Element and molecular mass spectrometry—an emerging analytical dream team in the life sciences
Mathias Wind† and Wolf D. Lehmann*
Central Spectroscopy, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, 69120 Heidelberg, Germany. E-mail: wolf.lehmann@dkfz.de; Fax: 0049 - 6221 - 424554; Tel: 0049 - 6221 - 424563
First published on 10th December 2003
Abstract
The recent literature on the combined applications of element (ICP, inductively coupled plasma) mass spectrometry and molecular (ESI, electrospray ionisation and MALDI, matrix assisted laser desorption ionisation) mass spectrometry in the life sciences is reviewed. Emphasis is put on investigations where both techniques have been used in a synergistic way with the aim of an analytical result with molecular specificity. Multi-element specificity and quantification are the key features of element MS, whereas molecular weight determination and structural information are those contributed by ESI and MALDI MS. The analytical applications cover the study of selenoproteins, phosphoproteins, metalloproteins, unmodified proteins, metallothioneins, phytochelatins, nucleic acids, arsenosugars, thyroid hormones, cobalamines, drugs and drug metabolites. Supported by on-line or off-line coupling to chromatographic or electrophoretic separation methods, the combined application of element and molecular mass spectrometry in all these studies has created the basis for new structural and/or quantitative insights, demonstrating the analytical excellence of this approach.
Introduction
Inductively coupled plasma mass spectrometry (ICP-MS), also termed element MS, is an established tool for quantitative element trace analysis and for speciation of elements in biological samples. In the last decade, innovative applications of ICP-MS coupled to chromatography are accumulating in life sciences projects where this technique is applied in combination with molecular MS techniques (ESI and MALDI-MS). This emerging combination of element and molecular mass spectrometry opens new possibilities for the spotting, identification and quantification of biomolecules with previously unknown structure. Classically, this field is often referred to as bioinorganic analysis. Earlier reviews on this area are focused on methodological aspects, such as the coupling of the various chromatographic methods to ICP-MS,1–4 and the bulk of applications is found in the area of metal speciation.In recent years, an innovative trend can be observed towards joint applications of element and molecular mass spectrometry for specific detection and molecular characterization of complex biomolecules containing an ICP-detectable element (ICP-tag). Another innovative feature in many of these studies is the application of ICP-MS for the analysis of metalloid or non-metal elements. The main constituents of organic molecules—C, H, N and O—can be considered as elements which are outside the scope of ICP-MS (hydrogen is not detectable and analysis of C, N, O requires elaborate efforts, so far preventing a routine application). However, all other elements of the biosphere can be detected by ICP-MS and can therefore be denominated as ICP-tag. In a number of biomolecules one or more of these ICP-tags are naturally present. If desired, they can also be introduced intentionally by a suitable derivatization. This concept opens a spectrum of new analytical options and strategies, since these ICP-tags comprise P, S, Se, Si, Cl, Br, I, As, and all metals. Bioanalytes must exhibit a certain thermodynamic and kinetic stability to be preserved in their native compositional and coordinative structure during their chromatographic separation and their molecular detection by MS. Therefore, this approach is most successful when applied to molecules, which contain their ICP-tag bound in a covalent or covalent-like form.
In contrast, metal ions weakly coordinated to organic ligands so far are difficult to analyze by this methodology,5 and if results are obtained, their biological impact has to be discussed with caution. In addition, ICP-sensitivity covers a broad range, e.g. P and S are detected with significantly lower sensitivity than are metals.
In spite of these somewhat limiting features, element and molecular mass spectrometry represent a truly supplementary team of techniques for exploratory analytical tasks in the life sciences. To underline this statement, this review gives reference to a collection of recent studies in the life sciences to which both techniques have provided significant input, finally leading to new structural or quantitative insights at the molecular level. According to this focus, ICP-MS studies without strong reference to individual molecular aspects were not considered, among these are for instance numerous studies centered on the speciation of metals in biological samples. This review is structured along compound classes, which comprise (i) amino acids, peptides and proteins, (ii) nucleotides and nucleic acids, (iii) arsenosugars, (iv) small biomolecules like hormones and vitamins and (v) drugs and drug metabolites.
1. Amino acids, peptides and proteins
Due to the stable incorporation of Se in Se-Cys, selenoprotein P isolated from rat plasma could be characterized by a standard analytical proteomics procedure. Such a procedure is schematically outlined in Fig. 1.
 | ||
| Fig. 1 Typical strategy for identification of Se in proteins: the Se-containing high molecular weight fraction is spotted by size-exclusion LC coupled to ICP-MS, this fraction is collected and digested by a protease. The digest is analyzed for Se-containing peptides by reversed-phase LC-ICP-MS. Se-containing fractions are collected and characterized by MALDI-MS for their molecular ion and by ESI-MS/MS for their structure (e.g.ref. 13). | ||
In this analysis, three truncated isoforms of selenoprotein P were found besides the intact form.23 Since all three forms ended with an amino acid preceding a Se-Cys residue (exactly before the 2nd, 3rd, and 7th residue) it was concluded that the UGA codons in this subgroup have a dual function, either encoding for the incorporation of Se-Cys or—as normal—encoding for chain termination. A somewhat lower chemical stability of Se-Cys residues compared to Cys residues under the standard analytical proteomics conditions was observed. For selenoprotein P, partial loss of SeH2 resulting in dehydroalanine formation was observed under conditions which do not affect the stability of normal Cys residues.24
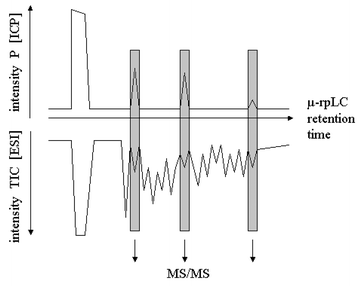 | ||
| Fig. 2 Typical strategy for analysis of protein phosphorylation. A phosphoprotein is isolated and digested. The analysis of this digest is displayed. Reversed phase capillary LC coupled to ICP-MS with phosphorus detection and to ESI-MS is used. Phosphopeptides are recognized in the 31P trace and characterized by ESI-MS and MS/MS. The latter technique can be performed either in the LC-MS mode, by nanoESI of the total digest or by nanoESI of isolated phosphopeptide fractions. | ||
The quantitative character of ICP-MS can be used for the determination of the degree of phosphorylation. When the phosphopeptide/protein contains at least one Cys or Met residue then the ratio of P over S can be determined. Based on the sensitivity factors for P and S, the experimental P/S ratio can be converted into the stoichiometric P/S ratio and thus into the degree of phosphorylation.33,34 Spectral interferences on m/z 31 and m/z 32 were overcome by HR-ICP-MS using medium resolution (4000).33 In the other approach, quadrupole ICP-MS was used to measure the phosphorylation degree of phosphoproteins in combination with a dynamic reaction cell.34 For further reduction of polyatomic interferences, sulfur and phosphorus were detected as their oxides SO+ and PO+ generated by ion–molecule reactions in this case. The introduction of the collision and reaction cell technology has expanded the applicability of quadrupole instrumentation, since it effects a significant reduction of polyatomic background interference and introduces a new dimension of methodological optimization35,36 (selection of collision/reaction gases, of pressure and collision energy).
When applied to purified metalloproteins, the use of LC-ICP-MS alone (without molecular MS methods) can also lead to new, quantitative insights into protein structure. One aspect is the combination of metal and protein quantification, and the other is the quantification of more than one metal. In a study of two bacterial metalloproteases, an LC-ICP-MS system was set up, which provided sufficient sensitivity for detection of a single metal ion in a protein and chromatographic conditions could be established with sufficiently low metal background.38 Zn could be identified as the natural metal in two bacterial membrane proteases at the molar ratio of metal/protein of 3 and 1, respectively. Using the same system, the absence of a metal cofactor could be demonstrated for another isolated bacterial protease.38
In a study of recombinant endothelial nitric oxide synthase, zinc and iron was quantified in the dimeric form of this enzyme. The presence of two atoms of heme iron and one atom of zinc per subunit was verified.39 In a subsequent functional study LC-ICP-MS was used to prove that Zn is necessary for the dimerization of this enzyme but that it is not functionally important.40
Three studies using only molecular or element mass spectrometry, respectively, for the analysis of MTs will be mentioned here in addition, since they highlight a particular potential of the corresponding techniques.
(i) In an ESI-MS/MS study noncovalent complexes between a Zn-loaded metallothionein and a set of glutathione-related compounds were analyzed by ESI-MS/MS. A partial transfer of Zn from metallothionein to glutathione was only observed for oxidized (S-S-linked) derivatives,48 a phenomenon which might be correlated with the function of metallothioneins in cellular Zn-transport.
(ii) A generic, analyte-independent isotope dilution analysis was applied in combination with CE-ICP-MS for metallothionein quantification: The sheath liquid supplied at in the CE-ICP interface is spiked with isotopically enriched metals and isotopically enriched sulfur (32S or 33S). Then continuous isotope ratio monitoring of the corresponding element isotopes is performed with ICP-MS, whereupon the various MT species are detected by alterations in this isotope ratio, since their elution introduces sulfur and metals with their natural isotope abundances.49,50 In this way, quantification of selected chelated metals and of the MT ligands is performed separately, so that both the absolute amounts and the degree of saturation of individual MT subforms are accessible.
(iii) Molecular specificity is conferred to ICP-MS by labelling antibodies with gold nanoparticles, which results in a stable-isotope immunoassay with a sensitivity comparable to the classical radio-immunoassay.51,52
Phytochelatins are small cysteine-containing metal-binding peptides, by which plants regulate their metal homeostasis. Biosynthetically, they are derived from glutathione and exhibit the core composition of (γ-Glu-Cys)n = 2–7 with different end groups. Following Cd-induction, phytochelatin–Cd complexes were recognized by LC-ICP-MS53 and the phytochelatin part of these complexes was characterized by ESI-MS and MS/MS.53–55 Intact phytochelatin–metal complexes have not yet been detected due to their low stability under standard electrospray conditions, nevertheless, ESI-MS and tandem MS studies so far have greatly contributed to unravel the molecular microheterogeneity of plant phytochelatins.
2. Nucleic acids
In the quantitative analysis of covalent nucleic acid adducts with mutagenic agents, a principle problem is the quantification of so far uncharacterized products. This problem has been solved for the quantitative estimation of styrene oxide adducts to nucleotides via their analysis by LC-ICP-MS with phosphorus detection.56 DNA was covalently modified, hydrolyzed and the monomers analyzed by LC-ICP-MS. Covalently modified monomeric units were spotted and quantified by ICP-MS. For these DNA adducts, comparison with parallel investigations by LC-ESI-MS revealed a relatively uniform electrospray ionization efficiency. Adducts of the anticancer drug cisplatin with mononucleotides were also studied by LC-ICP-MS.57 In this study the covalently modified reaction products could be spotted and quantified by simultaneous detection of phosphorus and platinum. Investigations by ESI-MS were conducted on selected adducts of cisplatin with selected dinucleotides,58 showing that these intact metal–organic compounds can be detected intact and that the corresponding ESI-MS/MS spectra give insight into their covalent structure.3. Arsenosugars
Arsenic is occurring in the marine environment and in marine organisms in inorganic form (arsenite/arsenate) and in various organic forms, including methylated arsenic species, such as dimethylarsinic acid, quaternized species such as arsenocholine/arsenobetaine and a variety of arsenoribosides summarized under the term of arsenosugars (e.g.ref. 59). In these ribosides, arsenic is located at the position of the 5-hydroxy group of ribose. This class of compounds has been carefully investigated both by LC-ICP, LC-ESI-MS and ESI-MS/MS. With ESI-MS both molecular, structural and sometimes also element information can be obtained, since the arsenosugars give abundant molecular ion signals in ESI, structurally significant fragment ions upon CID include an As+ fragment signal at m/z 75.60 This fragment ion seems to be absent in normal product ion spectra of arsenosugars,60 but was observed following collisional activation in the ion transfer region.61 Both element and molecular ion information by ESI MS was demonstrated also for organotin compounds.62 These findings highlight the ‘dual mode’ analytical potential of ESI, provided dedicated instrumental conditions are established. Nevertheless, in the majority of studies on arsenosugars, ICP-MS was used for As detection and quantification. These studies refer to the detection and structural assignment of arsenosugars in kelp,63 algae,64,65 and seaweed,66–69 as well as in urine from sheep70 and humans71 following ingestion of arsenosugars. In the application to arsenosugars the analytical strategy has repeatedly proven its capability for detection and structural characterization of novel metabolites. A selenium-containing hexose was also identified by a similar strategy.724. Hormones and vitamins
5. Drugs and drug metabolites
Metabolic and pharmacokinetic studies of drugs require sensitive and robust methods, among which ICP-MS is attracting increasing attention, since a variety of drugs contain ICP-detectable elements, such as chlorine, bromine, iodine, sulfur, phosphorus or covalently bound metals. Moreover, unknown compounds may also be quantified by ICP-MS on the basis of such an ‘element tag’ in case the stoichiometry of its presence can be verified. For this task ESI-MS spectra can provide valuable data. For investigations on the metabolic fate of 4-bromoanaline80,81 and of a bromo-acetanilide82 derivative in rats, LC-ICP- and LC-ESI-MS have been successfully used in combination. Also, the metabolic influence of human and rat plasma on brominated bradykinin was studied in an in vitro assay by the same methodology.83 The natural isotopic doublet of 79Br and 81Br is particularly beneficial for the spotting of Br-containing metabolites in the corresponding ESI-MS spectra. Quantification of the chlorine-containing drugs diclofenac and chlorpromazine on the basis of 35Cl detection was shown to be independent of the solvent composition in gradient LC-ICP-MS,84 and simultaneous detection of 35Cl and 32S allowed the quantitative detection of sulfur-containing metabolites such as a sulfate ester and an N-acetyl-cysteinyl conjugate.85 Sulfur-32 was also demonstrated to be appropriate for the determination of impurities in a drug preparation at a level below 0.1%.86 The innovative potential of ICP-MS in metallodrug studies was demonstrated in two different ways. (1) In a study of a 14C labelled organoplatinum drug, Pt and 14C were simultaneously monitored by ICP-MS.87 Monitoring of 14C by its mass instead of its radioactive decay is a new way to gain insight into the integrity of the organic part of the drug. (2) In a study of metallodrug/protein interactions with Ru- and Pt-containing drugs, these elements were monitored in combination with size-exclusion chromatography to study their binding kinetics to human serum proteins.88 Finally, first attempts on the quantitative detection of phospholipids by phosphorus-31 detection have been reported within an article summarizing pharmaceutical applications of element mass spectrometry.89Quantitative aspects
The sensitivity of the core analytical techniques reviewed here, LC-MS either in the ICP-MS or ESI-MS mode, is defined by the signal-to-noise ratio of an analyte in its corresponding transient chromatographic peak. The main factors governing the sensitivity are the recovery of the analytes from the LC column, the MS ionization efficiency and the background signal level. Biomolecules show nonspecific absorption on all types of chromatographic materials used for separation in liquid chromatography, an effect which usually is corrected by co-injection of an appropriate internal standard and the recording of calibration curves.The signal response in ESI-MS is strongly dependent on the chemical nature of the analyte and of the solvent. Due to the pronounced dependency of the signal intensity on analyte and matrix, quantitative results with ESI require the presence of an internal standard for each analyte, such as an isotopically labelled analogue or a homologous compound. The requirements of quantitative measurements by MALDI-MS are similarly strict. Background signals normally are not limiting the sensitivity in the detection of biomolecules by MS, due to the specificity of the relatively high molecular weight of the individual analytes (>500 Da).
The ionization efficiency in ICP-MS for the metals at one side and semi- or non-metallic elements on the other, varies between one and two orders of magnitude. The variation in corresponding detection limits is much larger. This is mainly caused by the much higher background for some nonmetallic elements, for instance P and S, compared to the extremely low background for some metals, for instance of the lanthanides. Quantitative measurements by LC-ICP-MS require attention mainly with respect to the matrix. In contrast, the chemical nature of individual analytes on the signal intensity is of minor importance, so that usually a single standard can be used as generic reference for a whole class of analytes.
For most applications reviewed here employing both element and molecular mass spectrometry, detection limits are typically in the range of 0.1–10 pmol (eluting from the LC column). Depending on the particular application, either ICP or ESI/MALDI may show more favourable detection limits. For instance, for the detection of lanthanides LC-ICP-MS will provide the lower detection limits, and for the detection of halogenated biomolecules, ESI-MS will probably be more sensitive. As demonstrated in the reviewed studies, the sensitivity ranges of both approaches exhibit a broad overlap allowing their combined application to a palette of problems.
Summary and outlook
In view of the numerous recent articles on the combined application of element and molecular mass spectrometry referenced here one can conclude that an active field of research is identified. The high level of analytical excellence achieved may trigger the development of dedicated instruments to further improve the impact of this combined approach. A simple variant would be a set-up optimized for simultaneous analysis of an LC-eluate by ICP- and ESI-MS. Laser ablation ICP-MS or secondary ion mass spectrometry (SIMS) can provide mass-specific surface images based on element ions (ICP, SIMS) or molecular fragment ions (SIMS), and in fact they are emerging as attractive tools for the characterization of tissue slices or for the read-out of microarrays and of chromatographic or electrophoretic systems. Hybrid element/molecular mass spectrometers, if developed, would offer a significant improvement in analysis speed and probably also in sensitivity over current instrumentation. It appears possible that the traditionally separated development and application of element and molecular mass spectrometry may be overcome in view of the current stimulating results. The implementation of both capabilities into one instrument would fit perfectly to the analytical demands in the life sciences, since the study of the principles and phenomena of life requires an interdisciplinary approach.Acknowledgements
We are indebted to N. Jakubowski for encouraging support and valuable discussions.References
- H. Chassaigne, V. Vacchina and R. Lobinski, Trends Anal. Chem., 2000, 19, 300–313 CrossRef CAS.
- J. Szpunar and R. Lobinski, Anal. Bioanal. Chem., 2002, 373, 404–411 CrossRef CAS.
- B. Michalke, TrAC Trends Anal. Chem., 2002, 21, 142–153 CrossRef CAS.
- B. Michalke, TrAC Trends Anal. Chem., 2002, 21, 154–165 CrossRef CAS.
- R. Lobinski and J. Szpunar, Anal. Chim. Acta, 1999, 400, 321–332 CrossRef.
- B. Michalke and P. Schramel, Biol. Trace Element Res., 1997, 59, 45–56 Search PubMed.
- M. Kotrebai, M. Birringer, J. F. Tyson, E. Block and P. C. Uden, Analyst, 2000, 125, 71–78 RSC.
- S. McSheehy, W. Yang, F. Pannier, J. Szpunar, R. Lobinski, J. Auger and M. Potin-Gautier, Anal. Chim. Acta, 2000, 421, 147–153 CrossRef CAS.
- M. Kotrebai, M. Birringer, J. F. Tyson, E. Block and P. C. Uden, Anal. Commun., 1999, 36, 249–252 RSC.
- C. Casiot, V. Vacchina, H. Chassaigne, J. Szpunar, M. Potin-Gautier and R. Lobinski, Anal. Commun., 1999, 36, 77–80 RSC.
- T. Lindemann and H. Hintelmann, Anal. Chem., 2002, 74, 4602–4610 CrossRef CAS.
- J. R. Encinar, R. Ruzik, W. Buchmann, J. Tortajada, R. Lobinski and J. Szupnar, Analyst, 2003, 128, 220–224 RSC.
- J. R. Encinar, L. Querdane, W. Buchmann, J. Tortajada, R. Lobinski and J. Szpunar, Anal. Chem., 2003, 75, 3765–3774 CrossRef.
- S. McSheehy, S. Pohl, J. Szpunar, M. Potin-Gautier and R. Lobinski, J. Anal. At. Spectrom., 2001, 16, 68–73 RSC.
- S. McSheehy, F. Pannier, J. Szpunar, M. Potin-Gautier and R. Lobinski, Analyst, 2002, 127, 223–229 RSC.
- D. Fagegaltier, A. Lescure, R. Walczak, P. Carbon and A. Krol, Nucleic Acids Res., 2000, 28, 2679–2689 CrossRef CAS.
- V. Mostert, Arch. Biochem. Biophys., 2000, 376, 433–438 CrossRef CAS.
- K. Ishida, T. Morino, T. Takagi and Y. Sukenaga, Nucleic Acids Res., 1987, 15, 10051 CAS.
- D. L. St. Germain and V. A. Galton, Thyroid, 1997, 7, 655–668 CAS.
- H. Koyama, K. Omura, A. Ejima, Y. Kasanuma, C. Watanabe and H. Satoh, Anal. Biochem., 1999, 267, 84–91 CrossRef CAS.
- S. Yoneda and K. T. Suzuki, Biochem. Biophys. Res. Commun., 1997, 231, 7–11 CrossRef CAS.
- U. Sidenius, O. Farver, O. Jons and B. Gammelgaard, J. Chromatogr., B, 1999, 735, 85–91 CrossRef CAS.
- S. Ma, K. E. Hill, R. M. Caprioli and R. F. Burk, J. Biol. Chem., 2002, 277, 12749–12754 CrossRef CAS.
- S. Ma, R. M. Caprioli, K. E. Hill and R. F. Burk, J. Am. Soc. Mass Spectrom., 2003, 14, 593–600 CrossRef CAS.
- M. Wind, A. Wegener, R. Kellner and W. D. Lehmann, Angew. Chem. Int. Ed. Engl., 2003, 115, 3425–3427 CrossRef CAS.
- P. Blume-Jensen and T. Hunter, Nature, 2001, 411, 355–365 CrossRef CAS.
- M. Wind, M. Edler, N. Jakubowski, M. Linscheid, H. Wesch and W. D. Lehmann, Anal. Chem., 2001, 73, 29–35 CrossRef CAS.
- M. Wind, D. Gosenca, D. Kübler and W. D. Lehmann, Anal. Biochem., 2003 Search PubMed.
- M. Wind, O. Kelm, E. Nigg and W. D. Lehmann, Proteomics, 2002, 2, 1516––1523 Search PubMed.
- O. Kelm, M. Wind, W. D. Lehmann and E. A. Nigg, J. Biol. Chem., 2002, 277, 25247–25256 CrossRef CAS.
- M. Wind, I. Feldmann, N. Jakubowski and W. D. Lehmann, Electrophoresis, 2003, 24, 241276–241280 CrossRef CAS.
- J. S. Becker, S. F. Boulyga, J. S. Becker, C. Pickhardt, E. Damoc and M. Przybylski, Int. J. Mass Spectrom., 2003, 228, 985–997 CrossRef CAS.
- M. Wind, H. Wesch and W. D. Lehmann, Anal. Chem., 2001, 73, 3006–3010 CrossRef CAS.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Anal. Chem., 2002, 74, 1497–1502 CrossRef CAS.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Spectrochim. Acta, B, 2002, 57, 1361–1452 CrossRef.
- S. E. O'Brien, B. W. Acon, S. F. Boulyga, J. S. Becker, H. J. Dietze and A. Montaser, J. Anal. At. Spectrom., 2003, 18, 230–238 RSC.
- J. Wang, D. Dreessen, D. R. Wiederin and R. S. Houk, Anal. Biochem., 2001, 288, 89–96 CrossRef CAS.
- I. Leopold and B. Fricke, Anal. Biochem., 1997, 252, 277–285 CrossRef CAS.
- A. Leber, B. Hemmens, B. Klösch, W. Goessler, G. Raber, B. Mayer and K. Schmidt, J. Biol. Chem., 1999, 274, 37658–37664 CrossRef CAS.
- B. Hemmens, W. Goessler, K. Schmidt and B. Mayer, J. Biol. Chem., 2000, 275, 35786–35791 CrossRef CAS.
- R. Lobinski, H. Chassaigne and J. Szpunar, Talanta, 1998, 46, 271–289 CrossRef CAS.
- S. McSheehy and Z. Mester, TrAC Trends Anal. Chem., 2003, 22, 311–326 CrossRef CAS.
- H. Chassaigne and R. Lobinski, J. Chromatogr., A, 1998, 829, 127–136 CrossRef CAS.
- K. Polec, O. Garcia-Arribas, M. Perez-Calvo, J. Szpunar, B. Ribas-Ozonas and R. Lobinski, J. Anal. At. Spectrom., 2000, 15, 1363–1368 RSC.
- K. Polec, M. Perez-Calvo, O. Garcia-Arribas, J. Szpunar, B. Ribas-Ozonas and R. Lobinski, J. Inorg. Biochem., 2002, 88, 197–206 CrossRef CAS.
- A. N. Richarz and P. Brätter, Anal. Bioanal. Chem., 2002, 372, 412–417 CrossRef CAS.
- A. Prange and D. Schaumlöffel, Anal. Bioanal. Chem., 2002, 373, 441–453 CrossRef CAS.
- C. Alfonso, Y. Hathout and C. Fenselau, J. Mass Spectrom., 2002, 37, 755–759 CrossRef CAS.
- D. Schaumlöffel, A. Prange, G. Marx, K. G. Heumann and P. Brätter, Anal. Bioanal. Chem., 2002, 372, 155–163 CrossRef CAS.
- Z. Wang and A. Prange, Anal. Chem., 2002, 74, 626–631 CrossRef CAS.
- C. Zhang, Z. Zhang, B. Yu, J. Shi and X. Zhang, Anal. Chem., 2002, 74, 96–99 CrossRef CAS.
- V. I. Baranov, Z. Quinn, D. R. Bandura and S. D. Tanner, Anal. Chem., 2002, 74, 1629–1636 CrossRef CAS.
- V. Vacchina, R. Lobinski, M. Oven and M. H. Zenk, J. Anal. At. Spectrom., 2000, 15, 529–534 RSC.
- V. Vacchina, H. Chassaigne, M. Oven, M. H. Zenk and R. Lobinski, Analyst, 1999, 124, 1425–1430 RSC.
- H. Chassaigne, V. Vacchinas, T. M. Kutchan and M. H. Zenk, Phytochemistry., 2001, 56, 657–668 CrossRef CAS.
- C. Siethoff, C. I. Feldmann, N. Jakubowski and M. Linscheid, J. Mass Spectrom., 1999, 34, 421–426 CrossRef CAS.
- S. Hann, A. Zenker, M. Galanski, T. L. Bereuter, G. Stingender and B. K. Keppler, Fresenius’ J. Anal. Chem., 2000, 370, 581–586 CrossRef CAS.
- T. Hagemeister and M. Linscheid, J. Mass Spectrom., 2002, 37, 731–747 CrossRef CAS.
- J. J. Corr and E. H. Larsen, J. Anal. At. Spectrom., 1996, 11, 1215–1224 RSC.
- S. A. Pergantis, S. Wangkarn, K. A. Francesconi and J. E. Thomas-Oates, Anal. Chem., 2000, 72, 357–366 CrossRef CAS.
- D. Kuehnelt, W. Goessler and K. A. Francesconi, Rapid Commun. Mass Spectrom., 2003, 17, 654–659 CrossRef CAS.
- J. J. Corr, J. Anal. At. Spectrom., 1997, 12, 537–546 RSC.
- P. A. Gallagher, X. Wei, J. A. Shoemaker, C. A. Brockhoff and J. T. Creed, J. Anal. At. Spectrom., 1999, 14, 1829–1834 RSC.
- A. D. Madsen, W. Goessler, S. N. Pedersen and K. A. Francesconi, J. Anal. At. Spectrom., 2000, 15, 657–662 RSC.
- S. McSheehy, P. Pohl, D. Velez and J. Szpunar, Anal. Bioanal. Chem., 2002, 372, 457–466 CrossRef CAS.
- S. McSheehy, M. Marcinek, H. Chassaigne and J. Szpunar, Anal. Chim. Acta, 2000, 410, 71–84 CrossRef CAS.
- P. A. Gallagher, J. A. Shoemaker, X. Wei, C. A. Brockhoff-Schwegel and J. T. Creed, Fresenius’ J. Anal. Chem., 2001, 369, 71–80 CrossRef CAS.
- M. Miguens-Rodriguez, R. Pickford, J. E. Thomas-Oates and S. A. Pergantis, Rapid Commun. Mass Spectrom., 2002, 16, 323–331 CrossRef CAS.
- M. Van Hulle, C. Zhang, X. Zhang and R. Cornelis, Analyst, 2002, 127, 634–640 RSC.
- K. A. Francesconi, R. Tanggard, C. J. McKenzie and W. Goessler, Clin. Chem., 2002, 48, 92–101 CAS.
- H. R. Hansen, A. Raab and J. Feldmann, J. Anal. At. Spectrom., 2003, 18, 474–479 RSC.
- Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, J. Chromatogr., B, 2002, 767, 301–312 CrossRef CAS.
- B. Michalke and P. Schramel, Electrophoresis, 1999, 20, 2547–2553 CrossRef CAS.
- K. Takatera and T. Watanabe, Anal. Chem., 1993, 65, 759–762 CrossRef CAS.
- B. Michalke, P. Schramel and P. Witte, Biol. Trace Element Res., 2000, 78, 81–91 Search PubMed.
- M. Wind, A. Eisenmenger and W. D. Lehmann, J. Anal. At. Spectrom., 2002, 17, 21–26 RSC.
- W. Buchberger, B. Czizsek, S. Hann and G. Stingeder, J. Anal. At. Spectrom., 2003, 18, 512–514 RSC.
- S. A. Baker and N. J. Miller-Ihli, Spectrochim. Acta, B, 2000, 55, 1823–1832 CrossRef.
- H. Chassaigne and R. Lobinski, Anal. Chim. Acta, 1998, 359, 227–235 CrossRef CAS.
- J. K. Nicholson, J. C. Lindon, G. Scarfe, I. D. Wilson, F. Abou-Shakra, J. Castro-Perez, A. Eaton and S. Preece, Analyst, 2000, 125, 235–236 RSC.
- F. R. Abou-Shakra, A. B. Sage, J. Castro-Perez, J. C. Lindon, G. B. Scarfe and I. D. Wilson, Chromatographia Suppl., 2002, 55, S-9–S-13 Search PubMed.
- J. K. Nicholson, J. C. Lindon, G. B. Scarfe, I. D. Wilson, F. Abou-Shakra, A. B. Sage and J. Castro-Perez, Anal. Chem., 2001, 73, 1491–1494 CrossRef CAS.
- P. Marshall, O. Heudi, S. Mckeown, A. Amour and F. Abou-Shakra, Rapid Commun. Mass Spectrom., 2002, 16, 220–228 CrossRef CAS.
- C. J. Duckett, N. J. C. Bailey, H. Walker, F. Abou-Shakra, I. D. Wilson, J. C. Lindon and J. K. Nicholson, Rapid Commun. Mass Spectrom., 2002, 16, 245–247 CrossRef CAS.
- O. Corcoran, J. K. Nicholson, E. M. Lenz, F. Abou-Shakra, J. Castro-Perez, A. B. Sage and I. D. Wilson, Rapid Commun. Mass Spectrom., 2000, 14, 2377–2384 CrossRef CAS.
- E. H. Evans, J. C. Wolff and C. Eckers, Anal. Chem., 2001, 73, 4722–4728 CrossRef CAS.
- C. J. Smith, I. D. Wilson, F. Abou-Shakra, R. Payne, H. Griesdale, A. Long, D. Roberts and M. Malone, Chromatographia Suppl., 2002, 55, S-151–S-155 Search PubMed.
- J. Szpunar, A. Makarov, T. Pieper, B. K. Keppler and R. Lobinski, Anal. Chim. Acta, 1999, 387, 135–144 CrossRef CAS.
- B. O. Axelsson, M. Joernten-Karlsson, P. Michelsen and F. Abou-Shakra, Rapid Commun. Mass Spectrom., 2001, 15, 375–385 CrossRef CAS.
Footnote |
| † Current address: Basilea Pharmaceutica Ltd, Grenzacherstrasse 487, 4005 Basel, Switzerland. |
| This journal is © The Royal Society of Chemistry 2004 |