enriched sample provided evidence for the presence of three types of hydrocarbyl ligands around Re: a neopentyl (46 ppm, CH2tBu), a neopentylidene (246 ppm, CHtBu), and a neopentylidyne (292 ppm, CtBu).54 Moreover, 2D HETCOR (heteronuclear correlation) solid state NMR using magic angle spinning (MAS) clearly allowed these assignments to be confirmed.56 Firstly, by using short contact times during the pulse sequence, it is possible to selectively detect the protons directly linked to a carbon. For example, the two signals at 2.6 and 3.0 ppm attributed to the two diastereotopic protons are indeed borne by the same carbon, while that at 11 ppm in the H dimension correlates with that at 246 ppm in the C dimension, confirming their assignments to the carbenic fragment. Using longer contact times (>1 ms), it is possible to observe extra correlation peaks, which arise from longer range dipolar through-space interactions, and which allow the full 13C NMR spectrum of 1
(including all quarternary carbons) to be reconstituted. Combining the information given by 1D and 2D solid state NMR spectroscopy provides a relatively complete NMR data set for the surface compound, which is fully consistent and indicates that the reaction of [Re(CtBu)(CHtBu)(CH2tBu)2] with SiO2−(700) leads to the formation of [(SiO)–Re(CtBu)(CHtBu)(CH2tBu)], 1, as the sole surface species.
As in solution chemistry, one would like to have more information on the coordination sphere of the metal, which is typically obtained by using X-ray crystallography. Silica is amorphous, but EXAFS can nonetheless allow the coordination sphere of surface complexes to be probed and identifies the first neighbours of the heavy element and their distances. EXAFS data from the Re–surface complex (Scheme 9) agree with what has been proposed from the NMR data, that is two carbons at relatively short distances (not resolved at 1.79 Å), corresponding to the (CHtBu) and (CtBu) ligands, another carbon and an oxygen atom at larger distances (not resolved at 2.01 Å), corresponding to the (CH2tBu) and (SiO) ligands. However, EXAFS data point to the presence of an extra ligand, an oxygen at 2.42 Å, probably arising from a dative bond from a siloxane bridge to the rhenium centre. Furthermore, in solution chemistry, it has been shown that measuring J(C–H) coupling constants can provide further insight into the way perhydrocarbyl ligands interact with the metal since they are directly correlated with the hybridisation of the carbon atoms.57 Scalar information are difficult to obtain in solid state NMR since dipolar interactions overshadow scalar information, nonetheless it is possible to extract this through J-resolved 2D NMR spectroscopy.58 Therefore, the corresponding spectrum presents the following features (Scheme 9): a quadruplet [q, JC–H=126 Hz, 29 ppm, CH3
(tBu)], a triplet (t, JC–H=126 Hz, 46 ppm, CH2tBu), a doublet (d, JC–H=110 Hz, 246 ppm, CHtBu) and a singlet (s, 292 ppm, CtBu). While all the splitting patterns and most of the JC–H values correspond to what is expected for each type of coupling system, the JC–H value of the doublet is quite low for an sp2 carbon, indicating that this C–H bond is probably elongated and that the (Re–C–H) bond angle is distorted from the typical 120° value.59,60 In fact, the JC–H value has been correlated to the bond angle and in this case would correspond to a bond angle on the order of 85°
(Scheme 9). Such a type of interaction points towards a syn configuration of the carbene ligands (low coupling constant)53,61,62 and the presence of an extra ligand (pentacoordinated complex).63
In conclusion, by assembling all the data, it is possible to have a molecular view of surface complex as in homogeneous catalysis.
Comparison with molecular organometallic chemistry: the use of molecular models
Another important tool to obtain further insight into surface organometallic chemistry is the use of soluble molecular models of silica, like alkyl silanols or polyoligomeric silsesquisioxanes (POSS).13–16 Upon reaction of the molecular complex with either [Ph3SiO–H] or [SiPOSSO–H] in benzene solution (Scheme 10) the corresponding monosiloxy complexes [Ph3SiO–Re(CtBu)(CHtBu)(CH2tBu)] and [SiPOSSO–Re(CtBu)(CHtBu)(CH2tBu)] are formed, like those observed for the silica surface.54,56 Both of these products are, however, obtained as a 10-to-1 mixture of the syn and the anti rotamers, contrary to the surface complex, which is obtained stereoselectively as the syn rotamer according to the NMR data. Note that the chemical shift data for the syn rotamers of the molecular complex exactly match those obtained for the surface complex, while the anti rotamers have quite different chemical shifts.53 Rotational isomers (rotamers) are common in the organometallic chemistry of alkylidene complexes. Some alkylidene organometallic syntheses yield only one rotamer (usually the syn one), which can be generally converted into a mixture of the syn and the anti rotamers either thermally or photochemically. Upon heating the surface complex syn-1 at 120°C under Ar for 30 min, new sets of NMR signals in the 1H and 13C NMR spectra appear, which correspond to those of the anti rotamers of the molecular complex. These assignments were further confirmed by 2D HETCOR and J-resolved solid state NMR spectroscopies. The syn/anti ratio is approximately 2.0±0.5, which corresponds to a ΔG° of about 2±1 kJ mol−1 at 120°C. This ratio does not evolve upon cooling to room temperature. Similarly, upon exposure of the syn complex to daylight for a couple of days, a syn-anti isomerisation is also observed (syn/anti=1.5). As in solution chemistry, carbene on surfaces can be present as syn and anti rotamers. Yet, upon grafting, the syn isomer is the sole isomer obtained, which shows that the grafting step is stereospecific.
 |
| | Scheme 10 Comparison between molecular and surface complex equivalents. | |
Understanding of the grafting mechanism.56
Based on these data, the grafting was further investigated in order to understand the origin of the stereospecificity of grafting. First, grafting of [Re(CtBu)(CHtBu)(CH2tBu)2], specifically 13C-enriched at the neopentyl and neopentylidene carbons, onto SiO2-(700) provided a sample that displayed no carbynic resonance in both the CP/MAS and direct excitation 13C solid state NMR spectra. The absence of scrambling of the 13C-label into the carbynic position shows that the neopentylidyne is a spectator ligand during grafting.64 Therefore, the silanols must react either with the neopentylidene and/or the neopentyls, which can readily be probed by using deuterated silica (prepared via exchange of the surface silanols with D2O). The grafting onto deuterated SiO2-(700) yielded undeuterated and monodeuterated neopentane, whose isotopic composition was consistent with a grafting mechanism that took place via direct electrophilic cleavage (61±10%) and a two-step mechanism (39±10%) involving addition followed by α-H abstraction (Scheme 11). Since the molecular precursor contains two alkyl ligands for one alkylidene ligand, the reactivity towards silanols of the alkyl and alkylidene moieties are in fact similar. In solution chemistry, the reaction of [Re(CtBu)(CHtBu)(CH2tBu)2] with Brønsted acids (HX, X=Cl, OC6F5, BF4, OTf, …) gives the products resulting exclusively from protonation onto the alkylidene moiety, [Re(CtBu)(CH2tBu)3X],53 which can be isolated in fairly good yields (60–80%), and their transformation into the corresponding alkylidene complexes only takes place upon the addition of donor ligands such as pyridine. By comparison, its reaction with silica directly gives [(SiO)–Re(CtBu)(CHtBu)(CH2tBu)(SiOSi)]
(1), which shows that the surface plays a role. The presence of a siloxane bridge (donor ligand) in 1 as observed by EXAFS suggests that the surface induces the spontaneous extrusion of the neopentyl ligand in [(SiO)–Re(CtBu)(CH2tBu)3] to form 1.
![Reaction of [Re(CtBu)(CHtBu)(CH2tBu)2] with the silica surface: mechanisms of grafting.](/image/article/2004/NJ/b310662b/b310662b-s11.gif) |
| | Scheme 11 Reaction of [Re(CtBu)(CHtBu)(CH2tBu)2] with the silica surface: mechanisms of grafting. | |
Catalytic reactivity
Preliminary catalyst testing.
After preparing and characterising a well-defined rhenacarbene on silica, the next step has been to evaluate its reactivity towards olefins. This is typically carried out by contacting propene onto the catalyst in a batch reactor at 25°C. Using 0.2% of catalyst, the thermodynamic equilibrium (30% conv.) is reached within 50–60 min, with an initial rate on the order of 0.25 mol mol−1 s−1.54,65 Note that the molecular precursor is not active in olefin metathesis, which shows the dramatic effect of the siloxy substituent, probably via an increase of electrophilicity of the metal centre.3 A 3∶1 E∶Z ratio of butene stereoisomers (thermodynamic mixture) is obtained at the end of the reaction, while it is slightly lower (2.5∶1) at the very beginning of the reaction, which probably indicates the intrinsic selectivity of the catalyst (Scheme 12). Such a selectivity—in favour of the E olefin—is expected at zero conversion for the metathesis of terminal olefins like propene since the stereochemical outcome of the reaction is usually related to the relative stability of the metallacyclobutane intermediate, that is by minimizing [1,2] interactions. Therefore, formation of E-2-butene through an intermediate having two substituents in equatorial arrangements is favoured over Z-2-butene, which involves an intermediate having the substituents in both equatorial and axial arrangements.66 Beside the self-metathesis products of propene, 0.7 equiv of neopentyl-containing products are also detected, namely 3,3-dimethylbutene and 4,4-dimethyl-2-pentene as a 3∶1 mixture. They correspond to cross-metathesis products and therefore identify the initiation step (Scheme 13). The relative ratio in cross-metathesis products is also explained by the relative stabilities of the metallacyclobutane intermediates, for which [1,2] interactions are avoided and [1,3] interactions are minimised.66 Note that, in contrast, the initiation step is still not fully understood for classical heterogeneous catalysts such as Re2O7/Al2O3.
![Initial stereoselectivity in propene metathesis (minimisation of [1,2] interactions).](/image/article/2004/NJ/b310662b/b310662b-s12.gif) |
| | Scheme 12 Initial stereoselectivity in propene metathesis (minimisation of [1,2] interactions). | |
 |
| | Scheme 13 Initiation step with a well-defined metallocarbene. | |
Similarly, Z-3-heptene is converted efficiently into an 1∶1 mixture of 3-hexenes and 4-octenes with an initial rate of 0.7 mol (mol Re)−1 s−1.65 The initial ratios for 3-hexenes and 4-octenes are 3∶4 and 6∶10. They are constant up to 45% conversion and then reach 7∶1 (thermodynamic equilibrium) as conversion proceeds. In the case of internal olefins, the partial retention of configuration depends on the minimisation of [1,3] interactions in the metallacyclobutane intermediates (Scheme 14).66 Furthermore, these constant E∶Z ratios show that 3-hexenes and 4-octenes are primary products.
The metathesis of bulky olefins is also possible with this system. For example, isobutene is converted into 2-3-dimethyl-2-butene and propene–isobutene mixtures lead to 2-methyl-2-pentene, besides the self-metathesis product of propene. Finally, the reactivity of 1 towards functionalised olefins is also noteworthy, and currently up to 900 TON can be reached in the metathesis of methyl oleate.
![Initial stereoselectivity for the metathesis of Z-olefins (minimisation of [1,3] interactions).](/image/article/2004/NJ/b310662b/b310662b-s14.gif) |
| | Scheme 14 Initial stereoselectivity for the metathesis of Z-olefins (minimisation of [1,3] interactions). | |
Comparison with other systems.
First of all, by using SOMC, well-defined group 5 and 6 metallocarbenes and carbynes have also been prepared and fully characterised using the same rigorous methodology (Scheme 15).55,67,68 The activities (initial rates in mol mol−1 s−1) in propene metathesis under identical experimental conditions (Table 1) are as follows: Mo (0.53)∼Mo=NH (0.47)>Re (0.25)∼W (0.23)≫Ta (inactive). Moreover, in the case of the metathesis of methyl oleate, the following order of stability towards the ester functional group has been observed: Re∼W>Mo.69 Both of these results parallel quite well what has been observed for related homogeneous systems.
 |
| | Scheme 15 First generation of olefin metathesis catalysts prepared via SOMC. | |
Table 1 Initial rates for the metathesis of propene with various heterogeneous catalysts
| Catalyst |
TOFa |
R
b
|
|
Initial or steady state rates in mol mol−1 s−1 at the temperature indicated in parentheses.
Substrate-to-catalyst ratio, for which the reaction reaches at least the thermodynamic equilibrium.
Not readily calculated from the published data.
|
| [SiO–Ta(CHtBu)(CH2tBu)2] |
Inactive(25) |
103 |
| [SiO–Mo(CtBu)(CH2tBu)2] |
0.53(25) |
500 |
| [SiO–Mo(NH)(CHtBu)(CH2tBu)] |
0.47(25) |
870 |
| [SiO–W(CtBu)(CH2tBu)2] |
0.23(25) |
1700 |
| [SiO–Re(CtBu)(CHtBu)(CH2tBu)] |
0.25(25) |
500 |
| MoO3/SiO2 |
0.004(400) |
–c |
| WO3/SiO2 |
–(400)c |
–c |
| Re2O7/SiO2 |
<0.002(150) |
–c |
| [Re(CO)3OH]4/SiO2 |
7,4.10−4(150) |
–c |
| MoO3/Al2O3 |
0.05–0.5(200) |
–c |
| WO3/Al2O3 |
0.1–1.0(300) |
–c |
| Re2O7/Al2O3 |
0.05–0.1(35) |
–c |
Secondly, this catalyst, 1, is as active and more robust than Re-based homogeneous systems. For example, [SiPOSSO–Re(CtBu)(CHtBu)(CH2tBu)] decomposes rapidly in the presence of olefins while [{RF6O}2Re(CHtBu)(CtBu)] reaches a TON of 25 in the metathesis of methyl oleate.70 On the other hand, it is still far from the current best homogeneous systems such as [{RO}2Mo(CHtBu)(NAr)], which reaches initial rates of 2–8 mol mol−1 s−1 and TON on the order of 10000 (terminal and internal olefins)71,72 or [Cl2Ru(CHR)(PCy3)L], with TON exceeding ½ million for various substrates (diallylmalonate, octene and methyl oleate), depending on the substituent on the heterocyclic carbene ligand L.40
Finally, classical heterogeneous catalysts are comparatively less active, even when using high temperature conditions (Table 1). Moreover, highly branched or functionalised olefins are still a problem and, for example, the metathesis of methyl oleate is typically in the 25–160 TON range, after activation by tin reagents, depending on the support.25 In the case of the surface complex 1, cross-metathesis is clearly the initiation step and therefore its behaviour is identical to that of well-defined homogeneous catalysts. It is, therefore, easier to understand this system than classical heterogeneous catalysts such as the Re2O7/Al2O3 system, in which less than 2% of the Re is active.73,74 A new generation of catalysts has appeared, such as methyl trioxorhenium supported on silica–alumina, which are compatible with a variety of functional groups, but again the nature of the active sites is still ill-defined.75–77 Note also that well-defined silica-supported systems, such as [SiO–ReO3], are nearly inactive.78–81 As shown with 1, for which the carbene is already set, silica is not a problem per se in terms of activity in olefin metathesis, however, the formation of the necessary carbene intermediate from oxo ligands is probably the most difficult step for [SiO–ReO3]
(Scheme 16). In contrast, Re oxo species on alumina are readily converted into carbene intermediates, but the initiation process has yet to be clarified.18,82–84 It is fair to say that catalysts prepared via surface organometallic chemistry provide a molecular insight into heterogeneous catalysts, which is a new way to think about these systems. Further developments in olefin metathesis catalysed by heterogeneous catalysts are currently in progress.
 |
| | Scheme 16 Comparison of Re-based heterogeneous catalysts. | |
Future and outlook
In conclusion, these types of heterogeneous catalysts display a level of molecular understanding and activities unprecedented in the field of heterogeneous catalysis. The active site is well-defined, the initiation step understood and the activity quite high (initial rates and TON). Preliminary results are encouraging and emphasise a further need to understand such types of systems. Several questions already arise: what is the fate of the catalysts? Does it deactivate and how? Can we understand its pathway of deactivation and prevent it? Is it possible to regenerate such types of catalysts? Through these studies, it should be possible to transfer the knowledge accumulated on this “model” system to more complex systems used in industry and to potentially give answers to unsolved problems, which have typically been difficult to address due to the complexity of classical heterogeneous catalysts (low number and numerous types of active sites). We are currently developing methodologies to test these catalysts under industrially relevant conditions and also to probe the structure of the catalysts under working conditions.85 This obviously calls for the development of better spectroscopic tools (in situ techniques), the large scale production of these catalysts and a better understanding of the reactivity of catalysts through the combined use of experimental evidence and molecular modelling. The gap between homogeneous catalysis and heterogeneous catalysis is closing, but there are still many open questions, which should be solved in the near future.
Acknowledgements
This work is dedicated to three mentors for their training and inspiration: J.-M. Basset, E. Negishi and K. B. Sharpless. This project has been conducted with talented younger co-workers, more directly M. Chabanas and A.-M. Leduc, whom I deeply thank for their contributions. Other co-workers not directly involved in this research should also be acknowledged; their names are cited within the references and my web page: www.cpe.fr/lcomsnew/coperet. My deepest thanks are directed to S. Hediger, A. Lesage, W. Lukens, Prof. L. Emsley and my LCOMS colleagues; they have been essential to the success of this project. Prof. R. A. Andersen and Dr. Y. Chauvin should also be acknowledged for numerous and insightful discussions. I would also like to thank Prof. A. K. Smith for polishing the manuscript. I am also grateful to the CNRS, the ESCPE Lyon, the French Ministry of Research and Education, the Rhône-Alpes district and BASF AG for financial support.
References
-
Applied Homogeneous Catalysis with Organometallic Compounds, eds. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim and New York, 2nd edn., 2002, vol. 1, pp. 598 Search PubMed.
-
Applied Homogeneous Catalysis with Organometallic Compounds, eds. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim and New York, 2nd edn., 2002, vol. 2, pp. 432 Search PubMed.
- D. G. H. Ballard, Adv. Catal., 1973, 23, 263 CAS.
- Y. I. Yermakov, B. N. Kuznetsov and V. A. Zakharov, Stud. Surf. Sci. Catal., 1981, 8, 522.
-
Surface Organometallic Chemistry: Molecular Approaches to Surface Catalysis, eds. J. M. Basset, B. C. Gates, J. P. Candy, A. Choplin, M. Leconte, F. Quignard, and C. Santini, NATO ASI Series, 1988, vol. 231, pp. 330 Search PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef CAS.
- M. Jezequel, V. Dufaud, M. J. Ruiz-Garcia, F. Carrillo-Hermosilla, U. Neugebauer, G. P. Niccolai, F. Lefebvre, F. Bayard, J. Corker, S. Fiddy, J. Evans, J.-P. Broyer, J. Malinge and J.-M. Basset, J. Am. Chem. Soc., 2001, 123, 3520 CrossRef CAS.
- D. Meunier, A. Piechaczyk, A. De Mallmann and J.-M. Basset, Angew. Chem., Int. Ed., 1999, 38, 3540 CrossRef CAS.
- C. Lécuyer, F. Quignard, A. Choplin, D. Olivier and J. M. Basset, Angew. Chem., Int. Ed. Engl., 1991, 30, 1660 CrossRef.
- V. Vidal, A. Theolier, J. Thivolle-Cazat and J.-M. Basset, Science, 1997, 276, 99 CrossRef CAS.
- V. Dufaud and J. -M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806 CrossRef CAS.
-
Handbook of Heterogeneous Catalysis, eds. G. Ertl and H. Knoezinger, Wiley-VCH, Weinheim, 1997, pp. 2800 Search PubMed.
- R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Chem. Rev., 1996, 96, 2205 CrossRef CAS.
- H. C. L. Abbenhuis, Chem. Eur. J., 2000, 6, 25 CrossRef CAS.
- B. Marciniec and H. Maciejewski, Coord. Chem. Rev., 2001, 223, 301 CrossRef CAS.
- R. Duchateau, Chem. Rev., 2002, 102, 3525 CrossRef CAS.
- R. L. Banks and G. C. Bailey, Ind. Eng. Chem. Prod. Res. Develop., 1964, 3, 170 Search PubMed.
-
Olefin Metathesis and Metathesis Polymerization, eds. K. J. Ivin and J. C. Mol, 2nd edn., 1996, pp. 496 Search PubMed.
-
E. J. Howman and L. Turner, Preparation of Rhenium Oxide-Alumina Catalyst for Disproportionation of Alkenes, Nl 6605328, 1966 Search PubMed.
- J. C. Mol, Catal. Today, 1999, 51, 289 CrossRef CAS.
- J. C. Mol, Catal. Today, 1999, 52, 377 CrossRef CAS.
- J. Cosyns, J. Chodorge, D. Commereuc and B. Torck, Hydrocarbon Process., Int. Ed., 1998, 77, 61 Search PubMed.
- E. Verkuijlen, F. Kapteijn, J. C. Mol and C. Boelhouwer, J. Chem. Soc., Chem. Commun., 1977, 198 RSC.
- M. Sibeijn and J. C. Mol, Appl. Catal., 1991, 67, 279 Search PubMed.
- J. C. Mol, Green Chem., 2002, 4, 5 RSC.
- J. L. Herisson and Y. Chauvin, Makromol. Chem., 1971, 141, 161 CrossRef CAS.
- R. R. Schrock, J. Chem. Soc., Dalton Trans., 2001, 2541 RSC.
- R. R. Schrock, Chem. Rev., 2002, 102, 145 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1986, 19, 342 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1990, 23, 158 CrossRef CAS.
- R. R. Schrock and J. Feldman, Prog. Inorg. Chem., 1991, 39, 1.
- J. L. Couturier, C. Paillet, M. Leconte, J. M. Basset and K. Weiss, Angew. Chem., 1992, 104, 622 CAS.
- R. R. Schrock, Polyhedron, 1995, 14, 3177 CrossRef CAS.
- R. R. Schrock, Top. Organomet. Chem., 1998, 1, 1 Search PubMed.
- A. H. Hoveyda and R. R. Schrock, Chem. Eur. J., 2001, 7, 945 CrossRef CAS.
- S. T. Nguyen, L. K. Johnson, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1992, 114, 3974 CrossRef CAS.
- S. T. Nguyen, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1993, 115, 9858 CrossRef CAS.
- T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, Angew. Chem., Int. Ed., 1998, 37, 2490 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS.
- M. B. Dinger and J. C. Mol, Adv. Synth. Catal., 2002, 344, 671 CrossRef CAS.
- W. C. P. Tsang, K. C. Hultzsch, J. B. Alexander, P. J. Bonitatebus, Jr., R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 2652 CrossRef CAS.
- D. Commereuc, J. Chem. Soc., Chem. Commun., 1995, 791 RSC.
- K. Weiss and G. Loessel, Angew. Chem., Int. Ed. Engl., 1989, 28, 62 CrossRef.
- R. Buffon, M. Leconte, A. Choplin and J. M. Basset, J. Chem. Soc., Chem. Commun., 1993, 361 RSC.
- R. Buffon, M. Leconte, A. Choplin and J.-M. Basset, J. Chem. Soc., Dalton Trans., 1994, 1723 RSC.
- B. A. Morrow, Stud. Surf. Sci. Catal., 1990, 57, A161 CAS.
- M. E. Bartram, T. A. Michalske and J. W. Rogers, Jr., J. Phys. Chem., 1991, 95, 4453 CrossRef CAS.
-
B. A. Morrow and I. D. Gay, in Adsorption of Silica Surfaces, ed. E. Papirer, Surfactant Science Series, Dekker, 2000, vol. 90, p. 9 Search PubMed.
- J. A. N. Ajjou and S. L. Scott, Organometallics, 1997, 16, 86 CrossRef CAS.
- J. A. N. Ajjou, G. L. Rice and S. L. Scott, J. Am. Chem. Soc., 1998, 120, 13436 CrossRef.
- L. Lefort, M. Chabanas, O. Maury, D. Meunier, C. Copéret, J. Thivolle-Cazat and J.-M. Basset, J. Organomet. Chem., 2000, 96, 593.
- D. S. Edwards, L. V. Biondi, J. W. Zillen, M. R. Churchill and R. R. Schrock, Organometallics, 1983, 2, 1505 CrossRef CAS.
- R. Toreki, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1992, 114, 3367 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Copéret and J.-M. Basset, J. Am. Chem. Soc., 2001, 123, 2062 CrossRef CAS.
- R. Petroff Saint-Arroman, M. Chabanas, A. Baudouin, C. Copéret, J.-M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2001, 123, 3820 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Copéret, J.-M. Basset, W. Lukens, A. Lesage, S. Hediger and L. Emsley, J. Am. Chem. Soc., 2003, 125, 492 CrossRef CAS.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS.
- A. Lesage, L. Emsley, M. Chabanas, C. Copéret and J.-M. Basset, Angew. Chem., Int. Ed., 2002, 41, 4535 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1979, 12, 98 CrossRef CAS.
-
Metal-Ligand Multiple Bonds, ed. W. A. Nugent and J. M. Mayer, Wiley, New York, 1988, pp. 134 Search PubMed.
- J. H. Oskam and R. R. Schrock, J. Am. Chem. Soc., 1993, 115, 11831 CAS.
- A. M. LaPointe and R. R. Schrock, Organometallics, 1995, 14, 1875 CrossRef CAS.
- R. R. Schrock, W. E. Crowe, G. C. Bazan, M. DiMare, M. B. O'Regan and M. H. Schofield, Organometallics, 1991, 10, 1832 CrossRef CAS.
- R. Toreki, R. R. Schrock and M. G. Vale, J. Am. Chem. Soc., 1991, 113, 3610 CrossRef CAS.
- M. Chabanas, C. Copéret and J.-M. Basset, Chem. Eur. J., 2003, 9, 971 CrossRef CAS.
- J. L. Bilhou, J. M. Basset, R. Mutin and W. F. Graydon, J. Am. Chem. Soc., 1977, 99, 4083 CrossRef CAS.
- W. A. Herrmann, A. W. Stumpt, T. Priermeier, S. Bogdanovic, V. Dufaud and J.-M. Basset, Angew. Chem. Int. Ed. Engl., 1996, 35, 2803 CrossRef CAS.
- M. Chabanas, E. A. Quadrelli, B. Fenet, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage and L. Emsley, Angew. Chem., Int. Ed., 2001, 40, 4493 CrossRef CAS.
-
M. Chabanas, PhD thesis, Université Claude Bernard, Lyon, 2001.
- R. Toreki, G. A. Vaughan, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1993, 115, 127 CrossRef CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875 CrossRef CAS.
- F. J. Feher and T. L. Tajima, J. Am. Chem. Soc., 1994, 116, 2145 CrossRef CAS.
- F. Kapteijn, H. L. G. Bredt, E. Homburg and J. C. Mol, Ind. Eng. Chem., Prod. Res. Dev., 1981, 20, 457 Search PubMed.
- Y. Chauvin and D. Commereuc, J. Chem. Soc., Chem. Commun., 1992, 462 RSC.
- W. A. Herrmann, W. Wagner, U. N. Flessner, U. Vokhardt and H. Komber, Angew. Chem., Int. Ed. Engl., 1991, 30, 1636 CrossRef.
- R. Buffon, A. Choplin, M. Leconte, J. M. Basset, R. Touroude and W. A. Herrmann, J. Mol. Catal., 1992, 72, L7 CrossRef CAS.
- W. A. Herrmann and F. E. Kuehn, Acc. Chem. Res., 1997, 30, 169 CrossRef CAS.
- A. W. Aldag, C. J. Lin and A. Clark, Recl. Trav. Chim. Pays-Bas, 1977, 96, 27 CAS.
- N. Tsuda and A. Fujimori, J. Catal., 1981, 69, 410 CAS.
- L. G. Duquette, R. C. Cieslinski, C. W. Jung and P. E. Garrou, J. Catal., 1984, 90, 362 CrossRef CAS.
- P. S. Kirlin and B. C. Gates, J. Chem. Soc., Chem. Commun., 1985, 277 RSC.
- X. Zhang and P. Chen, Chem. Eur. J., 2003, 9, 1852 CrossRef CAS.
- X. Chen, X. Zhang and P. Chen, Angew. Chem., Int. Ed., 2003, 42, 3798 CrossRef CAS.
- A. M. Santos, C. C. Romao and F. E. Kuehn, J. Am. Chem. Soc., 2003, 125, 2414 CrossRef CAS.
-
A.-M. Leduc, work in progress.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.