The fate of fipronil in modular estuarine mesocosms†
Spencer S.
Walse
a,
Paul L.
Pennington
b,
Geoffrey I.
Scott
c and
John L.
Ferry
*a
aDepartment of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208, USA. E-mail: ferry@mail.chem.sc.edu; Fax: (803) 777-9521; Tel: (803) 777-2646
bDepartment of Environmental Health Sciences, Arnold School of Public Health, University of South Carolina, Columbia, SC 29208, USA
cNOAA/NOS/NCCOS Center for Coastal Environmental Health and Biomolecular Research, 219 Ft. Johnson Road, Charleston, SC 29412, USA
First published on 26th November 2003
Abstract
The degradation and corresponding product manifold for the pesticide fipronil was determined in three replicate estuarine mesocosms. Aqueous fipronil concentrations rapidly decreased over the 672 h timescale of the experiment (95% removal). Loss was apparently first-order in fipronil, although there appeared to be a change in the removal mechanism after 96 h that corresponded to a dramatic slowdown in its disappearance. The reduction product of fipronil, fipronil sulfide, was not detected in the water column; however, it formed rapidly in sediments and was identified as the major product of fipronil degradation in the system (20% yield at 672 h, with respect to initial fipronil concentration). Fipronil sulfone is thought to form primarily via biological oxidation; and, although it was generated rapidly in the water column (10% yield), only trace amounts were detected in the sediment (1% yield). The direct photolysis product of fipronil, fipronil desulfinyl, was present in all samples; it formed rapidly in the water column (4% yield) and partitioned into the sediment phase (7% yield) over the course of the experiment. The mass balance on fipronil and associated products was 42% at 672 h.
Introduction
Fipronil [5-amino-1-[2,6-dichloro-4-(trifluoromethyl) phenyl]-4-[(trifluoromethyl) sulfinyl]-1H-pyrazole-3-carbonitrile], a widely used phenylpyrazole insecticide, is applied in granular or bait form for residential and commercial control of turf grass pests and as a seed treatment or aerial spray for agricultural pest control.1,2The mechanism of fipronil toxicity, non-competitive inhibition of the γ-aminobutyric acid (GABA) regulated chloride channel,3,4 operates with similar efficacy for many invertebrates.5 Accordingly, runoff from sites of fipronil application can impact non-target aquatic organisms,1,2,6 particularly crustaceans. For example, fipronil is acutely toxic to red swamp (Procambarus clarki) (96 h LC50 = 14.3 µg L−1) and white river crayfish (Procambarus zonangulus) (96 h LC50 = 19.5 µg L−1),7 adult grass shrimp (Palaemonetes pugio) (96 h LC50 = 0.32 µg L−1),8 and the estuarine copepod (Amphiascus tenuiremis) (96 h LC50 = 6.8 µg L−1).9 Furthermore, Chandler et al.9 reported that fipronil decreased Amphiascus tenuiremis reproduction by 94% at concentrations as low as 0.42 µg L−1. A more recent study by Cary et al.10 reported similar inhibition that was linked to sex-specific male reproductive dysfunction.
Fipronil desulfinyl (2), fipronil sulfide (3), and fipronil sulfone (4) are the three major products of fipronil (1) degradation in the environment (Fig. 1) and are all products of reactions involving the sulfinyl group. Although in general these products exhibit similar lethality to that of 1 within a single species,11–14 they do vary, sometimes widely, between different species (1, 2). Accordingly, an understanding of the fate and transport of 1–4 is essential to constructing a model of the potential risk associated with the use of 1 toward non-target species.
 | ||
| Fig. 1 The abbreviations and structures of fipronil and its products. | ||
Previous studies on equilibrium descriptors of environmental transport report that 1 and 2 have a low volatility (Henry's law constants (HLC) at 25 °C: 1, 8.5 × 10−10; 2, 1.6 × 10−8).15 Estimated HLC's indicate that 4 (1.7 × 10−8) has a similar volatility, while 3 (2.2 × 10−6) has slightly greater volatility than 1 and 2.16 Consistent with reports on other neutral multifunctional polar compounds,171–4 are moderately soluble in water (mg L−1: 1, 1.6; 2, 0.4; 3, 0.2; 4, 1.0)16 and have high affinities for organic carbon (OC) containing soils, sediments, and biota (log Kow: 1, 4.01 (3.56) ; 2, 4.63; 3, 4.77; 4, 3.68).1–3,16,18–20
On crops,11 soils,21,22 and in aqueous solution4,15,18,23–251 is readily converted to 2via photo-initiated sulfinyl (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) extrusion; this process appears to be the only route for its formation.11 Two mechanisms have been postulated to account for the loss of SO in aqueous solution, a concerted mechanism (simultaneous loss of SO while generating a new C–C bond between atoms α to the sulfinyl),26 or homolytic α-cleavage to form a sulfinyl/trifluoromethyl radical pair.11 There is experimental evidence to support both. The initial photoproduct, 2 photolyzes only slowly in solution26 and is stable toward photolysis on leaf surfaces.11
O) extrusion; this process appears to be the only route for its formation.11 Two mechanisms have been postulated to account for the loss of SO in aqueous solution, a concerted mechanism (simultaneous loss of SO while generating a new C–C bond between atoms α to the sulfinyl),26 or homolytic α-cleavage to form a sulfinyl/trifluoromethyl radical pair.11 There is experimental evidence to support both. The initial photoproduct, 2 photolyzes only slowly in solution26 and is stable toward photolysis on leaf surfaces.11
The reduction of 1 to 3 is rapid in flooded soils,23,26,27 pond sediments,7 and in modeled aquatic ecosystems,18 yet the relative importance of biological or abiotic processes has not been measured. Rapid oxidation of 1 to 4 has been reported in vivo,12,13,28,29 suggesting that there is a strong biological component to 4 formation in aqueous ecosystems,7,18,23 soils,21,22,24 and on crops.14,21,24 Nigm et al.26 identified only trace amounts (<5%) of 4 and its oxidation product, fipronil sulfonate, in thermal and photochemical studies on the in vitro degradation of aqueous 1.
Field studies on soils21,22 and in solution15,18 report that hydrolysis to fipronil amide (5) represents only a minor pathway of 1 degradation in the environment. Laboratory studies25,26,30 suggest that hydrolysis of 1 is specific base-catalyzed; the proposed reaction mechanism25 involves nucleophilic attack at the nitrile carbon yielding an unstable hydroxy imine that rapidly tautomerizes into an amide. This transformation was not observed in solutions containing 2, 3, or 4.26 Nitrile hydrolysis by soil microorganisms has been observed for several other pesticides,18 however, it has not been reported for 1. 5 appears to be stable toward photolysis and hydrolysis.11 In comparison to 1–4, little data exists on the toxicity of 5 toward non-target organisms.
The purpose of this study was to examine the fate and transport of 1 in an estuarine environment. Estuarine aquatic organisms, including many commercially and recreationally important fish and shellfish species, have been repeatedly impacted by agricultural and urban runoff in the Southeastern US.31,32 We report the loss of aqueous 1 in modular estuarine mesocosms designed to closely approximate coastal conditions typical of the Southeastern US. The distribution of the degradation products of 1 in treated mesocosms was of particular interest during this study; as several recent studies suggest that without reproducibility in the spatio-temporal dynamics of pollutant fate across treatments, there is large uncertainty associated with the validity of biological effect data.33,34 A distinction was made between the degradation products of 1 found in solution versus sediment, to aid in the development of species–specific bioconcentration (BCFs) and bioaccumulation factors (BAFs) that are used to predict non-target toxicity.35,36
Experimental section
Materials
Barnstead E-pure™ water (18 MΩ cm) was used for all solutions. OmniSolv® GC2 grade methyl tert-butyl ether (MTBE) was purchased from EM Science (Darmstadt, GR). Mirex (93%), 4-bromoanisole (99+%), and tetrabutylammonium hydrogensulfate (TBA)(97%) were acquired from Aldrich. NaCl, Na2SO3, Na2SO4, MgSO4, and Florisil® were used as received from Fischer Scientific. All stock solutions were prepared in analytical grade 2-propanol (Mallinkrodt), unless otherwise noted, and stored at 4 °C in the dark. 1 (98%) was obtained from Chem Service (West Chester, PA). Analytical standards (15 mg) of 2, 3, and 4 were donated by CCEHBR, NOAA (Charleston, SC) and were used to validate their respective syntheses. 2 was synthesized with a modified method of Hainzl et al.;11 a Pyrex® filtered 1000 W Xe arc lamp (vide infra) was used for irradiation (2.5 h). 3 was synthesized with the method of Beeler et al.;374 was prepared using the method of Schlenk et al.7Synthesis of fipronil amide (5)
5 was prepared by a modified method of Feichtinger.38 Water (5 mL), adjusted to pH 12 with 0.01 M NaOH, was added drop-wise over 20 min to a solution of 100 mg of purified 1 (0.23 mmol) in 25 mL THF at 35 °C. Reaction progress was monitored hourly via the dilution of 5 µL aliquots into 1.5 mL MTBE for subsequent analysis with gas chromatography–ion trap mass spectrometry (GC-ITMS). After ∼60% conversion to 5 (≅3 h), the reaction mixture was concentrated via rotary evaporation (Büchi R 110). The residue was dissolved in 25 mL MTBE and washed with 3 × 10 mL H2O/NaCl. The MTBE extract was then dried with anhydrous MgSO4 and concentrated to a residue that was then dissolved in a minimal amount of 5∶1 hexane/ethyl acetate. 5, retained on the stationary phase, was easily separated from unreacted 1via the column chromatography scheme employed for the purification of 1 (vide infra). 5 was eluted with a 4∶6 acetone/hexane mobile phase. After recrystallization from MTBE, the purity of 5 was determined to be >99.5% via gas chromatography-electron capture detection (GC-ECD).Separation/purification of fipronil and related products
1–4 were isolated via column chromatography utilizing a 5∶1 hexane/ethyl acetate mobile phase and a SiO2, 60 Å/230–400 Mesh ASTM stationary phase. This scheme was employed for the separation of crude residue from syntheses and in the purification of Chem Service 1 (∼2% 3 impurity). Eluant fractions were characterized by matching substrate retention times (GC-ECD and GC-ITMS) and mass spectra (GC-ITMS) against those of purchased or synthesized standards. Thin layer chromatography, utilizing a similar stationary phase (Riedel-de-Haën® SiO2 60F 254), yielded the following retention factors: 5, 0.06; 4, 0.17; 1, 0.22; 3, 0.25; 2, 0.29. 1–4 were recrystallized from 5∶1 hexane/ethyl acetate and stored at 4 °C in the dark; their purities were determined to be >99.9% via GC-ECD.Experimental design
Six replicate modular estuarine mesocosms, open to the atmosphere, were established at the Center for Coastal Environmental Health and Biomolecular Research (CCEHBR) in a greenhouse. These systems were based on an original design by Lauth et al.,39 however, each mesocosm was modified to contain its own reservoir for tidal water storage. This design modification allowed for the assumptions of parametric statistics to be met as outlined by Zar.40The mesocosms contained raised sediment trays representing the low or fringe marsh, mid or meadow marsh, and high marsh. Whole sediment plots (containing rooted vegetation as well as epibenthic, benthic, and meiobenthic flora and fauna) were collected from a pristine control site31 located along the West Branch of Leadenwah Creek on Wadmalaw Island, South Carolina. The organic carbon content of this sediment, determined via C∶H∶N analysis, ranged from 3.8–4.1%. Sediment plots (27.3 cm wide by 29.8 cm long by 15.2 cm deep) were removed (using a device of roughly the same dimensions made of sharpened landscape edging), transported back to CCEHBR, and immediately placed into sediment trays that were then transferred into the mesocosms. Each mesocosm had an approximate marsh surface area of 0.5 m2 and a total sediment volume of 75 L. The concentrations of 1–5 in the top ∼0.5 cm of sediment throughout each mesocosm (∼2.5 L) were estimated via the quantitation of a single sediment sample of known density (vide infra).
Each mesocosm contained approximately 300 L of seawater that was collected from Cherry Point Boat Landing on Wadmalaw Island, South Carolina, USA. The collected water (unfiltered) was diluted to a salinity of 20‰ and homogenized prior to mesocosm introduction.
Submerged magnetic impeller pumps, set to a central timer, controlled tidal variation in the mesocosm. The tides occurred at the same time everyday (flood, 10:00–16:00 and 22:00–04:00; ebb, 16:00–22:00 and 04:00–10:00) and the tidal flow rate was consistent across all mesocosms.41
Experimental procedure
The modular estuarine mesocosms were maintained for 5 months prior to fipronil addition. The following water quality parameters were monitored daily at high tide (13:00): temperature (°C), [O2] (mg L−1), dissolved O2% saturation, salinity (‰), and pH. Temperature and dissolved O2 measurements were made using a YSI 85 dissolved oxygen meter (YSI, Yellow Springs, OH 45387), salinity was measured via a refractometer, and pH was measured with a portable pH meter (Orion, Beverly, MA 01915). Total organic carbon (TOC) was measured weekly with a Shimadzu TOC5000A analyzer equipped with an auto sampler. Solar irradiation was measured continuously 0.5 m above the water's surface with a LI-190SA Quantum Sensor from LI-COR® (Lincoln, NE) and was recorded as Photosynthetic Photon Flux Density (PPFD) (photons s−1m−2).Acetone stock solutions of 1 (Chem Service) were introduced at 5000 ng L−1 (nominal) into the tidal reservoir of the mesocosms 1 h prior to flow tide (∼09:00, 06/05/02); no detectable fipronil photolysis or hydrolysis occurred (in the acetone stock solutions) prior to its addition. Three mesocosms were treated; three were left as untreated controls (randomized selection). Initial 1 concentrations (ca. 2 × 10−7 M) were below estimated/published water solubilities,16 these concentrations/conditions were chosen in an attempt to minimize the volatilization and adsorption processes of 1 that more readily occur above solubility limits and emulate the aqueous fraction of 1 that could enter the aquatic environment via runoff, vapor deposition, and spray drift based on field observation. Significantly, this concentration is consistent with those measured by Demcheck et al. in LA surface waters.27
Aqueous sampling
Over the course of 28 d, 17 samples were withdrawn from the mesocosms for quantitative analysis; four 5 mL aliquots were removed with a spring-loaded 10 mL Manostat syringe and combined in Fisherbrand™ 40 mL amber EPA vials to represent a single sample (20 mL). Mirex was then introduced (5 µL) via isopropanolic stocks (4 µM) as an external (recovery) standard. The EPA vials were charged with 4 mL of MTBE + internal standard (4-bromoanisole). Samples were rapidly mixed for 2 min on a vortex Genie® mixer followed by 20 min of sonication (Branson 2510). Emulsions were broken by the addition of ∼300 mg NaCl and the MTBE layer was removed for subsequent GC-ECD or GC-ITMS analysis.At the conclusion of each sampling period, 500 mL samples were taken from a representative of each treatment and extracted with 2 × 200 mL of MTBE. To minimize water loss and to insure consistency, these relatively large samples were alternately removed from similarly treated mesocosms. MTBE extracts were dried with anhydrous MgSO4, concentrated to ∼2 mL, and used for qualitative product analysis (GC-ITMS).
Sediment sampling
Samples were removed from the top ∼0.5 cm of sediment from the mid marsh trays (∼20 g total) in each mesocosm prior to the introduction of 1 and again at 12 h, 24 h, 7 d, 14 d, 21 d, and 28 d. Sampling occurred 1 h prior to flow tide (09:00 or 21:00). Samples were immediately placed into 200 mL Teflon®-lined screw-cap glass jars and stored at −20 °C (CCEHBR, NOAA, Charleston, SC) until they could be analyzed.All sediment extraction and clean-up procedures were performed in the dark. Samples were allowed to equilibrate to room temperature before being homogenized. The density (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ±
s; 0.89 ± 0.05 g mL−1) and moisture content (
±
s; 47.5 ± 0.5%) of each sediment sample was determined by gravimetric analysis of wet and dry samples. Additional 10 g sub-samples were removed from each sediment sample, transferred to a mortar, and a recovery standard, endosulfan sulfate, was added (50 µL of 2.5 × 10−9 M (∼10 ppb) in DMSO). After 30 min, the sub-samples (10 g) were macerated with 12 g anhydrous Na2SO4, immediately transferred to solvent-cleaned cellulose thimbles (Whatman® #2810339), and extracted (Soxhlet) for 36 h with 400 mL MTBE. The extracts were then reduced to 1 mL with a gentle stream of N2 gas.
±
s; 0.89 ± 0.05 g mL−1) and moisture content (
±
s; 47.5 ± 0.5%) of each sediment sample was determined by gravimetric analysis of wet and dry samples. Additional 10 g sub-samples were removed from each sediment sample, transferred to a mortar, and a recovery standard, endosulfan sulfate, was added (50 µL of 2.5 × 10−9 M (∼10 ppb) in DMSO). After 30 min, the sub-samples (10 g) were macerated with 12 g anhydrous Na2SO4, immediately transferred to solvent-cleaned cellulose thimbles (Whatman® #2810339), and extracted (Soxhlet) for 36 h with 400 mL MTBE. The extracts were then reduced to 1 mL with a gentle stream of N2 gas.
A modified method of Cotham and Bidleman42 was used to remove interferences from the extracts. Elemental sulfur was removed by rapidly mixing the extracts for 2 min on a Genie® vortex mixer after the addition of 1 mL TBA-sulfite reagent (0.01 mol TBA in Na2SO3 saturated solution) and 1 mL of 2-propanol (containing 2.0 × 10−8 M (∼11 ppb) mirex as an external standard).43 The extract/solution was then rapidly mixed for 1 min after the addition of water (5 mL); emulsions were broken by the addition of ∼600 mg NaCl.
The organic phase was removed and passed through a Florisil® column for final clean-up; a 14.5 cm disposable glass Pasteur pipette was plugged with glass wool and then filled with 5 cm of activated Florisil® (∼1 g) followed by 0.5 cm of anhydrous Na2SO4. The column was prepared by washing with 5 mL ethyl acetate and then 10 mL pentane. The MTBE extract (1 mL) was added to the column and eluted successively with: 5 mL of pentane, 2 mL of 5∶1 hexane/ethyl acetate, and then 2 mL of 4∶6 acetone/hexane. The eluant was then reduced to 1.5 mL with a gentle stream of N2 gas and used for quantitative and qualitative product analyses.
Gas chromatography-electron capture detector and gas chromatography-mass spectrometry
All MTBE extracts were transferred by Pasteur pipette to 11 mm amber crimp-top GC vials and stored at 4 °C until analyses. Products were verified by matching retention times (GC-ECD and GC-ITMS) and mass spectra (GC-ITMS) against those of purchased or synthesized standards. The retention times, RT, of all analytes are presented in supporting information (see the table in ESI†) as are their mass spectra (see Figs 1–5 in the ESI†).A modified method of Nigm et al. was used for the gas chromatographic analyses.26 Holox (Charlotte, NC) high purity helium was used as a carrier gas (1.3 mL min−1). Splitless injections of 1 µL were made at an injector port temperature of 230 °C. The oven program was as follows: isothermal at 100 °C for 1 min, followed by heating from 100 °C to 270 °C at 10 °C min−1, then held isothermally at 270 °C for 10 min. The analytical column was a J&W DB-5MS column (L = 30 m, ID = 0.25 mm, df = 0.25 µm).
Quantitative analysis was accomplished with a Hewlett Packard (Palo Alto, CA) 5890 series II gas chromatograph equipped with a 63Ni electron-capture detector (GC-ECD) utilizing nitrogen as the make-up gas (1.5 mL min−1). The detector temperature was maintained at 310 °C. Data was processed with a Shimadzu integrator. Method detection limits for all analytes were: 1, 2.7 × 10−11 M; 2, 2.3 × 10−11 M; 3, 3.2 × 10−11 M; 4, 8.7 × 10−11 M; 5, 4.7 × 10−10 M.
Qualitative analysis was performed on a Varian 3800 gas chromatograph equipped with a Varian Saturn 2000 ion trap mass spectrometer (GC-ITMS) in electron impact (EI) ionization mode (70 eV). Full scan spectra were acquired over the ranges m/z 40 to 650 at 0.85 s per scan. Method detection limits for all analytes were: 1, 3.0 × 10−6 M; 2, 4.0 × 10−6 M; 3, 2.5 × 10−6 M; 4, 3.4 × 10−6 M; 5, 4.6 × 10−6 M. The method limits of detection were improved approximately three orders in magnitude via acquiring spectra over the scan range m/z 350 to 395 and “extracting” ions of interest with select ion monitoring (SIM): 1, 367–369 m/z; 2, 388–390 m/z; 3, 351–353 m/z; 4, 383–385 m/z; 5, 386–387 m/z.
Treatment of error
Prior to the addition of 1, aqueous and sediment samples were removed from the mesocosms. After the addition of 50 µL of a stock solution containing 1.25 × 10−9 M each of 1–5 in either 2-propanol (aqueous) or DMSO (sediments), the samples were processed as described above. The standard error of the mean (SEM) from eight replicate additions was used to estimate error in the mesocosm aqueous and sediment GC concentration measurements.The standard deviation associated with triplicate injections was used to assess error in all GC concentration measurements; in all cases, it was negligible compared to the SEM associated with replicate sampling.
Results and discussion
Mesocosm studies
Prior to the addition of 1, bacterial density and microalgae community composition were consistent across all mesocosms.41 Water quality parameters (
±
s) remained relatively steady over the timescale of the experiment (28 d): temperature (28.6 ± 2.4 °C), [O2]
(6.2 ± 2.9 mg L−1), dissolved O2% saturation (91.5 ± 45.3%), salinity (22.3 ± 1.0‰), pH (7.8 ± 0.5), and TOC (5.1 ± 0.4 mg L−1). 1–5 were not detected in aqueous or sediment samples from any of the three control mesocosms.
Aqueous concentrations of 1 decreased rapidly (96.2–98.8% loss) in the modular estuarine mesocosms over the timescale of this experiment (Fig. 2). The loss of aqueous 1 appeared to occur in distinct phases (periods); fitting straight-lines to (ln[1]t/ln[1]0) plotted versus time before and after 96 h (t < 96 h, r2
(
±
s)
= 0.97 ± 0.04); t > 96 h, r2
(
±
s)
= 0.96 ± 0.01) indicated that the initial rate of 1 loss was ∼ fourfold faster (t1/2(<96)
= 47.3 ± 4.2 h; t1/2(>96)
= 203.1 ± 31.9 h). Bi-phasic (i.e., two-period) loss of insecticides from aqueous systems due to rapid phase transfer (e.g. volatilization and partitioning) following introduction has been observed in many studies.44,45 Even though volatilization is not expected to be a dominant pathway of 1 loss,15,16 the mesocosms contain many compartments that it could quickly partition into from the aqueous phase, such as the surface-microlayer, biota, bio-films, and sediment.
![We obtained high coefficients of determination (r2) for fitting straight-lines through ln [fipronil]t/[fipronil]0
(aqueous) plotted versus time before 96 h and after 96 h.; in all cases the rate of fipronil removal before 96 h was ∼a factor of four greater, suggesting that the loss of aqueous fipronil in the mesocosms occurred in two distinct phases.](/image/article/2004/EM/b307304a/b307304a-f2.gif) | ||
| Fig. 2 We obtained high coefficients of determination (r2) for fitting straight-lines through ln [fipronil]t/[fipronil]0 (aqueous) plotted versus time before 96 h and after 96 h.; in all cases the rate of fipronil removal before 96 h was ∼a factor of four greater, suggesting that the loss of aqueous fipronil in the mesocosms occurred in two distinct phases. | ||
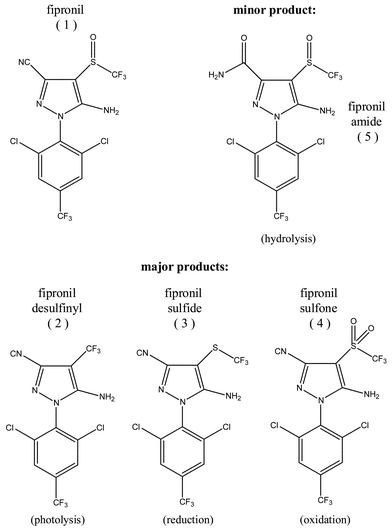
At t = 672 h, an average mass balance of 42.2 ± 9.5% (grand mean) was obtained for 1 loss in the mesocosms via the quantification of analytes in aqueous (grand mean: 14.2 ± 3.6%) and sediment samples (grand mean: 28.0 ± 8.7%). Product distributions (aqueous and sediment) at t = 672 h in relative% [1]0 are presented for the three treated mesocosms in Table 1. The average concentration of 1–5 across all treated mesocosms was reported as the grand mean ± estimated SEM (N = 3).46 The concentrations of products (aqueous and sediment) in all three treated mesocosms were statistically equivalent based on the error associated with their determination (SEM from eight replicate fortified samples). This observation, in combination with the small standard deviations associated with average half-life estimates, clearly identifies the reproducibility of fipronil loss in these mesocosms.
| Products (relative% of [1]0 @ 672 h) | Mesocosm treatment (5000 ng L−1) | ||||
|---|---|---|---|---|---|
| Rep. 1 | Rep. 2 | Rep. 3 | Grand mean ± SEM | ||
| a Within each mesocosm, the error associated with product quantitation was estimated from the SEM obtained from eight replicate fortified samples. | |||||
| % 1 | Sediment aqueous Σ aq., sed. | 0.3 ± 2.5 | 0.5 ± 2.5 | 0.1 ± 2.5 | 0.3 ± 4.3 |
| 2.2 ± 1.0 | 3.8 ± 1.0 | 2.2 ± 1.0 | 2.7 ± 1.7 | ||
| 2.5 ±2.7 | 4.3 ± 2.7 | 2.3 ± 2.7 | 3.0 ± 4.7 | ||
| %2 | Sediment aqueous Σ aq., sed. | 7.1 ± 2.5 | 7.8 ± 2.5 | 7.1 ± 2.5 | 7.3 ± 4.3 |
| 4.3 ± 1.0 | 3.1 ± 1.0 | 4.9 ± 1.0 | 4.1 ± 1.7 | ||
| 11.4 ± 2.7 | 10.9 ± 2.7 | 12.0 ± 2.7 | 11.4 ± 4.7 | ||
| %3 | Sediment aqueous Σ aq., sed. | 18.5 ± 2.5 | 19.0 ± 2.5 | 20.9 ± 2.5 | 19.5 ± 4.3 |
| — | — | — | — | ||
| 18.5 ± 2.5 | 19.0 ± 2.5 | 20.9 ± 2.5 | 19.5 ± 4.7 | ||
| %4 | Sediment aqueous Σ aq., sed. | 0.8 ± 2.5 | 1.1 ± 2.5 | 1.1 ± 2.5 | 1.0 ± 4.3 |
| 10.0 ± 1.0 | 10.9 ± 1.0 | 9.3 ± 1.0 | 10.4 ± 1.7 | ||
| 10.8 ± 2.7 | 11.9 ± 2.7 | 10.5 ± 2.7 | 11.1 ± 4.7 | ||
| %5 | Sediment aqueous Σ aq., sed. | — | — | — | — |
| — | — | — | — | ||
| — | — | — | — | ||
| Σ 1–5 | Sediment aqueous Σ aq., sed. | 26.6 ± 5.1 | 28.3 ± 5.1 | 29.2 ± 5.1 | 28.0 ± 8.7 |
| 14.3 ± 2.1 | 14.0 ± 2.1 | 14.2 ± 2.1 | 14.2 ± 3.6 | ||
| 40.9 ± 5.5 | 42.3 ± 5.5 | 43.5 ± 5.5 | 42.2 ± 9.5 | ||
Through time, sediment product distributions also varied little across treated mesocosms (Fig. 3). The combined sediment concentrations of 1–4 (Σ1–4) increased over the timescale of the experiment, however, approximately 45% of the combined sediment concentrations at 672 h in each mesocosm were achieved before 96 h had passed. This observation provides evidence to suggest that 1 rapidly partitioned into the sediment and that this process could have contributed to the fourfold faster initial rate of aqueous 1 loss.
 | ||
| Fig. 3 Combined sediment concentrations of 1–4 (Σ1–4) steadily increased over the experimental timescale, however, approximately 45% of the combined sediment concentrations at 672 h in each mesocosm were achieved before 96 h had passed. | ||
The manifold of aqueous degradation products included 1, 2 and 4 (ESI Figure 6†). Although 3 was not detected in aqueous samples, it formed rapidly in the sediment and was identified as the major product of 1 degradation (grand mean: 19.5 ± 4.7% of [1]0 at 672 h). 3 represented ∼50–70% of the combined sediment concentrations of 1–4 and increased steadily over the course of the experiment, suggesting it will continue to play a major role in the environmental effects of fipronil application for at least one growing season. These results, supported by previous work,15,23 imply that 3 formation occurs in sediments, and its remobilization to solution (i.e., pore water or surface water) was minimal. The complexity of the experimental design precluded resolution of abiotic versus biological processes in the reduction of the sulfinyl group. The observation of only trace amounts of 1 and 4 in sediment (pore water inclusive) after 96 h suggests that potential oxidation processes do not effectively compete with the reduction of 1 in estuarine sediment typical of the Southeastern US.
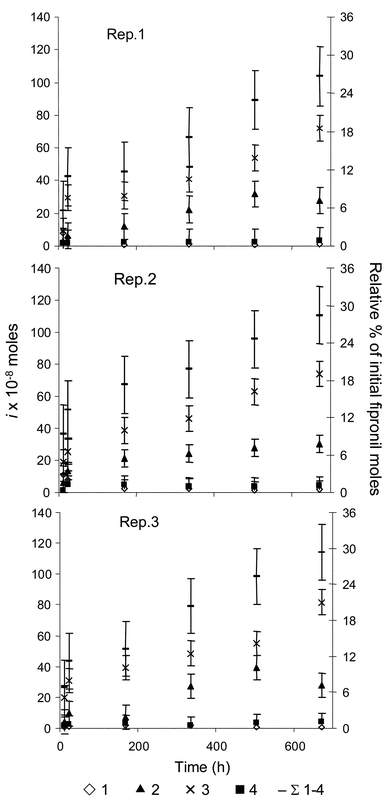
Aqueous concentrations of 4 increased rapidly over the initial 96 h of the experiment, accounting for ∼85% of [4]aq at 672 h (Fig. 4), and then remained relatively constant over the remainder of the experiment (major product in the water column). This observation is compelling and suggests that aqueous 1 is subject to rapid biological processing in an estuarine environment, or at least in these mesocosms. The concentration of 4 in the sediment remained relatively constant over the course of the experiment; aqueous concentrations of 4 were consistently higher by approximately a factor of ten. This was surprising given its log Kow (3.68), however, it is consistent with field data reported by Schlenk et al.7 This observation suggests that 4 does not readily partition into sediments, or that 4 is consumed in the sediments by an unknown process (possibly, the reduction of 4, through an intermediate of 1, to 3). Several studies have reported that sulfate-reducing bacteria in the sediments and rhizosphere of Spartina alterniflora, the dominant flora in the mesocosms, are efficient reductants of not only inorganic sulfoxides47,48 but also organic sulfoxides, (sulfones) such as endosulfan sulfate.49 Regardless, the discrepancy between experimental and predicted results (based on the log Kow of 4) highlights the value of using model ecosystems for evaluating aqueous-sediment pollutant distribution.
![Aqueous concentrations of fipronil sulfone (4), the biological oxidation product of fipronil, increased rapidly over the initial 96 h of the experiment, accounting for ∼85% of [4]aq at 672 h. This observation suggests that aqueous fipronil is subject to rapid biological processing in an estuarine environment.](/image/article/2004/EM/b307304a/b307304a-f4.gif) | ||
| Fig. 4 Aqueous concentrations of fipronil sulfone (4), the biological oxidation product of fipronil, increased rapidly over the initial 96 h of the experiment, accounting for ∼85% of [4]aq at 672 h. This observation suggests that aqueous fipronil is subject to rapid biological processing in an estuarine environment. | ||
Even though the aqueous formation of 4 was favored by a factor of two over 2, the trend in their quantitation was similar over the course of the experiment (Fig. 5). Unlike 4, sediment concentrations of 2 gradually increased through time, representing ∼15–25% of the combined sediment concentrations of 1–4. Given the known photodegradation of 1, this was interpreted as a result of 2 forming first in the water column followed by partitioning into the sediments. At 672 h, aqueous concentrations of 2 (grand mean: 4.1 ± 1.7% of [1]0) were less than those in sediments (grand mean: 7.3 ± 4.3% of [1]0). Nevertheless, these results suggest that both BCFs (aqueous exposure) and BAFs (sediment exposure) should be developed and considered when predicting the non-target toxicity of 2 in estuarine systems.
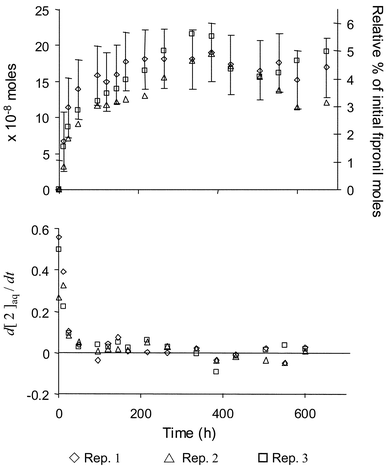 | ||
| Fig. 5 Aqueous concentrations of fipronil desulfinyl (2), formed via the direct photolysis of fipronil, increased rapidly over the initial 96 h of the experiment. Although its concentration in the water column leveled off after the initial period, it increased steadily in sediment samples over the course of the experiment. | ||
Acknowledgements
This work was supported by the US Environmental Protection Agency Grant # R827397. We would like to extend our gratitude to Dr. G. Thomas Chandler, Department of Environmental Health Science, University of South Carolina for the use of his GC-ECD. We would also like to deeply thank Dr. Mike H. Fulton and Dr. Edward F. Wirth, Center for Coastal Environmental Health and Biomolecular Research-NOAA, for allowing the use of their facilities.References
- C. C. D. Tingle, J. A. Rother, C. F. Dewhurst, S. Lauer and W. J. King, Rev. Environ. Contam. Toxicol., 2003, 176, 1–66 Search PubMed.
- U.S.E.P.A., Fipronil environmental fate and ecological effects assessment and characterization for broadcast treatment with granular product to control turf insects and fire ants (addendum), Environmental Fate and Effects Division, 2001 Search PubMed.
- L. M. Cole, R. A. Nicholson and J. E. Casida, Pestic. Biochem. Physiol., 1993, 46, 47–54 CrossRef CAS.
- D. B. Gant, A. Chalmers, M. Wolff, H. Hoffman and D. Bushey, Rev. Toxicol., 1998, 2, 147–156 Search PubMed.
- T. N. Robinson and R. W. Olsen, in Comparative Invertebrate Neurochemistry, ed. G. G. Lunt and R. W. Olsen, Cornell University Press, Ithaca, NY, 1988, pp. 90–123 Search PubMed.
- P. Connelly, Environmental Fate of Fipronil, Environmental Monitoring Branch, Department of Pesticide Regulation, California Environmental Protection Agency, 2001 Search PubMed.
- D. Schlenk, D. B. Huggett, J. Allgood, E. Bennet and J. M. Rimoldi, Arch. Environ. Contam. Toxicol., 2001, 41, 325–332 CrossRef CAS.
- P. B. Key, K. W. Chung, A. D. Opatkiewicz, E. F. Wirth and M. H. Fulton, Bull. Environ. Contam. Toxicol., 2003, 70(3), 533–540 CrossRef CAS.
- G. T. Chandler, T. L. Carey, D. C. Volz, S. S. Walse, J. L. Ferry and S. L. Klosterhaus, Environ. Toxicol. Chem., 2003 Search PubMed , in press.
- T. L. Cary, G. T. Chandler, D. C. Volz, S. S. Walse and J. L. Ferry, Environ. Sci. Technol., 2003 Search PubMed , in press.
- D. Hainzyl and J. E. Casida, Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 12764–12767 CrossRef.
- D. Hainzyl, L. M. Cole and J. E. Casida, in 215th ACS National Meeting, American Chemical Society, Dallas, TX, 1998 Search PubMed.
- D. Hainzyl, L. M. Cole and J. E. Casida, Chem. Res. Toxicol., 1998, 11, 1529–1535 CrossRef CAS.
- J. E. Mulrooney and D. Goli, Field and Forage Crops, 1999, 92, 1364–1368 Search PubMed.
- K. K. Nigm and D. G. Crosby, Environ. Toxicol. Chem., 2001, 20, 972–977.
- U.S.E.P.A. EPI Suite, v3.10 ed., 2002.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, John Wiley & Sons Inc., New York, 1993 Search PubMed.
- A. Aajoud, P. Ravanel and M. Tissut, J. Agric. Food Chem., 2003, 51, 1347–1352 CrossRef CAS.
- G. G. Ying and R. S. Kookana, J. Environ. Sci. Health, B, 2001, 36, 545–558 CrossRef CAS.
- A. Bobe, C. M. Coste and J.-F. Cooper, J. Agric. Food Chem., 1997, 45, 4861–4865 CrossRef CAS.
- H. Fenet, E. Beltran, B. Gadji, J. F. Cooper and C. M. Coste, J. Agric. Food Chem., 2001, 49, 1293–1297 CrossRef CAS.
- A. Bobe, J.-F. Cooper, C. M. Coste and M. A. Muller, Pestic. Sci., 1998, 52, 275–281 Search PubMed.
- R. C. Biever, J. R. Hoberg, B. Jacobson, E. Dionne, M. Sulaiman and P. McCahon, Environ. Toxicol. Chem., 2003, 22, 167–174 CAS.
- G. Zhu, J. Wu, Q. Liu and J. Sun, Nongyaoxue Xuebao, 2000, 21(2), 52–56 Search PubMed.
- A. Bobe, P. Meallier, J.-F. Cooper and C. Coste, J. Agric. Food Chem., 1998, 46, 2834–2839 CrossRef CAS.
- K. K. Nigm, S. A. Mabury and D. G. Crosby, J. Agric. Food Chem., 2000, 48, 4661–4665 CrossRef.
- D. K. Demcheck and S. C. Skrobialowski, Fipronil and degradation products in the rice-producing areas of the Mermentau River Basin, Louisiana, February-September 2000, U.S.G.S, 2003 Search PubMed.
- M. E. Scharf, B. D. Siegfried, L. J. Meinke and L. D. Chandler, Pest. Manage. Sci., 2000, 56, 757–766 Search PubMed.
- G. Brookhard and D. Bushey, in Eighth International Union of Pure and Applied Chemistry, International Congress of Pesticide Chemistry, Washington, DC, 1994, abstract 189 Search PubMed.
- A. Ramesh and M. Balasubramanian, J. Agric. Food Chem., 1999, 47, 3367–3371 CrossRef CAS.
- G. I. Scott, M. H. Fulton, M. C. Crosby, P. B. Key, J. W. Daugomah, J. T. Waldren, E. Strozier, C. J. Louden, G. T. Chandler, T. F. Bidleman, K. L. Jackson, T. W. Hampton, T. Huffman, A. Shulz and M. Bradford, Agricultural Insecticide Runoff Effects on Estuarine Organisms: Correlating Laboratory and Field Tests, Ecophysiology, Bioassays, and Ecotoxicological Biomonitoring. Research Grant Final Report, 1992 Search PubMed.
- D. B. Finley, G. I. Scott, J. W. Daugomah, S. L. Layman, L. Reed, M. Sanders, S. K. Sivertsen and E. D. Strozier, in Ecotoxicology and Risk Assessment for Wetlands, ed. M. A. Lewis, F. L. Mayer, R. L. Powell, M. K. Nelson, S. J. Klaine, M. G. Henry and G. W. Dickson, SETAC Press, 1995, pp. 242–273 Search PubMed.
- S. J. Maund, T. N. Sherratt, T. Stickland, J. Biggs, P. Williams, N. Shillabeer and P. C. Jepson, Pestic. Sci., 1997, 49, 185–190 Search PubMed.
- H. Sanderson, Environ. Sci. Pollut. Res., 2002, 9, 429–435 Search PubMed.
- H. Lee, II, in Sediment Toxicity Assessment, 1988, pp. 267–291 Search PubMed.
- H. Lee II, in Organic Substance in Sediment and Water, ed. J. F. McCarthy, Lewis Publishers, Chelsea, MI, 1991, pp. 73–93 Search PubMed.
- A. B. Beeler, D. K. Schlenk and J. M. Rimoldi, Tetrahedron Lett., 2001, 42, 5371–5372 CrossRef CAS.
- H. Feichtinger and H. Linden, W. Chem. Ber., 1967, 100, 855–862 Search PubMed.
- J. R. Lauth, G. I. Scott, D. S. Cherry and A. L. Buikema, J. Environ. Toxicol. Chem., 1996, 15, 630–637 Search PubMed.
- J. H. Zar, Biostatistical Analysis, Prentice Hall, Upper Saddle River, NJ, USA, 4th edn., 1999 Search PubMed.
- Ph. D. Thesis, School of Public Health, University of South Carolina, Columbia, SC, 2002.
- W. E. Cotham and T. F. Bidleman, J. Agric. Food Chem., 1989, 37, 824–828 CrossRef CAS.
- S. Jensen, L. Renberg and L. Reutergardh, Anal. Chem., 1977, 49, 316–318 CrossRef CAS.
- T. F. Guerin, Environ. Pollut., 2001, 115, 219–230 CrossRef CAS.
- Y. Sudhakar and A. K. Dikshit, J. Environ. Sci. Health (B), 1999, 34, 587–615 Search PubMed.
- D. Skoog and J. Leary, Principles of Instrumental Analysis, 4th edn., 1992 Search PubMed.
- M. E. Hines, R. S. Evans, B.R. Sharack-Genthner, S. G. Willis, S. Friedman, J. N. Rooney-Varga and R. Devereux, Appl. Environ. Microbiol., 1999, 65, 2209–2216 CAS.
- G. M. King, Limnol. Ocean., 1988, 33, 376–390 Search PubMed.
- B. B. Harvey, Endosulfan Sulfate: Effects of Sulfate-Reducing Bacteria (SRB) in Tidal Marsh Sediments, Colorado School of Mines, Department of Environmental Science and Engineering, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: table of GC-ECD and GC-ITMS retention times, RT, for fipronil analytes; mass spectra of fipronil, fipronil desulfinyl, fipronil sulfide, fipronil sulfone and fipronil amide; and aqueous product distribution of fipronil in estuarine mesocosms over the experimental timescale. See http://www.rsc.org/suppdata/em/b3/b307304a/ |
| This journal is © The Royal Society of Chemistry 2004 |