Study of chemical speciation of trace elements by molecular activation analysis and other nuclear techniques
Z. F.
Chai
*a,
Z. Y.
Zhang
a,
W. Y.
Feng
a,
C. Y.
Chen
a,
D. D.
Xu
a and
X. L.
Hou
b
aLaboratory of Nuclear Analytical Techniques, Institute of High Energy Physics, Chinese Academy of Sciences, P. O. Box 918, Beijing 100039, China. E-mail: chaizf@ihep.ac.cn
bRisø National Laboratory, NUK-202, DK-4000 Roskilde, Denmark
First published on 16th October 2003
Abstract
The information on the chemical speciation of trace elements in biological and environmental systems is much needed to evaluate their biological and environmental significance. Albeit a number of atomic behavior-based analytical techniques are available for the analysis of chemical speciation of trace elements, nuclear analytical techniques, especially the molecular activation analysis method, can in many cases play a unique role. This review describes the methodology, merits and limitations of nuclear analytical techniques for chemical speciation study in biological and environmental samples. The emphases are focused on the chemical species and the environmental and biological significance of rare earth elements in natural plants and human liver, selenium in a mammalian organism, mercury in rat brain and liver, chromium in rat organs and Cr-rich yeast, organohalogens in pine needles and marine organisms, and iodine in sea-water, soil, atmosphere, marine plants and the thyroid gland for demonstration of the features of nuclear analytical techniques. The future perspectives of nuclear analytical techniques for the study of chemical species of trace elements will be briefly outlined as well.
Introduction
Trace elements, including the essential and the toxic, play an important role in life and environmental sciences, which, however, is highly related to their chemical species. An implicit fact is that the bulk contents or concentrations of trace elements in systems of interest are often meaningless in judging their biological and environmental significance. Therefore, in recent years, more and more attention has been paid to the study of the chemical species of trace elements, e.g. refs. 1–3. A critical issue for this study is the definition of the chemical species and speciation. Various definitions were put forward before 2000,4 when the IUPAC made an attempt to define: what is the chemical species? what is speciation? and what is the speciation analysis?5 The IUPAC's effort was admirable and at least it provides a basis for further discussion. But some expressions of its definitions are difficult to understand. For example, the IUPAC states that the chemical species for chemical elements is the specific form of an element defined in terms of its isotopic composition, electronic or oxidation state, and/or complex or molecular structure, and that the speciation of an element is the distribution of an element amongst defined chemical species in a system.5 The question is why the isotopic composition, a nuclear property, is regarded as a chemical species of elements, a chemical property, although the two parameters have a certain relationship. Further, the definition that the speciation means the distribution of an element amongst chemical species seems a bit narrow in sense. The “distribution” is only one aspect of the speciation, which should also include the processes of transformation, alteration and interaction amongst the chemical species. The above fact fully demonstrates that speciation study is now just at the preliminary stage of its developmental process.Methodology of nuclear analytical techniques for chemical speciation study of trace elements
All the available non-nuclear analytical techniques (non-NATs) for chemical speciation study are, per se, based on a combination of analytical methods and separation techniques. Thus, the term “hybrid” is often assigned to these non-NATs, of which the best known are HPLC-ICP-MS and GC-ICP-MS. However, the NATs for chemical speciation study can be divided into two types: direct and indirect. It should be clearly pointed out that the NATs for chemical speciation include a number of techniques, such as molecular activation analysis (MAA), proton induced X-ray emission technique (PIXE), synchronous radiation-based analytical techniques, Mössbauer spectrometry and others.2–4 MAA, PIXE and X ray fluorescence based on synchronous radiation (SRXRF) need to combine with the chemical separation techniques, so that they are a hybrid technique, similar to the non-NATs. Another type, so called the direct one, includes extended X-ray absorption fine structure (EXAFS), X-ray absorption near edge structure (XANES), Mössbauer spectrometry and others, which can directly study the chemical species of elements without any chemical manipulation, by use of which on line and in situ analysis for chemical species are achievable. In comparison with the non-nuclear techniques, the NATs possess unique features, e.g., high sensitivity, good accuracy and precision, non-destructiveness, and no or reduced matrix effect, etc. It should be mentioned that NATs are only adopted in a few nuclear laboratories in the world for the speciation study of trace elements, simply because of the difficulty, for most users, in accessing the expensive nuclear facilities such as a nuclear reactor and accelerator. The previous reviews on NATs for chemical speciation study of trace elements can be referred to.2–4,6 The available NATs for chemical speciation study are listed in Table 1, along with their essentials and features, where ICP-MS is also included for comparison.| Methoda | Accuracy | Sensitivity | Space resolution | In situ analysis | Non-destructive analysis | Matrix effect | Time response |
|---|---|---|---|---|---|---|---|
| a MAA, molecular activation analysis, which, in fact, is a combination of conventional neutron activation analysis with chemical or biological separation; PIXE, proton induced X-ray emission and its derivative SPM, scanning proton microscopy; SRXRF, synchronous radiation X-ray fluorescence; EXAFS, X-ray absorption fine structure spectrometry; XANES, X-ray absorption near edge spectrometry; Möss.Sp., Mössbauer spectrometry; IT, isotopic tracer. | |||||||
| MAA | Excellent | High | Low | No | No | Less | Slow |
| PIXE | Middle | Middle | Middle–Good | Possible | No | Yes | Fast |
| SRXRF | Middle | Middle | Good | Possible | No | Yes | Fast |
| EXAFS | Middle | Poor | Good | Yes | Yes | Yes | Slow |
| XANES | Middle | Poor | Good | Yes | Yes | Yes | Slow |
| Möss.Sp. | Limited | Poor | Poor | Yes | Yes | Yes | Fast |
| IT | Good | High | Possible | Possible | Yes | Less | Fast |
| ICP-MS | Good | High | Low | No | No | Severe | Slow |
Chemical speciation study of rare earth elements (REEs) in organism by MAA
China is very rich in REE resources and its industrial reserve has been estimated to be five times the total of other countries. In recent years, REEs have been widely applied not only in industry, but also as fertilizer and feed in agriculture, forestry and animal husbandry. As a result of their usage, more and more REEs are getting into the environment as well as the food chain in various ways. However, information on their chemical speciation and the resulting biological effects in the organism are scarce. For this reason, research on the REEs' absorption, subcellular distribution and REE-binding macromolecules, e.g., protein, polysaccharides and DNA, in plants, livers of rats and human beings, have been carried out by MAA based on instrumental neutron activation analysis (INAA) combined with chemical and biochemical separation techniques. Fig. 1 shows a schematic diagram for chemical speciation study of REEs in plant. | ||
| Fig. 1 Scheme for chemical speciation study of REEs in plant–soil system by MAA. | ||
It was found that REEs existing as water-soluble forms in soils were the most assimilable species for plant roots.7 Most of REEs in plant roots were firmly bound with cell wall materials,8 whereas the subcellular distributions of REEs in mesophyll protoplasts of Brassia napus clearly indicated that REEs were accumulated in chloroplasts and that every 2000 chlorophyll molecules contained one REE atom. This is one possible reason to explain why REEs are able to enhance the photosynthetic rates of crops.9–11 Moreover, the presence of two REE-binding proteins, four REE-binding polysaccharides and one REE-binding DNA in the leaves of a highly REE-enriched fern, Dicranopteris dichotoma, was demonstrated by MAA.12,13 The molecular weights (MWs) of the two REE-binding proteins were determined to be 800 kD and less than 12.4 kD, respectively. Their SDS-PAGE graphs showed that both contained two proteins subunits, which were likely glycoproteins with different glyco-units. All the four REE-binding polysaccharides were low MW polysaccharides (10–20 kD). Less than 0.1% of total REE in the plant leaves was combined with DNA. To our knowledge, it was the first time that REE-binding biological macromolecules in plants were reported.
REEs have a preferential uptake in liver after they have been absorbed into the bloodstream of a human or animal body.14 The distributions in subcellular fractions of liver cells of both rats and human beings were similar for all REEs, i.e., the highest concentration was found in the microsomal fraction, while the lowest in the cytosolic fraction.15,16 However, in cultured rat cells that were treated with rare earth ions, it was discovered by PIXE that La, Ce, and Gd existed mostly in the nuclear fraction, and then in the cell membrane.17 By means of size-exclusion chromatography (SEC), two soluble REE-binding proteins with MWs of about 68 and more than 40 kD were found in rat livers after the rats were intravenously injected by enriched stable isotope tracer of 152Sm and 168Yb. The elution profiles of 152Sm and proteins were shown in Fig. 2. With the same MAA method, at least three La-binding proteins (MW 335 ± 50, 94.5 ± 15.4, 13.6 ± 3.8 kD), three Ce-binding proteins (MW 335 ± 50, 85 ± 12, 22.8 ± 6.3 kD) and about four Sm-binding proteins (MW 335 ± 70, 82.1 ± 5.4, 32.3 ± 5.8 and 13.6 ± 4.5 kD) were found in the supernatant fraction of human liver samples, which were obtained from normal subjects who had an accidental death. Most of La, Ce and Sm were found in the high-molecular-weight protein region.16
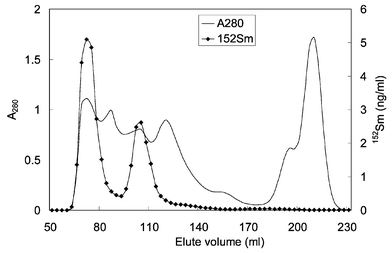 | ||
| Fig. 2 Sephadex G-150 column chromatography of liver cytosol from rats injected by 152SmCl3. | ||
Selenium (Se) speciation study in mammalian organism by NATs
As an essential trace element, Se plays important nutritional roles. Se is needed for the proper functioning of the immune system and is relevant to many kinds of human diseases, being involved in the prevention of certain types of cancer, Alzheimer's disease, cardiovascular disease, inhibiting HIV progression to AIDS, etc.18–22 Since the bioavailability of Se is a consequence of its individual chemical species, there is considerable interest in the speciation of Se in the mammalian organism to define its biological function. However, since Se is primarily present in the form of selenoproteins, studies of its chemical speciation are not only involved with its low molecular weight species, but also mostly in Se-containing proteins.23–25Various analytical methods have been developed for the determination of total Se content in biological samples, such as AAS, AFS, electrochemical detection, ICP-MS, radiochemical and instrumental NAA, PIXE, etc. NATs and the isotopic tracer technique are often imperative in this aspect because of their advantages of high accuracy and precision and freedom from contamination from acids or reagents used in analysis. The main application examples based on NATs for Se speciation are summarized below.
The bioavailability of anionic, cationic and neutral Se compounds and selenoamino acids such as selenocystine, selenocysteine, selenomethionine and Se-methylselenocysteine is of much clinical interest, but low-level determinations of selenium are very difficult. Blotcky et al. determined the Se metabolites, including total Se, trimethylselenonium (TMSe+) ion, selenite, and selenoamino acids in urine and plasma by using MAA combined with anion exchange chromatography.26–28
The basis of selenium’s nutritional role lies in its active involvement with glutathione peroxidase (GPx), iodothyronine deiodinases, thioredoxin reductase (TR) and other selenoenzymes in the form of selenocysteine, which has been confirmed as the 21st amino acid. Until now, about 16 selenoproteins with specific functions have been recognized,18,29,30 which, however, cannot explain all Se biological functions. Thus, there is an increasing interest in characterizing new selenoproteins in different biological species.
For most biological materials, such as tissues, cells, cell fractions and protein, etc., from rats, cattle and humans, the Se content can be determined by the conventional INAA via the short-lived 77mSe (T1/2 = 17.5 s) and long-lived 75Se (T1/2 = 121 d)31–34 with different irradiation conditions. Generally, the detection limits are 5 ng for 77mSe and 0.1 ng for 75Se. The incorporation of Se into protein as peroxidase isozymes in wheat seedlings was studied by an optimized INAA after electrophoretic separation.35 After sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), the gel sections containing corresponding protein bands were cut off and subjected to INAA. The lower detection limit of 0.02 ng was achieved through longer irradiation (8.3 d) with higher neutron flux and longer radioactivity counting time. Direct determination of metals in protein bands on the electrophoretic gels by SRXRF after SDS–PAGE or isoelectric focusing separation was also developed in our laboratory.36–38 Recently, an innovative technique for the detection of Se in selenoprotein bands by using monochromator with SRXRF has been established.
Combined with biochemical separation, INAA was applied to selenium speciation in normal human tissues, human hepatocellular carcinoma and their normal liver-adjacent tissues.39–43 The distribution patterns of selenium species in healthy and patient subjects could help to identify the respective physiological roles of various Se species in human liver, heart, kidney and their compartments.31,39 In general, Se was mainly enriched in mitochondria, nuclei and cytosol. In human liver specimens, 45% of total Se was in nuclei and 25% in mitochondria and cytosol, respectively. In human heart specimens, about 70% of the total Se existed in nuclei and 20% in cytosol, while for kidney specimens about 70% was in nuclei and 14% in cytosol and mitochondria, respectively. These new findings indicate the long-term accumulation of Se in human bodies.39,40 Human hepatocellular carcinoma and their normal liver-adjacent tissues were collected from 7 patients. It is of interest to find that the Se level was higher in carcinoma tissues than in the normal adjacent tissues, while the mitochondrial and cytosolic GPx and TR were also higher.
Speciation and subcellular distribution of the Se-containing proteins in human liver were further separated by SEC and SDS–PAGE followed by INAA and HG-AFS. Eight kinds of Se-containing proteins with MWs of 335 ± 20, 249 ± 15, 106 ± 11, 84.6 ± 5.8, 70.5 ± 5.4, 45.6 ± 1.5, 14.8 ± 2.6 and 8.5 ± 1.2 kD were found in the subcellular fractions of human liver.40 The more accurate molecular weight and better resolution were obtained by SDS–PAGE than SEC. Another important advantage of SDS–PAGE was that Se weak binding or non-specific incorporation to protein would be removed after electrophoresis. It was found that about 24 kinds of Se-containing proteins existed in subcellular fractions of normal human liver.41 The MWs of their subunits were mostly in the range of 20–30 kD and 50–80 kD. The 61, 21 and 54 kD proteins were identified as the known selenoproteins such as selenoprotein P, GPx and TR, respectively. Most of the proteins are yet to be identified. The specific subcellular distributions of different Se-containing proteins suggest that they could play specific and important biological roles in each organelle of human liver.41
For a tracer study, both an enriched stable isotope, e.g., 82Se, and a radioactive isotope, 75Se, were used to study the metabolic pathway and the chemical speciation of Se in the organism. The labeled Se-containing proteins were separated by using electrophoretic or chromatographic methods, subsequently analyzed by autoradiography44–49 and ICP-MS.50,51 Selenoprotein P was found firstly in rat liver and plasma by the use of 75Se tracer.31 More than 20 selenoproteins or subunits in liver organelles and other tissues of Se-deficient rats after replenishment with 75Se-labeled selenite were reported by Behne et al.44,45
The advantage of HPLC-ICP-MS with the use of enriched stable isotopes over the conventional use of radioisotopes is that the simultaneous detection of the speciation of exogenous (labeled with enriched stable isotope) and endogenous Se are possible without a requirement for special and expensive radiotracer facilities.50,51 However, one disadvantage of the above method is that it more or less loses the structural information during the element detection process. An alternative is to use tandem mass spectrometry (MS-MS) as a detector to provide the structural and molecular information, allowing the identification of species.52 The applicability of the optimized HPLC-MS-MS system was demonstrated by the analysis of a mixture containing Se-methyl-selenocysteine, selenomethionine, selenocystine, selenoethionine and selenocystamine. For selenoprotein, the structural analysis of selenopeptide after enzymatic digestion was conducted by MALDI-TOF-MS and electrospray ionization triple quadrupole MS.53,54
Mercury (Hg) speciation study in biological samples by NATs
Mercury is a toxic element to human beings. Its derivatives could cause irreversible brain damage and react with the important receptors in the nervous system. However, the toxicity of different Hg species is extremely different. Generally, Hg in its organic species, usually as methylmercury (MeHg), is more hazardous than its inorganic species.The analytical methods for Hg determination are diverse. Nevertheless, NAA is usually regarded as a “reference method”, because of its excellent accuracy and high sensitivity. The detection limit for total Hg is as low as 0.1 ng g−1 by RNAA in any medium.55 Therefore, this method is very useful for small amount and rare sample analysis. In order to study the affinity of Hg to metallothionein (MT) in the brain tissue of rats that were exposed to Hg0, after gel filtration separation, Falnoga et al.56 used RNAA to analyze the concentration of Hg in rat brain supernatant. They found that about 80% of the Hg in it was in the fraction of low-molecular-weight proteins. Furthermore, a MT-like protein Hg–Cu–Zn–thionein was isolated and partially characterized.
Although the toxicities of Hg and MeHg have been extensively studied, their poison mechanisms to humans still remain unclear. For the latter study, the isotopic tracer technique would become a very powerful method. 203Hg has a suitable half life (t1/2 = 46.9 d), therefore, it is often chosen as an isotopic tracer in the study. Bhattacharya et al.57 studied the specific binding of Hg2+ to ouabain-sensitive Na(+)–K(+)–ATPase of rat liver plasma membrane using isotopic tracer 203Hg2+. They found that the binding of Hg to the enzyme caused the significant inhibition of the enzyme and that the capability of Hg2+ binding to reduced glutathione (GSH) was stimulated by GSH-S-transferase (GST). Therefore, they proposed that the transport of Hg2+ inside the cell occurred by increased dissociation of Hg2+ from the membrane due to the greater avidity of Hg2+ towards cytosolar GSH binding. The GSH–Hg complex entered the nucleus to induce MT transcription.
EXAFS is a powerful method for structural analysis, which, in recent years, has been widely used for the analysis of protein structures, especially for metalloproteins. Gailer et al.58 reported a study of the mutual detoxification of Hg–Se–S-containing species in rabbit plasma by EXAFS after chromatographic purification. The EXAFS spectroscopy revealed the presence of 4 coordinated Se and Hg entities separated by 2.61 Å. The Hg and Se near-edge X-ray absorption spectra of erythrocytes, plasma and bile of rabbits injected with sodium selenite and mercuric chloride solutions showed that Hg and Se in plasma existed as a Hg–Se–S-species.
Chromium (Cr) speciation study in biological samples by MAA
Chromium, as a trivalent organic or inorganic species, has been proposed to act as a potentiator of insulin action in animals and human beings. The Cr deficiency induces symptoms resembling diabetes, such as glucose intolerance impairment with the requirement of increasing insulin, etc., and Cr supplement can alleviate these symptoms. However, up to now, the biological function of Cr in an organism is still unclear. For the Cr speciation study, different kinds of chemical and biological separation methods are needed. Unfortunately, as the concentration of Cr in many biological materials is usually at ultratrace level (∼ng g−1), it is almost impossible to avoid the exogenous Cr contamination resulting from separation and determination processes. Therefore, the analysis is usually so complicated and difficult that a highly sensitive and accurate analytical method is desirable. Using an enriched stable isotope 50Cr tracer technique combined with different kinds of chemical or biochemical separation methods and highly sensitive NAA could overcome the above difficulty.Feng et al.59 used 50Cr3+ tracer combined with differential centrifugation and NAA to study the intracellular distribution of Cr(III) in the liver, pancreas, testes and kidney homogenates of diabetic and normal rats. They found that the nuclear fraction contained the highest Cr concentration in the liver cells of both normal and diabetic rats. The diabetic rats retained more Cr in the mitochondrial and lysosomal fractions of liver homogenate than the normal. The concentrations of Cr in the subcellular fractions of pancreas, testes and kidneys in the normal rats were higher than those in the diabetic rats. Additionally, as an advantage of the multi-element analysis ability for NAA, the significant alterations of the levels of other essential trace elements in subcellular fractions of the observed diabetic organs could be simultaneously observed as well.60 These results suggest that the hormone level change may interfere with some trace elements accumulation both in the organs and the subcellular fractions of rats. Furthermore, after intravenous injection of enriched stable isotope 50Cr tracer solution, Feng et al.61,62 separated Cr-containing proteins in the diabetic and normal rat liver cytosol, serum and urine by Sephadex G-25 gel filtration chromatography. The elution fractions were determined by NAA via50Cr(n, γ)51Cr. It was found that Cr was mainly combined with a high-molecular-weight protein in the liver cytosol and serum. A low-molecular-weight, Cr-containing compound (LMWCr) was found in all the observed liver, serum and urine samples of normal and diabetic rats. They concluded that Cr was excreted chiefly as LMWCr in urine. Recently, the fundamental distribution patterns of the Cr-containing proteins in the nucleic, mitochondrial, lysosomal, microsomal and cytosolic subcellular fractions of the rat liver were investigated by means of stable isotopic tracer technique, Sephadex G-100 gel chromatography and NAA.63 A total of 9 kinds of Cr-containing proteins were found in the five subcellular fractions and the relative MWs were identified as 96.6 ± 6.2, 68.2 ± 1.4, 57.9 ± 4.7, 36.6 ± 1.2, 24.2 ± 1.8, 14.0 ± 1.5, 8.8 ± 0.6, 6.9 ± 0.4 and 4.2 ± 0.4 kD. In this research, about 64.5% of Cr proteins were found in the cytosolic fraction. The LMW Cr proteins (<4.2 kD) were mainly observed in mitochondria, lysosomes and microsomes. More than 69% Cr-containing proteins were present in the liver cytosolic fraction of ≥57.9 kD.
As a useful and convenient chromium supplement, Cr-rich yeast is widely used for diabetics. The study of chromium speciation in the yeast is imperative to understand its biological function. Ding et al.64 assessed the Cr distribution in a Cr-rich yeast cell by NAA and found that 80.9% of Cr was accumulated in the protoplast. Furthermore, Ding et al.65 and Liu66 have studied the Cr species combined with biological macromolecular compounds, i.e., DNA, RNA, and proteins, in Cr-rich yeast by gel chromatography and electrophoretic separation followed by NAA. Liu found that in the Cr-rich yeast, most Cr-containing proteins were present as LMW Cr compounds.66
The Cr species in the Cr-rich yeast was analyzed using EXAFS and the results proved that Cr(III) was the main species in the yeast, which assured the safe intake of the high-Cr yeast.66 A new type detector was adopted to determine the XANES spectra of the yeast samples with different Cr(III)/Cr(VI) ratios, and the results showed that Saccharomyces cerevisiae strain could significantly reduce Cr(VI) to Cr(III).66
Organohalogens in biological and environmental samples by MAA
Organic halogens have long been recognized to be severe environmental pollutants, because of their persistence, bioaccumulation and potential hazardous impact on human health. Therefore, these compounds have been widely investigated in various environmental matrices, e.g., refs. 67–69. Recently, interest in using extractable organohalogens (EOX) as parameters for the quantification of total organohalogen content in sediment,70,71 biota,72–74 and water75 has dramatically increased. However, there is ever-growing evidence that traditional analytical methods, such as GC and GC-MS, can only provide information about the known organochlorines (OCs), which contributed to a minor part of total extractable organochlorinated compounds (EOCl) in samples and, therefore, the results could not reflect the actual OCs contamination levels, e.g., refs. 76,77. From an ecotoxicological point of view, organohalogens, including EOCl, EOBr and EOI, may be important to biota and human beings, because they have a harmful effect on organisms78 and persist in the environment long-term.79NAA is a very convenient method for halogen analysis and is also the only method currently available for simultaneously determining EOCl, EOBr and EOI in an extract.70,75,80–82 Another alternative for the analysis of total organohalogens is microcoulometric or potentiometeric titration,83–85 which, however, needs tedious and time-consuming manipulation steps: enrichment, conversion of organohalogens into halide ions and final determination. Further, it is not able to differentiate halogen elements. Thus, NAA is preferred to determine Cl, Br, I, and EOX in various biological and environmental matrices.75,80,82 The literature survey of this aspect is given in Table 2.
| Sample | Analyte | Converted into halide-ion | Method | Detection limit | Ref. |
|---|---|---|---|---|---|
| Water | TO-Cl, -Br, -I | No | NAA | Cl 5 µg L−1: Br, I 1 µg L−1 | 86 |
| TOX | Yes | Microcoulometric titration | 10 µg L−1 | 87 | |
| AOX | Yes | Microcoulometric titration | 3–30 µg L−1 | 75 | |
| EOCl | No | NAA | 17 µg L−1 | 88 | |
| EO-Cl, -Br, -I | No | NAA | Cl 20 µg L−1: Br 5 µg L−1: I 3 µg L−1 | 89 | |
| Rain and snow | EOCl | No | NAA | 20–80 µg L−1 | 90 |
| AO-Cl, -Br, -I | Yes | Capillary zone electrophoresis-UV | Cl 1–3 ng g−1: Br 2–5 ng g−1: I 4–8 ng g−1 | 91 | |
| Sediment | AOX, EOX | Yes | Microcoulometric titration | AOX 0.5 µg g−1: EOX 0.05 µg g−1 | 83 |
| EOCl | No | NAA | 5 µg g−1 | 75 | |
| EO-Cl, -Br, -I | No | NAA | 0.1 µg g−1 | 92 | |
| EO-Cl, -Br, -I | No | NAA | 0.2–0.4 µg g−1 | 70 | |
| EOX | Yes | Microcoulometric titration | 0.18 µg g−1 | 84 | |
| Soil | EOX | Yes | Microcoulometric/pyrolysis titration | 10 µg g−1 | 93 |
| Marine organism | EOX | Yes | Microcoulometric titration | 0.02–0.17 µg g−1 | 85 |
| Human tissue | EO-Cl, -Br, -I | No | NAA | 0.1 µg g−1 | 92 |
| Marine organism | EO-Cl, -Br, -I | No | NAA | 0.1–5 µg g−1 | 75,92 |
| Shrimp | EO-Cl, -Br, -I | No | NAA | Cl 16–85 ng g−1: Br 1.0–35 ng g−1: I 0.35–10 ng g−1 | 94 |
| Beluga whale | EO-Cl, -Br, -I | No | NAA | Cl 30 ng; Br 6 ng; I, 3 ng | 76 |
| Pine needles | EO-Cl, -Br, -I | No | NAA | Cl 50 ng; Br 8 ng; I 3.5 ng | 81 |
Using MAA based on NAA and organic solvent extraction, Xu et al.81,95 found that the concentrations of halogens were in the order of Cl ≫ Br > I in the pine needle samples from 17 Chinese regions, which was in agreement with their elemental abundances in nature. About 0.1–3.9% of total chlorine was EOCl, whereas 0.2–15% and 2–57% of total bromine and iodine were EOBr and EOI, respectively, which suggested that halogens in pine needles mainly existed as inorganic species and non-extractable organochlorinated compounds.81 Similar results were reported for yogurt and apples.82
Further, the extract was treated by different methods before NAA measurement. The results indicated that 69–93% of total EOCl, 20–58% and 20–90% of total EOBr and EOI were water-soluble polar compounds in pine needle extracts.88 It is known that this persistent EOX (EPOX) seems to be more important for organisms. For the INAA determination of EPOX, the extract was first treated with concentrated sulfuric acid. About 1.6–38% of EOCl survived as the species EPOCl in pine needles,81 in comparison with 11.7% in blue mussel,70 11.3–11.9% in the sediment,70 23–58% in yogurt and 29–35% in apples,82 17% in water75 and 0–39% in aerosol.98 It was also found that the EPOCl concentration increased with increasing age of pine needle.81
The MWs of EOCl have been studied by gel permeation chromatography (GPC) combined with NAA75 and about 90% of EOCl in bleach plant effluent were found to be associated with low MW compounds (MW < 300), whereas 60–70% of EOCl in fish and sediment were associated with MW > 300 compounds.75 Jokela et al.99 and Hemming and Lehtinen100 also reported similar results in bleached kraft mill effluent and in rainbow-trout liver effluent.
Known organochlorine compounds were reported to account for 1.4–30.4% of the EPOCl in pine needles from China,81 1–14% in bird, 48% in sediment from the USA97 and 2–18% in marine organism from the Osaka Bay.92 The identified compounds accounted for 10–15% in fish and 5% in sediment from Bornholm in the Baltic Sea,101 whereas 13–58% of EOCl was found in carp from the Buffalo river96 and 2–25% in fish from the USA.97 About 45% of EOCl reported in blubber lipid of beluga whales is attributed to the known compounds.76 Similarly, 25–50% of EOCl in herring gull eggs from Lake Ontario can be explained by the known compounds.102 In Japanese human adipose tissue, about 59% of EOCl can be accounted for by PCBs, DDTs, PCTs and HCHs.92 The above comparison indicates that the unknown proportion of EOCl was higher in almost all environmental and biological samples.
There is debate about the sources of EOCl and EPOCl in environmental matrices, since most compounds with heteroatoms (oxygen or nitrogen atom) or an unsaturated bond, such as most known naturally occurring OCs, can be protonated or destroyed and removed from the extracts after treatment with concentrated sulfuric acid.80,103 It is known that environmental factors have a heavy impact on EPOCl accumulation in pine needles. Thus, a conclusion can be drawn that EPOCl in pine needles is mainly originated from the ambient atmosphere.81 Kiceniuk et al.76 also found that it was impossible to account for the observed high levels of EOCl in tissues of beluga whales from natural compounds. However, in fish and sediment 60–80% of EOCl can be hydrolyzed by lipase and about 30% of EOCl were acidic material.91 By NATs Lunde and Steinnes104 showed that a certain amount of EOCl in marine organism oil was synthesized by natural processes in the marine environment. Grimvall105 found that the major part of organohalogens in freshwater and the marine environment came from the natural incorporation of halogens into humic substances or other macromolecules. Also, it was found that Cl and Br can be bound to aromatic rings in humic substances,106 a chloroperoxidase-like catalyst in soil.107 Until now, although many studies show that organohalogens have many natural sources, the quantitative contribution of the naturally produced organohalogens has not been estimated. Anthropogenic and atmospheric input have still been considered to be the main sources of organohalogens in the Baltic Sea environment.90 Further studies are needed to estimate the contributions of naturally produced and anthropogenic sources in various environmental samples, to characterize the potential toxicity of organohalogens in aquatic and terrestrial ecosystem, and to better understand the unknown EOCl, for which NATs are imperative.
Chemical speciation study of iodine (I) in biological and environmental samples by NATs
In biological and environmental samples, iodine exists as both inorganic and organic species, which, however, varies with the sample. In recent years, an epithermal NAA and various chemical and biochemical separation techniques has been established for study of the chemical species of stable iodine, as well as long-lived 129I, in various environmental and biological samples, such as water, seaweed, human liver, milk, urine and soil.Iodine in water exists as iodate, iodide and organic iodine.108 The distribution of iodide and iodate in sea-water can give a clue to understanding the marine environment and has been quite well studied. Hou et al. developed a MAA method based on NAA and chemical separation for the determination of iodide, iodate, organic iodine and total iodine in water sample.109 A similar technique was also used for the determination of 129I in iodide and iodate species, which can be used to investigate the geochemical cycle of stable iodine.110 It was found that in the coast water from the North Sea, 50–60% of iodine exists as iodate and 40–50% as iodide, while most of iodine exists as iodide (>80%) in the Baltic Sea water. A low concentration of organic iodine was observed in all sea-water samples measured. A similar result was also reported using iodimetric spectrophotometry for iodate, catalytic spectrophotometry for total inorganic iodine and irradiation with UV-light followed by catalytic spectrophotometry for organic iodine in the Baltic Sea.111 Reifanhauser and Heumann112 developed a method by combining isotope dilution mass spectrometry (IDMS) with anion exchange separation to investigate the chemical species of iodine in fresh water. Besides iodide and iodate, anionic organic iodine and non-elutable organic iodine were determined. They observed that most of iodine existed as organic iodine in lake and river water.
Iodine mainly exists as inorganic iodine in sea-water. In open sea-water most of iodine is iodate, while in coastal or anoxic sea-water (such as the Baltic Sea) the concentration of iodide is high. The concentration of organic iodine in lake and river water is higher than that in sea-water. Although the concentration of organic iodine in sea-water is low, it plays a very important role in the global geochemical cycle of iodine, because the transfer of iodine from the iodine rich ocean to the atmosphere, then to the terrestrial environment, is thought to occur primarily through the volatilization of organic iodine hydrocarbon in the sea-water.113 These volatile organic iodine species were also supposed to relate to ozone depletions in the stratosphere.114 Various volatile organic iodine compounds, and CH3I, CH2ClI, CH2I2 and CH3CH2CH2I have been observed in the sea-water.115
The concentration of iodine in the atmosphere ranges from 0.2 to 10 ng m−3; a high iodine concentration was observed in an urban area due to the combustion of oil and coal. In the atmosphere, iodine exists as particle-bound iodine (particulate iodine), inorganic gas iodine (I2, HI, HOI) and organic gas iodine (CHI3, CH2I2, CH3CH2CH2I), whose concentrations vary with various parameters, such as location, season and climate. The particulate iodine is usually separated and collected using Millipore filter or glass microfibre.116–119 I2 and HI can be separated by adsorption using an LiOH (NaOH) impregnated filter117,118 or silver screens.116 HOI is normally collected by tetrabutylammonium hydroxide (TBAH) impregnated filter117 or charcoal filter116 and organic iodine by charcoal bed116,117 or triethylenediamine (TEDA) impregnated charcoal bed.118 Then, the various iodine species can be determined by NAA or ICP-MS.
Due to high concentration of iodine in marine plants, the chemical species of iodine in a plant is mainly focused on seaweed. Hou et al. developed a MAA method for the determination of various chemical species of iodine in seaweed, such as water soluble iodine, soluble organic iodine, iodide, iodate, and protein-, pigment- polyphenol- or polysaccharide-bound iodine.120,121 The results show that 9–99% of iodine is water-soluble in seaweed. In addition, the percentage of water soluble iodine is the highest in brown algae and lowest in green algae. In the water leachate of seaweed, iodine exists mainly as iodide, the percentage of organic iodine makes up 5–40%, and the iodate is lower than 5% in all 30 species of seaweed investigated. In biological macromolecules, iodine is mainly bound with proteins, polyphenol and pigments, but little iodine is bound with polysaccharide. It is also found that iodine in seaweed also exists as iodinated amino acid and non-polar derivatives of secondary metabolites. In addition, iodinated compounds in seaweed can also exist as part of a complex of polyhalogented compounds with bromo- or chloro-analogues. Using alkaline hydrolysis, organic solvent extraction, GC, HPLC and gel chromatography, many iodine-containing compounds, such as monoiodothyrosine (MIT), diiodothyrosine (DIT), triiodothyroine (T3), thyroxine (T4) and 1-iododibromoheptanone were separated and identified from different seaweeds.122–124
Iodine is a normal component of thyroid gland, and mainly exists as iodo-amino acids such as MIT, DIT, T3, rT3, and T4, which are mainly bound with proteins in thyroid but function as free T3 and T4. Beside thyroid, iodine is also distributed in all tissues.125 The radioimmunoassay method is widely used for the determination of T3, T4 and rT3 in blood for diagnosis of thyroid diseases. Hou et al. investigated the distribution of iodine in various subcellular fractions of human liver using gradient centrifugation coupled with NAA126 and observed that the iodine content is in the order of nuclei > cytosol > mitochondria > lysosome > microsome. Furthermore, gel chromatography was used to investigate the combination of iodine with protein in cytosol of human liver, and observed 3 iodine proteins in which iodine is mainly bound with mid- and high-molecular weight proteins. Because most iodine is bound with macromolecular protein, the sample has to be enzymolysed, then iodine ion and various iodo-amino acids can be separated and measured for determination of the concentrations of iodo-amino acids. HPLC was successfully used to determine various chemical species of iodine, such as I, MIT, DIT, T3, T4, rT3 and iodopolypeptide in hydrolyzed solution of thyroid, serum, urine and milk.127–131
Soil is the main source of iodine for terrestrial plants, so that the chemical species of iodine in soil is directly related to the bioavailability of iodine to plant. From the radiation protection point of view, the chemical species of radioactive 129I is the most important issue to affect the transfer of 129I in the environment. Hou et al. studied the chemical fractionation of 129I in soil and sediment using NATs combined with sequential extraction techniques and observed that iodine was mainly bound with organic matter and oxides. Only 10–20% of iodine was in the readily available phase (water soluble and exchangeable fractions).132 The high level of iodine in the organic fraction is attributed to the ability of the humic substance to fix iodine,133 in which a microorganism was thought to participate.134 Solvent extraction and HPLC were used to study the chemical species of iodine in soil solution.112,134 In a similar way to in water, iodine exists as iodide, iodate and organic iodine, in which the iodine is mainly bound with humic substance.
Future perspectives
Although much progress has been made in the chemical speciation study of trace elements by NATs, quite a number of issues in this regard still remain to be addressed to elucidate the mechanism of trace elements in life activity, e.g., whether one or another element can combine the biological macromolecules, e.g., protein, DNA or receptor molecule? And if yes, how? Further, where is (are) the combination position(s) of trace elements bound to macromolecules? What is the molecular structure of metal-bound proteins? A new scientific frontier “metalloproteomics” focusing on the above issues is being formed. In this new field the nuclear analytical techniques with high sensitivity, good accuracy, high space resolution and in situ and on line analytical ability will play a unique role.Acknowledgements
This work is supported by National Natural Science Foundation of China (Major Project, 19935020 and 20071032) and Chinese Academy of Sciences (KJCX-N01). R. Lobinski is highly appreciated for his encouragement of our preparation of this manuscript.References
- C. K. Jayawickreme and A. Chatt, Biol. Trace Elem. Res., 1990, 30, 503–513 Search PubMed.
- Z. F. Chai, X. Y. Mao, Z. H. Hu, Z. Y. Zhang, C. Y. Chen, W. Y. Feng, S. M. Hu and H. Ouyang, Anal. Bioanal. Chem., 2002, 372, 407–411 CrossRef CAS.
- Z. F. Chai, Z. Y. Zhang, W. Y. Feng, N. Q. Liu, P. Q. Zhang, X. Y. Mao, C. Y. Chen, F. L. Li, W. K. Zhong, J. J. Zhao, H. Q. Xiao, F. Zhang, J. Liu, D. D. Xu, Y. X. Gao and H. Ouyang, in Proceedings of the International Symposium on Bio-Trace Elements, ed. S. Enomoto and Y. Seko, Riken and Yamanashi, Japan, 2003, pp. 22–26 Search PubMed.
- Z. F. Chai, X. Y. Mao, Y. Q. Wang, J. X. Sun, Q. F. Qian, X. L. Hou, P. Q. Zhang, C. Y. Chen, W. Y. Feng, W. J. Ding, X. L. Li, C. S. Li and X. X. Dai, Fresenius’ J. Anal. Chem., 1999, 363, 477–480 CrossRef CAS.
- D. M. Templeton, F. Ariese, R. Cornelis, L. G. Danielsson, H. Muntau, H. P. Van Leeuwen and R. Lobinski, Pure Appl. Chem., 2000, 72, 1453–1470 Search PubMed.
- Z. F. Chai, J. Nucl. Radiochem. Sci., 2000, 1, 19–22 Search PubMed.
- Z. Y. Zhang, Y. Q. Wang, F. L. Li, H. Q. Xiao and Z. F. Chai, J. Radioanal. Nucl. Chem., 2002, 252, 461–465 CrossRef CAS.
- Z. Y. Zhang, F. L. Li, L. Xu, N. Liu, H. Q. Xiao and Z. F. Chai, J. Radioanal. Nucl. Chem. Search PubMed , in the press.
- Z. Y. Zhang, Y. Q. Wang, F. L. Li and Z. F. Chai, J. Radioanal. Nucl. Chem., 2001, 247, 557–560 CrossRef CAS.
- Z. Y. Zhang, Y. Q. Wang, F. L. Li and J. X. Sun, Chinese J. Anal. Chem., 2001, 29, 494–498 Search PubMed.
- Z. Y. Zhang, Y. Q. Wang, J. X. Sun and F. L. Li, Chinese Sci. Bull., 2000, 45, 1497–1499 Search PubMed.
- Z. Y. Zhang, Y. Q. Wang, J. X. Sun and F. L. Li, Chinese Rare Earths, 2000, 21, 42–45 Search PubMed.
- Y. Q. Wang, J. X. Sun, Z. Y. Zhang, P. Jiang, F. Q. Guo and H. M. Chen, Nucl. Technol., 2001, 23, 721–726 Search PubMed.
- F. L. Li, Y. Q. Wang, Z. Y. Zhang, J. X. Sun and Z. F. Chai, J. Radioanal. Nucl. Chem., 2002, 251, 437–441 CrossRef CAS.
- F. L. Li, Y. Q. Wang, Z. Y. Zhang, J. X. Sun and Z. F. Chai, Acta Sci. Nat. Univ. Pekin., 2001, 37, 278–280 Search PubMed.
- C. Y. Chen, P. Q. Zhang and Z. F. Chai, Anal. Chim. Acta, 2001, 439, 19–27 CrossRef CAS.
- Z. H. Feng, X. Wang, S. X. Zhang, L. Z. An, J. X. Zhang and H. Y. Yao, Chin. J. Nucl. Med., 2001, 21, 111–114 Search PubMed.
- M. Rayman, The Lancet, 2000, 356, 233–241 Search PubMed.
- J. Neve, J. Cardiovas. Risk, 1996, 3, 42–47 Search PubMed.
- L. C. Clark, G. F. Combs, Jr., B. W. Turnbull, B. Dalkin and A. Krongrad, JAMA, 1996, 276, 1957–1963 Search PubMed.
- G. N. Schrauzer and J. Sacher, Chem.-Biol. Interact., 1994, 91, 199–205 Search PubMed.
- C. Y. Chen, J. Y. Zhou, H. B. Xu, Y. Jiang and G. F. Zhu, Biol. Trace Elem. Res., 1997, 59, 187–193 Search PubMed.
- C. D. Thomson, Analyst, 1998, 123, 827–831 RSC.
- D. Behne, C. Hammel, H. Pfeifer, D. Rothlein, H. Gessner and A. Kyriakopoulos, Analyst, 1998, 123, 871–873 RSC.
- R. Lobinski, J. S. Edmonds, K. T. Suzuki and P. C. Uden, Pure Appl. Chem., 2000, 72, 447–461 Search PubMed.
- A. J. Blotcky, A. Ebrahim and E. P. Rack, Anal. Chem., 1988, 60, 2734–2737 CrossRef CAS.
- A. J. Blotcky, G. T. Hansen, N. Borkar, A. Ebrahim and E. P. Rack, Anal. Chem., 1987, 59, 2063–2066 CrossRef CAS.
- A. J. Blotcky, G. T. Hansen, L. R. Opelanio-Buencamino and E. P. Rack, Anal. Chem., 1985, 57, 1937–1941 CrossRef CAS.
- P. C. Uden, Anal. Bioanal. Chem., 2002, 373, 422–431 CrossRef CAS.
- T. C. Stadtman, Annu. Rev. Biochem., 1996, 65, 83–100 CrossRef CAS.
- C. Y. Chen, P. Q. Zhang, X. L. Hou and Z. F. Chai, Biol. Trace Elem. Res., 1999, 71–72, 131–138 Search PubMed.
- D. Behne, C. Weiss-Nowak, M. Kalcklösch, C. Westphal, H. Gessner and A. Kyriakopoulos, Biol. Trace Elem. Res., 1994, 43–45, 287–297 Search PubMed.
- C. Y. Chen, X. L. Lu, P. Q. Zhang, X. L. Hou and Z. F. Chai, J. Radioanal. Nucl. Chem., 2000, 244, 199–203 CrossRef CAS.
- C. K. Jayawickreme and A. Chatt, J. Radioanal. Nucl. Chem., 1987, 110, 583–593 CAS.
- A. Peng, Y. Xu and Z. J. Wang, Biol. Trace Elem. Res., 1999, 70, 117–125 Search PubMed.
- Y. X. Gao, C. Y. Chen, Z. F. Chai, J. J. Zhao, J. Liu, P. Q. Zhang, W. He and Y. Y. Huang, Analyst, 2002, 127, 1700–1704 RSC.
- C. Y. Chen, P. Q. Zhang, Z. F. Chai, G. C. Li and Y. Y. Huang, Sci. China (English version), 2000, 43, 88–92 Search PubMed.
- Y. X. Gao, C. Y. Chen, P. Q. Zhang, Z. F. Chai, W. He and Y. Y. Huang, Anal. Chim. Acta, 2003, 485, 131–137 CrossRef CAS.
- C. Y. Chen, J. J. Zhao, P. Q. Zhang, Y. X. Gao and Z. F. Chai, J. Nucl. Radiochem., 2002, 24, 138–143 CAS (in Chinese).
- C. Y. Chen, P. Q. Zhang, X. L. Hou and Z. F. Chai, BBA, 1999, 1427, 205–215 Search PubMed.
- C. Y. Chen, J. J. Zhao, P. Q. Zhang and Z. F. Chai, Anal. Bioanal. Chem., 2002, 372, 426–430 CrossRef CAS.
- C. Y. Chen, P. Q. Zhang, X. L. Lu, X. L. Hou and Z. F. Chai, Fresenius’ J. Anal. Chem., 1999, 363, 512–516 CrossRef CAS.
- C. Y. Chen, Y. Liu, J. Y. Zhou, S. S. Qu and H. B. Xu, Biol. Trace Elem. Res., 1997, 60, 115–122 Search PubMed.
- D. Behne, S. Scheid, A. Kyriakopoulos and H. Hilmert, BBA, 1990, 1033, 219–225 Search PubMed.
- D. Behne, C. Hammel, H. Pfeifer, D. Rothlein, H. Gessner and A. Kyriakopoulos, Analyst, 1998, 123, 871–3 RSC.
- K. Hwang and J. A. Milner, Biol. Trace Element Res., 1996, 51, 133–147 Search PubMed.
- T. Kato, R. Read, J. Rozga and R. F. Burk, Am. J. Physiol., 1992, 262, G854–858 Search PubMed.
- R. F. Burk and P. E. Gregory, Arch. Biochem. Biophys., 1982, 213, 73–80 CrossRef CAS.
- R. F. Burk, K. E. Hill and A. K. Motley, J. Nutr., 2003, 133, 1517S–1520S CAS.
- C. Sasakura and K. T. Suzuki, J. Inorg. Biochem., 1998, 71, 159–162 CrossRef CAS.
- Y. Kobayashi, Y. Ogra and K. T. Suzuki, J. Chromatogr. B, 2001, 760, 73–81 CrossRef CAS.
- T. Lindemann and H. Hintelmann, Anal. Bioanal. Chem., 2002, 372, 486–90 CrossRef CAS.
- J. R. Encinar, R. Ruzik, W. Buchmann, J. Tortajada, R. Lobinski and J. Szpunar, Analyst, 2003, 128, 220–224 RSC.
- M. A. Quijano, P. Moreno, A. M. Gutierrez, M. C. Perez-Conde and C. Camara, J. Mass Spectrom., 2000, 35, 878–884 CrossRef CAS.
- A. R. Byrne and L. Kosta, Talanta, 1973, 21, 1083–1090 CrossRef.
- I. Falnoga, I. Kregar, M. Skreblin, M. Tusek-Znidaric and P. Stegnar, Biol. Trace Elem. Res., 1993, 37, 71–83 Search PubMed.
- S. Bhattacharya, S. Bose, B. Mukhopadhyay, D. Sarkar, D. Das, J. Bandyopadhyay, R. Bose, C. Majumdar, S. Mondal and S. Sen, Biometals, 1997, 10, 157–162 CrossRef CAS.
- J. Gailer, G. N. George, I. J. Pickering, S. Madden, R. C. Prince, E. Y. Yu, M. B. Denton, H. S. Younis and H. V. Aposhian, Chem. Res. Toxicol., 2000, 13, 1135–1142 CrossRef CAS.
- W. Y. Feng, Q. F. Qian, W. J. Ding and Z. F. Chai, Biol. Trace Elem. Res., 1999, 71–72, 121–129 Search PubMed.
- W. Y. Feng, Q. F. Qian, W. J. Ding and Z. F. Chai, Metabolism, 2001, 50, 1168–1174 CrossRef CAS.
- W. Y. Feng, Q. F. Qian, W. J. Ding and Z. F. Chai, J. Radioanal. Nucl. Chem., 2000, 244, 321–325 CrossRef CAS.
- W. Y. Feng, Q. F. Qian, W. J. Ding and Z. F. Chai, Chinese Nucl. Chem. Radiochem., 1999, 21, 147–152 Search PubMed.
- W. Y. Feng, B. Li, J. Liu, Z. F. Chai, P. Q. Zhang, Y. X. Gao and J. J. Zhao, Anal. Bioanal. Chem., 2003, 375, 363–368 CAS.
- W. J. Ding, Q. F. Qian, X. L. Hou, W. Y. Feng, C. Y. Chen, Z. F. Chai, B. R. Zhang and K. Wang, Biol. Trace Elem. Res., 2002, 88, 193–199 Search PubMed.
- W. J. Ding, Q. F. Qian, X. L. Hou, W. Y. Feng, Z. F. Chai and K. Wang, J. Radioanal. Nucl. Chem., 2000, 244, 259–262 CrossRef CAS.
- J. Liu, PhD Thesis, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 2003.
- S. Chen and J. Q. Gao, J. Assoc. Off. Anal. Chem., 1993, 76, 1193–1205 Search PubMed.
- P. Kömp and M. S. McLachlan, Environ. Sci. Technol., 1997, 31, 886–890 CrossRef.
- E. Atlas and C. S. Glam, Science, 1981, 211, 163–166 CAS.
- K. Gustavson and P. Jonsson, Mar. Pollut. Bull., 1999, 38, 723–736 CrossRef CAS.
- D. Haynes, P. Rayment and D. Toohey, Mar. Pollut. Bull., 1996, 32, 823–827 CrossRef CAS.
- W. H. Newsome, P. Andrews and H. B. S. Conacher, J. AOAC. Int., 1993, 76, 703–706 CAS.
- M. Kawano, Y. Tanaka, J. Falandysz and R. Tatsukawa, Bromatol. Chem. Toksykol., 1997, 30, 87–94 Search PubMed.
- D. Haynes, P. Mosse and G. Levay, Mar. Pollut. Bull., 1995, 30, 463–469 CrossRef CAS.
- K. Martinsen, A. Kringstad and G. E. Carlberg, Water Sci. Technol., 1988, 20, 13–24 CAS.
- J. W. Kiceniuk, J. Holzbecher and A. Chatt, Environ. Pollut., 1997, 97, 205–211 CrossRef CAS.
- I. Watanabe, T. Kashimono and M. Kawano, Chemosphere, 1987, 16, 847–857 CrossRef CAS.
- J. Pellinen and R. Soimasuo, Sci. Total. Environ. Suppl., 1993, 1247–1256 Search PubMed.
- M. Kähkönen, K. P. Suominen, P. K. G. Manninen and M. S. Salkinoja-Salonen, Environ. Sci. Technol., 1998, 32, 1741–1746 CrossRef.
- G. Lunde and J. Gether, AMBIO, 1976, 5, 180–183 Search PubMed.
- D. Xu, W. Zhang, L. Deng, Z. Chai and X. Mao, Environ. Sci. Technol., 2003, 37, 1–6 CrossRef CAS.
- D. Xu, Z. Chai, H. Zhang, X. Mao, H. Ouyang and H. Sun, Anal. Bioanal. Chem. Search PubMed , in the press.
- H. Palm and R. Lammi, Environ. Sci. Technol., 1995, 29, 1722–1727 CAS.
- H. T. Kankaanpää, M. A. Laurén, R. J. Saares, L. V. Heitto and Ü. K. Suursaar, Environ. Sci. Technol., 1997, 31, 96–104 CrossRef.
- A. Kostamo, M. Vilianen, J. Pellinen and J. Kukkonen, Chemosphere, 2000, 41, 1733–1740 CrossRef CAS.
- US Environmental Protection Agency, Method for total organic halides (TOX) by neutron activation analysis. Revision 0, 9022, September, 1986.
- US Environmental Protection Agency, Method for extractable organic halides (EOX). Revision 2, 9020, September, 1994.
- M. Concepción and C. López, Environ. Int., 2003, 28, 751–759 CrossRef.
- J. Gether, G. Lunde and E. Steinnes, Anal. Chim. Acta, 1979, 108, 137–147 CrossRef CAS.
- F. Wulff, L. Rahm, P. Jonsson, L. Brydsten, T. Ahi and A. Granmo, AMBIO, 1993, 22, 27–31 Search PubMed.
- K. Laniewski, J. Dahlén, H. Borén and A. Grimvall, Chemosphere, 1999, 38, 771–782 CrossRef CAS.
- I. Watanabe, T. Kashimoto, M. Kawano and R. Tatsukawa, Chemosphere, 1987, 16, 849–857 CrossRef CAS.
- US Environmental Protection Agency, Method for extractable organic halides (EOX) in soil. Revision 0, 9023, Dec. 1996.
- S. B. Christina, J. W. Kiceniuk and A. Chatt, Biol. Trace Elem. Res., 1999, 71-72, 149–166 Search PubMed.
- D. Xu, PhD Thesis, Institute of High Energy Physics, Chinese Academy Sciences, China, 2003.
- B. G. Loganathan, K. Kannan, I. Watanabe, M. Kawano, K. Irvine, S. Kumar and H. C. Sikka, Environ. Sci. Technol., 1995, 29, 1832–1838 CAS.
- K. C. Kannan, M. Kawano, Y. Kashima, M. Matsui and J. P. Giesy, Environ. Sci. Technol., 1993, 33, 1004–1008 CrossRef.
- D. Xu, W. Zhang, L. Deng, Z. Chai, X. Mao and Z. Zhang, J. Radiochem. Nucl. Chem., 2003 Search PubMed , in the press.
- J. K. Jokela and M. Salklnoja-Salonen, Environ. Sci. Technol, 1992, 26, 1190–1197 CAS.
- J. Hemming and K. J. Lehtinen, Nord. Pulp Pap. Res. J., 1988, 4, 185–190 Search PubMed.
- C. Wesén, G. E. Carlberg and K. Martinsen, AMBIO, 1990, 19, 36–38 Search PubMed.
- R. J. Norstrom, A. P. Glilman and D. J. Hallett, Sci. Total Environ., 1981, 20, 217–230 CrossRef CAS.
- G. W. Gribble, J. Chem. Educ., 1994, 71, 907–911 CAS.
- G. Lunde and E. Steinnes, Environ. Sci. Technol., 1975, 9, 155–157 CAS.
- A. Grimvall, in Proceedings of the International Conference on Naturally Produced Organohalogens, Formation, Occurrence, Characteristics and Environmental Significance, September 14–17, 1993, Delft, The Netherlands, p. 1.
- C. Johansson, I. Pavasars, H. Borén, A. Grimvall, O. Dahlman, R. Mörck and A. Reimann, Environ. Int., 1994, 20, 103–111 CrossRef CAS.
- V. Niedan, I. Pavasars and G. Öberg, Chemosphere, 2000, 41, 779–786 CrossRef CAS.
- G. T. F. Wong, Rev. Aquatic Sci., 1991, 4, 45–73 Search PubMed.
- X. L. Hou, H. Dahlgaard, B. Rietz, U. Jacobsen, S. P. Nielsen and A. Aarkrog, Anal. Chem., 1999, 71, 2745–2750 CrossRef CAS.
- X. L. Hou, H. Dahlgaard and S. P. Nielsen, Mar. Chem., 2001, 74, 145–155 CrossRef CAS.
- V. W. Truesdale, G. Nausch and A. Baker, Mar. Chem., 2001, 74, 87–98 CrossRef CAS.
- C. Reifenhauser and K. G. Heumann, Fresenius’ J. Anal. Chem., 1990, 336, 559–563 CrossRef.
- M. L. A. M. Campos, P. D. Nightingale and T. D. Jickelis, Tellus, 1996, 48B, 106–114 Search PubMed.
- S. Solomon, S. R. Garcia and A. R. Ravishankara, J. Geophys. Res., 1994, 99, 20491–20499 CrossRef.
- C. Schall, K. G. Heumann and G. O. Kirst, Fresenius’ J. Anal. Chem., 1997, 359, 298–305 CrossRef CAS.
- H. Noguchi and M. Murata, J. Environ. Radiat., 1988, 7, 65–74 Search PubMed.
- H. E. Gabler and K. G. Heumann, Fresenius’ J. Anal. Chem., 1993, 345, 53–59 CAS.
- S. Yoshida and Y. Muramatsa, J. Radioanal. Nucl. Chem., Articles, 1995, 196, 295–302 Search PubMed.
- K. A. Rahn, R. D. Borys and R. A. Duce, Science, 1976, 192, 549–550 CAS.
- X. L. Hou, C. F. Chai, Q. F. Qian, X. J. Yan and X. Fan, Sci. Total Environ., 1997, 204, 215–221 CrossRef.
- X. L. Hou, X. Y. Yan and C. F. Chai, J. Radioanal. Nucl. Chem., 2000, 245, 461–167 CrossRef CAS.
- C. B. Coulson, Chem. Ind., 1953, 38, 997–998 Search PubMed.
- X. J. Wang, Study on content, distribution and chemical state of iodine in Laminaria japonic, Masters Thesis, Institute of Oceanography, Chinese Academy of Science, China, 1995.
- J. S. Edomons and M. Morita, Pure Appl. Chem., 1998, 70, 1567–1584 CAS.
- X. L. Hou, C. F. Chai, Q. F. Qian, C. S. Li and Q. Chen, Biol. Trace Elem. Res., 1997, 56, 225–230 Search PubMed.
- X. L. Hou, C. Y. Chen, Q. F. Qian and C. F. Chai, Biol. Trace Elem. Res., 1999, 69, 69–76 Search PubMed.
- J. C. Meurizis, C. Nicoles and J. Michelet, J. Chromatogr., 1982, 150, 129–135 CrossRef CAS.
- B. Michalke, P. Schramel and H. Witte, Biol. Trace Elem. Res., 2000, 78, 67–79 Search PubMed.
- M. Leiterer, D. Truckenbrodt and K. Franke, Eur. Food Res. Technol., 2001, 213, 150–153 Search PubMed.
- K. Takatera and T. Watanabe, Anal. Chem., 1993, 65, 759–762 CrossRef CAS.
- X. L. Hou, H. C. L. Fogh, J. Kucera, K. G. Andersson, H. Dahlgaard and S. P. Nielsen, Sci. Total Environ., 2003, 308, 97–109 CrossRef CAS.
- G. Radlinger and K. G. Heumann, Environ. Sci. Technol., 2000, 34, 3922–3936 CrossRef.
- K. G. Heumann, R. Radlinger, M. Erbes, I. Heiber, U. Obst, Z. Filip and H. Claus, Acta Hydrochim. Hydrobiol., 2000, 28, 193–201 CrossRef CAS.
- K. Yuita, Soil Sci. Plant Nutr., 1992, 38, 281–287 Search PubMed.
| This journal is © The Royal Society of Chemistry 2004 |