Hyphenation of high performance liquid chromatography with sector field inductively coupled plasma mass spectrometry for the determination of ultra-trace level anionic and cationic arsenic compounds in freshwater fish†
Jian
Zheng‡
and
Holger
Hintelmann
*
Department of Chemistry, Trent University, 1600 West Bank Drive, Peterborough, Ontario, Canada K9J 7B8. E-mail: hhintelmann@trentu.ca
First published on 16th December 2003
Abstract
We describe a hyphenation technique between HPLC and ICP-SFMS for ultra-trace arsenic speciation analysis. Exceptional analytical performance was achieved using a MicroMist nebulizer preceded by a high-pressure splitter. Despite a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶7.5 flow splitting, the detection limits in the range of 1.2 to 2.4 pg mL−1 were about two times lower than those obtained with a concentric nebulizer without any flow splitting, demonstrating the applicability of coupling conventional HPLC system (1.5 mL min−1 eluent flow) with microflow nebulizer for ultra-trace arsenic speciation analysis. This set-up offers an advantage for on-line fraction collection for either multidimensional chromatographic separation of co-eluting As compounds or for structural identification of unknown compounds without sacrificing analytical sensitivity. In addition, this system showed good accuracy and repeatability. The method was applied to the determination of arsenic compounds in freshwater fish samples from an arsenic-rich lake, Moira Lake, Canada. Using cation-exchange chromatography, tetramethylarsonium ion (Tetra) was detected in freshwater fish samples for the first time. Moreover, in pumpkinseed, Tetra was found to be the major arsenic species, indicating that the biomethylation pathway in freshwater ecosystems may include the tetramethyl stage.
∶7.5 flow splitting, the detection limits in the range of 1.2 to 2.4 pg mL−1 were about two times lower than those obtained with a concentric nebulizer without any flow splitting, demonstrating the applicability of coupling conventional HPLC system (1.5 mL min−1 eluent flow) with microflow nebulizer for ultra-trace arsenic speciation analysis. This set-up offers an advantage for on-line fraction collection for either multidimensional chromatographic separation of co-eluting As compounds or for structural identification of unknown compounds without sacrificing analytical sensitivity. In addition, this system showed good accuracy and repeatability. The method was applied to the determination of arsenic compounds in freshwater fish samples from an arsenic-rich lake, Moira Lake, Canada. Using cation-exchange chromatography, tetramethylarsonium ion (Tetra) was detected in freshwater fish samples for the first time. Moreover, in pumpkinseed, Tetra was found to be the major arsenic species, indicating that the biomethylation pathway in freshwater ecosystems may include the tetramethyl stage.
Introduction
Arsenic speciation has been extensively studied, owing to its complex environmental chemistry, the diverse toxicity of compounds, and profound metabolism in humans. Although the environmental cycling of arsenic in marine ecosystem is well established,1,2 the fate of arsenic in the terrestrial environment is still to be elucidated. Our knowledge about the arsenic compounds in such systems is scarce, due largely to the lack of sensitive analytical methods. The much lower arsenic concentrations in terrestrial environment require analytical methods with detection limits at sub-ng mL−1 and pg mL−1 levels to identify minor arsenic compounds and to cover knowledge gaps about arsenic compounds in the terrestrial ecosystem.3Double focusing sector field ICP-MS (ICP-SFMS) can be operated at low mass resolution (LMR) or at high mass resolution (HMR), providing a general method to overcome limitations from spectroscopic interferences. In addition, in the low resolution mode, the sensitivity is about two orders of magnitude better than that of quadrupole ICP-MS.4 Therefore, the combination of HPLC with ICP-SFMS should be an ideal avenue for interference-free arsenic speciation at trace or ultra-trace level. However, very little work has been published for this combination.5
Given the large number of arsenic compounds present in the environment, it is difficult to separate all As compounds existing in a given sample. It is often necessary to collect the fractions of either unknown peaks or co-eluted As containing peaks for further chromatographic separation or for structural identification with electrospray mass spectrometry (ES-MS). Off-line fraction collection is time-consuming or even inaccurate in case of matrix induced retention time shift. Therefore, on-line fraction collection is preferred, which can be done by connecting a splitter between the HPLC column outlet and the inlet of ICP-MS nebulizer. However, the split of sample mass could result in the deterioration of sensitivity.
In this work, we report the coupling of a conventional HPLC system to ICP-SFMS for the speciation of eight of the most commonly encountered arsenic compounds, arsenite, arsenate, monomethylarsonic acid (MMA), dimethylarsinic acid (DMA), arsenobetaine (AB), arsenocholine (AC), trimethylarsine oxide (TMAO) and tetramethylarsonium ion (Tetra). The purpose of this study is to develop a sensitive and accurate analytical method for ultra-trace arsenic speciation in terrestrial environmental samples, which will also enable the on-line fraction collection of As containing peaks for multidimensional chromatographic separation or ES-MS detection. For this purpose, three nebulizers, namely, conventional concentric nebulizer, MicroMist nebulizer and an Aridus sample introduction system were investigated and compared in terms of the detection limits, precision and the band dispersion of the chromatographic peak. The developed method was applied to the study on arsenic species distribution in fresh water fish in an arsenic-rich lake, Moira Lake, in Canada.
Experimental
Chemical and reagents
All commercial chemicals were of analytical-reagent grade and were used without further purification. Sodium arsenite [As(III)], Na2HAsO4·7H2O [As(V)], and dimethylarsinic acid (DMA) were purchased from Sigma (St. Louis, MO, USA). Monosodium acid methane arsenate (MMA) was obtained from Chem Service (West Chester, PA, USA). Arsenobetaine bromide (AB), arsenocholine bromide (AC), trimethylarsine oxide (TMAO), and tetramethylarsonium ion (Tetra) were purchased from Tri Chemical Laboratory Inc. (Yamanashi, Japan).Instrumentation
The ICP-MS instrument used was a Finnigan sector field high resolution ICP-MS (Element2) (Thermo Finnigan, Bremen, Germany). The sample introduction system included a Scott-type spray chamber fitted with a concentric nebulizer. The chromatographic system employed was a Waters 2690 separation module (Mississauga, ON, Canada) with a variable sample loop (5–250 µl). Two HPLC columns were used in this work: (1) PRP-X100 anion exchange column (250 × 4.6 mm, particle size 10 µm, Hamilton, Reno, NA, USA); and (2) Zorbax 300-SCX cation exchange column (150 × 4.6 mm, particle size 5 µm, Agilent Technologies, Mississauga, ON, Canada). The chromatographic system was interfaced with the ICP-MS instrument using a 120 mm PEEK capillary tubing (0.25 mm id) to connect the column outlet to the nebulizer inlet. When an Aridus sample introduction system (with membrane desolvation) (CETAC, Technologies, Omaha, Nebraska, USA) was used, it was necessary to split the HPLC flow (1.5 mL min−1), to accommodate the 100 µl min−1 maximum uptake rate of the low-flow micronebulizer in the Aridus. Similarly, when a MicroMist nebulizer was used, the HPLC flow rate was split to 200 µl min−1. The columns were conditioned by passing at least 100 ml of mobile phase through the column before injection of the As standards and samples. The chromatograms were exported and processed using Grams 32 software. Quantifications were performed in the peak area mode. The operating conditions for HPLC-ICP-SFMS are summarized in Table 1.| Plasma parameters | |
| Rf power | 1250 W |
| Carrier gas flow rate | 0.93 l min−1 |
| Measurement parameters | |
| Acquisition mode | Escan |
| Monitored isotopes | 75As, 77Se, |
| Mass window | 10% for low resolution; 120% for high resolution |
| Sample time | 150 ms for low resolution; 20 ms for high resolution |
| Samples per peak | 100 for low resolution; 30 for high resolution |
| Passes | 1 |
| Integration type | Average for low resolution; integral for high resolution |
| Resolution (m/Δm) | 300, 10000 |
| HPLC | |
| 1. Anion-exchange column | Hamilton PRP-X100 (25 cm × 4.6 mm, 10 µm) 20 mM ammonium dihydrogenphosphate, pH 5.6 |
| Mobile phase | 1.5 ml min−1 |
| 2. Cation exchange column | Zorbax 300-SCX (15 cm × 4.1 mm, 5 µm) |
| Mobile phase | 20 mM pyridine, pH 2.31, 1.5 ml min−1 |
| Injection volume | 100 µl |
| Column temperature | Ambient |
Determination of total As in fish samples
Aliquots (ca. 0.5 g) of the fish tissues (muscle, liver, heart, etc.) were weighed into 20 ml glass tubes. A mixture of HNO3/H2SO4 (5 ml, 7∶3, v/v) was added and the glass tube was covered with a glass marble. After 30 min pre-digestion at room temperature the tube was placed into an aluminium heating block on a hot-plate. The digestion was carried out at 120 °C for ca. 4 h. After cooling to room temperature the digest was diluted to 20 ml, the internal standard was added (25 ng mL−1 germanium) and the sample analysed by ICP-SFMS. The procedure was validated using reference material NIST 1566b, oyster tissue. The measured value of 7.21 ± 0.38 µg g−1
(n
= 3) was statistically not different from the certified As concentration of 7.65 ± 0.65 µg g−1.
Extraction of arsenic species from fresh water fish
Fish tissues (muscle, liver, kidney, brain and heart) were dissected from individual fish collected from Moira Lake, Ontario, Canada. Fresh fish tissue samples were weighed, homogenized with methanol/water (1∶1, v/v; sample/solvent mixing ratio of approximately 1∶10, w/v) using an Omni-mixer (Ivan Sorvall, Inc., Norwalk, Conn. USA), sonicated for 20 min, and centrifuged (4000 rpm × 20 min) to obtain the extract. The extraction process was repeated three times, and the extracts were combined, evaporated to dryness with a Rotavapor under reduced pressure, and dissolved in 5 mL water. The samples were then centrifuged (5000 rpm, 15 min) and filtered through 0.45 µm cellulose nitrite filters. Total As concentration in the extracts was determined with ICP-SFMS operated at high resolution mode. Aliquots (100 µl) of these solutions were then directly chromatographed.. The extraction efficiencies in the four investigated fish species ranged from 67–89% of the total arsenic (Table 2). The remaining arsenic may be bound to the –SH groups of cytosolic proteins or may correspond to species such as arsenolipids, which would not be soluble in a MeOH–H2O mixture.
| Species | Common name | Composite samples | Length/cm | Weight/g | As in mg kg−1 (range) | Extraction yield (%) |
|---|---|---|---|---|---|---|
| Esox lucius | Northern pike | 2 | 48–51 | 750–1000 | 0.41 (0.38–0.45) | 70.5 ± 8.1 |
| Micropterus salmoides | Largemouth bass | 1 | 29 | 650 | 0.20 (---) | 88.7 ± 7.6 |
| Perca flavescens | Yellow perch | 4 | 12–16 | 19.8–50.1 | 0.09 (0.05–0.14) | 77.8 ± 7.2 |
| Lepomis gibbosus | Pumpkinseed | 4 | 12–15 | 45.3–50.3 | 0.39 (0.35–0.43) | 66.8 ± 5.9 |
Results and discussion
Two chromatographic separation systems (anion-exchange and cation-exchange) were employed in this study. The selection of the chromatographic system was based on its resolving power and good matrix tolerance. A PRP-X100 anion-exchange column with a phosphate buffered mobile phase was employed to separate As(III), DMA, MMA and As(V) (Fig. 1A). This system is in principle also capable of separating arsenosugars as demonstrated by Raber et al.6 The cationic arsenic compounds, such as arsenobetaine (AB), arsenocholine (AC), trimethylarsine oxide (TMAO) and tetramethylarsonium cation (Tetra) were resolved with a Zorbax 300 SCX cation-exchange column using 20 mM pyridine as the mobile phase (Fig. 1B). Details about the chromatographic systems were summarized in Table 1. For As species analysis in freshwater fish samples, the ICP-SFMS was operated in low resolution mode. | ||
| Fig. 1 Separation and determination of anionic and cationic arsenic compounds (0.5 ng ml−1) by HPLC-ICP-SFMS (with conventional concentric nebulizer) at low resolution (m/Δm = 300). A: anion-exchange chromatography; B: cation-exchange chromatography. Chromatographic conditions are summarized in Table 1. | ||
Coupling of HPLC to ICP-SFMS using different nebulizers
Considering the mobile phase flow rate of the selected HPLC separation systems (1.5 mL min−1) a conventional concentric nebulizer was initially investigated to couple the HPLC to the ICP-SFMS. The analytical figures of merit obtained with this standard set-up are provided in the electronic supplementary information.† Obtained detection limits for arsenic compounds ranged from 2.5 to 5.8 pg mL−1 or 0.25 to 0.58 pg absolute (peak area). The application of an Aridus desolvation system was investigated as an interface for coupling HPLC to ICP-MS by splitting the 1.5 ml min−1 flow of the HPLC to accommodate the 100 µl min−1 sample uptake limit of the Aridus. The signal to noise ratios significantly improved by a factor of two compared to the concentric nebulizer, even after a 1∶15 flow splitting. However, severe peak broadening for all investigated As compounds was observed as well and baseline separation was not obtained for all species. The deterioration in resolution was attributed to the much larger internal volume (ca. 400 mL) of spray chamber and desolvation tube of the Aridus system.
Eventually, a high efficiency nebulizer was evaluated as an interface between HPLC and ICP-SFMS. The reduced sample uptake rate of the MicroMist micro-uptake nebulizer (0.2 ml min−1) necessitated a solvent split of the HPLC mobile phase flow (1.5 mL min−1) at a 1∶7.5 split ratio. The nebulizer gas flow was re-optimized and set to a flow rate of 0.93 L min−1, identical to that established for the concentric nebulizer. Analytical figures of merit for the MicroMist nebulizer set-up are shown in Table 3. Excellent detection limits, ranging from 1.2 to 2.4 pg ml−1
(or 0.12 to 0.24 pg absolute) were obtained in peak area mode. These detection limits are approximately two times lower than those obtained with the concentric nebulizer (electronic supplementary information),† suggesting that the superior nebulization efficiency of the MicroMist nebulizer more than compensates for the analyte loss from the 1∶7.5 flow splitting. This set-up proved to be very stable and dependable. Excellent repeatability was found with RSDs of less than 2%
(peak area mode) and 2.5%
(peak height mode) for a standard solution at a concentration of 0.5 ng mL−1. Calibration curves were linear with correlation factors close to 1 for each arsenic compounds over the investigated work concentration range (0.05–20 ng mL−1).
| Analyte | LOD/pg mL−1 | LOD/pg | Repeatability (%) | Range/ng ml−1 | R 2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ab | Hc | A | H | A | H | A | H | ||
| a Detection limits were determined by making 6 repetitive injections (100 µl) of standards in the lower linear concentration range (0.05 ng mL−1 each) and were calculated from the standard deviations of the peak areas and peak heights (3σ), respectively. Repeatability was determined from peak areas and peak heights by calculating the relative standard deviation (RSD) of four successive analysis of a standard solution containing 0.5 ng mL−1 of each analyte. b Peak area. c Peak height. | |||||||||
| As(III) | 1.5 | 3.8 | 0.15 | 0.38 | 0.9 | 2.1 | 0.05–20 | 0.99998 | 0.99999 |
| DMA | 2.1 | 2.7 | 0.21 | 0.27 | 1.5 | 1.9 | 0.05–20 | 1.00000 | 1.00000 |
| MMA | 2.4 | 3.5 | 0.24 | 0.35 | 1.6 | 2.1 | 0.05–20 | 0.99999 | 0.99881 |
| As(V) | 2.2 | 3.8 | 0.22 | 0.38 | 1.1 | 1.8 | 0.05–20 | 0.99979 | 0.99992 |
| AB | 1.2 | 1.5 | 0.12 | 0.15 | 0.9 | 1.1 | 0.05–20 | 0.99992 | 0.99963 |
| TMAO | 1.3 | 1.8 | 0.13 | 0.18 | 1.1 | 1.3 | 0.05–20 | 0.99967 | 0.99963 |
| AC | 1.6 | 3.3 | 0.16 | 0.33 | 1.2 | 2.3 | 0.05–20 | 0.99982 | 0.99968 |
| Tetra | 1.6 | 1.9 | 0.16 | 0.19 | 1.2 | 1.3 | 0.05–20 | 1.00000 | 0.99960 |
As chloride may potentially interfere with the determination of As(V) using anion exchange chromatography, it may be necessary to determine some species in high resolution mode (m/Δm = 10,000) to resolve 40Ar35Cl+ from the 75As signal. To assess the value of high resolution acquisition, the cation-exchange and anion-exchange chromatographic systems were combined with ICP-SFMS operated in high resolution mode. The resulting analytical figures of merit can be found in the electronic supplementary information.† Compared with low resolution mode, the detection limits were ca. 30 to 80 times (with an average of 50 times) higher, but still at the sub-ppb level, which is comparable with most of the LODs reported for quadrupole ICP-MS. Therefore, this combination provides a viable and sensitive method for interference free trace level As speciation analysis.
Employing a MicroMist nebulizer has notable advantages. The use of conventional HPLC columns allows the injection of relatively large sample volumes (100 µl in this study), resulting in excellent concentration detection limits at the 1–2 pg mL−1 level, demonstrating that the necessary flow splitting does not compromise detection limits. This is, so far, the lowest detection limit reported for As speciation using HPLC based hyphenated techniques5,7–15 (details in electronic supplementary information),† allowing the speciation of As in terrestrial environmental samples at ultra-trace level without any sample pre-concentration. Due to the flow splitting, the solvent loading on the plasma was dramatically reduced, resulting in a better stability of the plasma, which makes it possible to use higher contents of organic solvent as the HPLC mobile phase, if needed. Another significant benefit using the solvent split is the possibility of on-line fraction collection. Only ca. 15% of the injected sample is diverted to the ICP-MS and 85% can be collected for further analysis, e.g., multidimensional chromatography or structure elucidation using ESI-MS/MS.
Determination of As compounds in freshwater fish
Due to historic mining operations, the Moira River system is contaminated with As, Cu and Co. This river supplies most of the water to the Moira Lake, resulting in a high As concentration in the lake. In our previous study,16 elevated levels of As in the surface water of up to 75 ng mL−1 in Moira River and 50 ng mL−1 in Moira Lake were detected, 98% of which was present as arsenate. Since no isobaric interferences were detected with the chromatographic system described, all fish and tissue samples were analyzed in low resolution mode. The total As concentrations in the muscle tissue of the 4 species of fish are summarized in Table 2 and were in the same range reported previously by Azcue and Dixon17, and comparable to results obtained in different freshwater environments.18–20Results of the speciation analysis are presented in Table 4. The percentage of each species represents the fraction of the total arsenic extracted with the methanol–water mixture. Representative chromatograms obtained from the extract of pumpkinseed were shown in Fig. 2. A variety of arsenic compounds, including As(III), As(V), MMA, DMA, AB, AC, TMAO and Tetra, are detected in the freshwater fish samples. Two unknown As containing species, one (U1) eluted after As(V) in AEC chromatogram, and the other (U2) eluted right after As(III) in CEC chromatogram, showing as a shoulder of As(III) peak, were also observed. Their identities remain unknown in the present study. Since As(III) and AB have the same retention time on the anion-exchange HPLC system (AEC) used, the first two peaks (cationic As and As(III)/AB) in the AEC chromatogram were collected employing the developed on-line fraction collection technique and subjected to a subsequent cation-exchange chromatography (CEC) analysis. The HPLC on-column recovery of the extracted arsenic, defined as the sum of individual arsenic compounds divided by the total arsenic in the extract ranged from 85 to 110% in the investigated fish samples.
 | ||
| Fig. 2 Chromatograms obtained from the extract of fresh water fish tissue (muscle), pumpkinseed, Lepomis gibbosus (Linnaeus). (A) Anion-exchange chromatography; (B) cation-exchange chromatography. The first two peaks in the anion-exchange chromatogram (A) were collected and subjected to a subsequent cation-exchange chromatography (B). | ||
| Species | As(III) | As(V) | MMA | DMA | AB | TMAO | AC | Tetra | Unknown |
|---|---|---|---|---|---|---|---|---|---|
| Esox lucius | 9.9 (9.3–10.4) | 2.4 (1.3–3.5) | 0.08 (0–0.17) | 46.0 (42.3–49.6) | 25.3 (21.4–29.1) | 0.25 (0–0.5) | 0.07 (0–0.14) | 10.2 (5.5–14.7) | 5.8 (1.6–10) |
| Micropterus salmoides | 16.5 | 8.8 | nd | 11.6 | 16.1 | 6.6 | 0.38 | 24.4 | 15.8 |
| Perca flavescens | 33.5 (28–39) | 39.6 (7.9–56.3) | 0.8 (0.16–1.2) | 4.1 (1.3–7.2) | 3.9 (0.6–8.2) | 5.0 (1.0–9.3) | 0.2 (0–0.7) | 6.6 (0.3–20.7) | 6.3 (0.8–18.2) |
| Lepomis gibbosus | 19.8 (17.1–22.4) | 18.9 (9.2–32.3) | 0.7 (0–2.2) | 4.6 (1.4–7.1) | 6.0 (4.0–9.4) | 6.9 (5.4–8.7) | 0.9 (0.8–1.1) | 34.9 (21.4–44.9) | 7.2 (3.2–11.6) |
It was found that the arsenic compound distribution patterns in Moira Lake fish were quite different from that in marine fish. In addition, differences are also shown among the investigated freshwater fish species. In contrast to the situation in marine organisms, where AB is the predominant species, in freshwater fish samples from Moira Lake, AB accounted for less than 10% of the total As in the muscle extracts of Yellow perch and Pumpkinseed. The distribution of As compounds in Largemouth bass is fairly even and although AB accounted for 16% of the total As in muscle extract, it cannot be regarded as a major species, because inorganic As [As(III)
+ As(V)], DMA, Tetra and the unknown species, respectively, accounted for 25.3, 11.6, 24.4 and 15.8% of the total As in the extract. In the yellow perch and Pumpkinseed, most of the extracted arsenic was present as inorganic arsenic compounds (39–73%). Interesting results are obtained with the predatory fish, Northern pike. It was found that DMA was the predominant As species (46%), although a relatively high concentration of AB (25.3%) was also detected. To shed more light in this unusual As distribution pattern, the distribution of arsenic species in different tissues (liver, kidney, heart and brain) of Northern pike was also investigated (electronic supplementary information).† It was shown that DMA was present as the major arsenic species not only in muscle tissue, but also in liver, kidney, heart and brain. A lower concentration of AB was detected in kidney, heart and brain tissues, compared with that in muscle and liver tissues. The ratio of liver∶muscle As concentration has been used to indicate the degree to which an organism is under As stress.17 In this work, we observed a ratio of 0.55 in Northern pike from Moira Lake, suggesting a relatively low level of As stress. Therefore, it seems that the extremely high content of DMA in Northern pike tissues is a result of natural metabolism, rather than an indication of detoxification. Fig. 3 shows the representative chromatograms obtained from the extract of kidney of Northern pike.
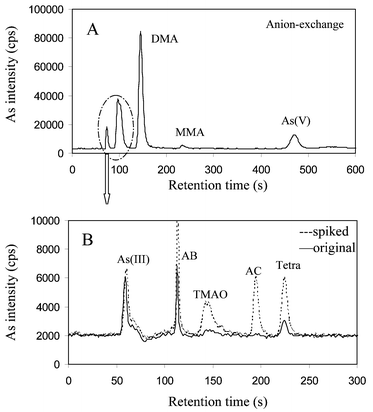 | ||
| Fig. 3 Chromatograms obtained from the extract of fresh water fish tissue (kidney), Esox lucius (Linnaeus). (A) Anion-exchange chromatography; (B) cation-exchange chromatography. The first two peaks in the anion-exchange chromatogram (A) were collected and subjected to a subsequent cation-exchange chromatography (B). For a clear identification of detected As compounds, a spike experiment with AB, TMAO, AC and Tetra (100 pg mL−1 each) was performed. | ||
As can be seen from Table 4, in addition to the common methylated arsenic compounds, such as MMA, DMA and TMAO, a per-methylated arsenic compound, the tetramethylarsinium ion (Tetra) was detected in all fish samples investigated. In particular, a high Tetra content was observed in the muscle tissue of pumpkinseed (34.9%) and largemouth bass (24.4%). To our best knowledge, this is the first report about the presence of Tetra in freshwater fish. In the published studies on arsenic compounds in freshwater fish,20–24 the As methylation pathway is only discussed up to trimethylated arsenic compounds. However, the results of the present study indicate that fresh water fish accumulated the inorganic arsenate from the lake water and transformed it into the tetramethylarsonium ion. Recently, a similar biotransformation of arsenate to the tetramethylarsonium ion in the marine polychaetes nereis diversicolor and nereis virens was reported by Geiszinger et al.25 Although there is a possibility that the observed Tetra may result from decarboxylation of arsenobetaine, it is more likely that the biosynthesis of Tetra in freshwater fish involves sequential reduction and oxidative methylation steps,26via MMA, DMA, and TMAO, because all these intermediates were detected. Definitely, more research is needed to completely understand the unusual arsenic distribution pattern, especially the widespread occurrence of tetramethylarsonium ion in freshwater fish.
Acknowledgements
The authors thank Mr M. S. Dzurko for his assistance in the fish sampling in Moira Lake.References
- K. A. Francesconi and J. S. Edmonds, Adv. Inorg. Chem., 1997, 44, 147–189 CAS.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef.
- D. Kuehnelt, J. Lintschinger and W. Goessler, Appl. Organomet. Chem., 2000, 14, 411–420 CrossRef CAS.
- J. M. Gonzalez LaFuente, J. M. Marchante-Gayon, M. L. Fernandez Sanchez and A. Sanz-Medel, Talanta, 1999, 50, 207–217 CrossRef CAS.
- G. Koellensperger, J. Nurmi, S. Hann, G. Stingeder, W. J. Fitz and W. W. Wenzel, J. Anal. At. Spectrom., 2002, 17, 1042–1047 RSC.
- G. Raber, K. A. Francesconi, K. J. Irgolic and W. Goessler, Fresenius’ J. Anal. Chem., 2000, 367, 181–188 CrossRef CAS.
- X. C. Le and M. S. Ma, Anal. Chem., 1998, 70, 1926–1933 CrossRef CAS.
- A. Chatterjee, Y. Shibata, J. Yoshinaga and M. Morita, Anal. Chem., 2000, 72, 4402–4412 CrossRef CAS.
- T. Nakazato, T. Taniguchi, H. Tao, M. Tominaga and A. Miyazaki, J. Anal. At. Spectrom., 2000, 15, 1546–1552 RSC.
- J. A. Day, M. Montes-Bayon, A. P. Vonderheide and J. A. Caruso, Anal. Bioanal. Chem., 2002, 373, 664–668 CrossRef CAS.
- J. Mattusch and R. Wennrich, Anal. Chem., 1998, 70, 3649–3655 CrossRef CAS.
- S. Londesborough, J. Mattusch and R. Wennrich, Fresenius’ J. Anal. Chem., 1999, 363, 577–581 CrossRef CAS.
- U. Kohlmeyer, J. Kuballa and E. Jantzen, Rapid Commun. Mass Spectrom., 2002, 16, 965–974 CrossRef CAS.
- J. Zheng, W. Goessler and W. Kosmus, Mikrochim. Acta, 1998, 130, 71–79 CAS.
- Q. Xie, R. Kerrich, E. Irving, K. Liber and F. Abou-Shakra, J. Anal. At. Spectrom., 2002, 17, 1037–1041 RSC.
- J. Zheng, H. Hintelmann, B. Dimock and M. S. Dzurko, Anal. Bioanal. Chem., 2003 Search PubMed , in the press.
- J. M. Azcue and D. G. Dixon, J. Great Lakes Res., 1994, 20, 717–724 Search PubMed.
- M. G. Johnson, Can. J. Fish. Aquat. Sci., 1987, 44, 3 CAS.
- M. K. Saiki and T. W. May, Sci. Total Environ., 1988, 74, 199 CrossRef CAS.
- T. Kaise, M. Ogura, T. Nozaki, K. Saitoh, T. Sakurai, C. Matsubara, C. Watanabe and K. Hanaoka, Appl. Organomet. Chem., 1997, 11, 297–304 CrossRef CAS.
- K. Shiomi, Y. Sugiyama, K. Shimakaru and Y. Nagashima, Appl. Organomet. Chem., 1995, 9, 105–109 CrossRef CAS.
- J. F. Lawrence, P. Michalik, G. Tam and H. B. S. Conacher, J. Agric. Food Chem., 1986, 34, 315–319 CrossRef CAS.
- Z. Slejkovec, A. R. Byrne, B. Smodis and M. Rossbach, Fresenius’ J. Anal. Chem., 1996, 354, 592–595 CAS.
- V. Devesa, M. A. Suner, V. W. M. Lai, S. C. R. Granchinho, J. M. Martinez, D. Velez, W. R. Cullen and R. Montoro, Appl. Organomet. Chem., 2002, 16, 123–132 CrossRef CAS.
- A. E. Geiszinger, W. Goessler and K. A. Francesconi, Environ. Sci. Technol., 2002, 36, 2905–2910 CrossRef CAS.
- W. R. Cullen, H. Li, G. Hewitt, K. J. Reimer and N. Zalunardo, Appl. Organomet. Chem., 1994, 8, 303 CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: analytical figures of merit obtained with a standard set-up, the LOD for the high resolution mode, LODs reported by other authors and tissue concentrations for the Northern pike. See http://www.rsc.org/suppdata/ja/b3/b304890j/ |
| ‡ Current address: Nakaminato Laboratory of Radioecology, National Institute of Radiological Sciences, 3609 Isozakicho, Hitachinaka, Ibaraki, 311-1202, Japan (jzheng@nirs.go.jp). |
| This journal is © The Royal Society of Chemistry 2004 |