DOI:
10.1039/B300976A
(Tutorial Review)
Chem. Soc. Rev., 2004,
33, 32-42
The Pauson–Khand reaction, a powerful synthetic tool for the synthesis of complex molecules
Received
11th June 2003
First published on 27th November 2003
Abstract
There are still some synthetic chemists who hesitate to use metal-mediated or -catalysed reactions. The Pauson–Khand reaction (PKR) is a powerful transformation that has now been sufficiently well developed to be routinely considered when planning a synthesis, especially of polycyclic complex molecules. This tutorial review aims to encourage the use of this process explaining the best ways of performing a PKR both in the stoichiometric and the catalytic version, showing the scope of the process and its limitations. Additionally, asymmetry can be introduced in the reaction using several strategies, which will be discussed. The most recent examples of the synthetic applications of the PKR in natural product synthesis will give the reader an idea of the great usefulness of this reaction.
 From left to right: Leticia Pérez-Serrano, Gema Domínguez, Javier Pérez-Castells, Jaime Blanco-Urgoiti, Loreto Añorbe | Jaime Blanco-Urgoiti was born in Madrid, Spain, in 1975. He studied Chemistry at the Universidad San Pablo CEU (Madrid)
(B.S. degree in 1997) and received his PhD in Organic Chemistry in 2003, under the supervision of Javier Pérez-Castells at the Universidad San Pablo CEU (Madrid). He is currently working in Lilly Research and Development Laboratories in Alcobendas (Madrid) as a Research Associate. |
| Loreto Añorbe was born in Tenerife, Spain, in 1973. She studied Chemistry at the Universidad Complutense de Madrid (B.S. degree in 1996) and received her PhD in Organic Chemistry in 2002, under the supervision of Miguel F. Braña at the Universidad San Pablo CEU (Madrid). In 1991 she enjoyed a three month stay in the group of Prof. Roberto Pellicciari (Universita di Perugia, Italy). She became Assistant Lecturer in 2002 and she is currently working on metal-mediated cyclization of allenes and enynoindoles. |
| Leticia Pérez Serrano was born in Madrid, Spain, in 1976. She studied Chemistry at the Universidad San Pablo CEU (Madrid)
(B.S. degree in 1998) and received her PhD in Organic Chemistry in 2002, under the supervision of Javier Pérez-Castells at the Universidad San Pablo CEU (Madrid). She is currently working in Lilly Research and Development Laboratories in Alcobendas (Madrid) as a Research Associate. |
| Gema Domínguez was born in Madrid, Spain, in 1960. She studied Chemistry at the Universidad Complutense de Madrid (B.S. in 1982), where she received her PhD in Organic Chemistry in 1986 under the supervision of Prof. Joaquín Plumet Ortega. She worked two years (1986–1987 and 1990) at the Consejo Superior de Investigaciones Científicas de Madrid (CSIC). She became Laboratory Demonstrator in 1987 at the Departament of Organic Chemistry of Universidad Complutense (Madrid) and Assistant Professor in 1994 at the University San Pablo CEU. She is currently working on metal-catalysed cyclization in PKR and metathesis. |
| Javier Pérez Castells was born in Madrid, Spain, in 1967. He studied Chemistry at the Universidad Complutense de Madrid (B.S. in 1990), where he received his PhD in Organic Chemistry in 1994 under the supervision of Profs. Miguel A. Sierra and Benito Alcaide. Since 1995 he has been assistant professor at the Universidad San Pablo-CEU, where he works on metal catalysed cyclization reactions. |
1 Introduction
The Pauson–Khand reaction (PKR) is formally a [2 + 2 + 1] cycloaddition in which a triple bond, a double bond and carbon monoxide form a cyclopentenone. This implies the formation of three new bonds and one or two cycles in the intermolecular or intramolecular versions, respectively (Scheme 1).
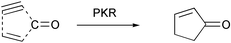 |
| | Scheme 1 Connectivity of the Pauson–Khand reaction. | |
The aim of this review will be to point out recent advances in this field and to encourage the use of this reaction for the synthesis of complex molecules.1 The most outstanding, recently reported examples of its use in natural product synthesis will also be shown. There are indeed few reactions that can compete with the Pauson–Khand in the construction of great molecular complexity in one step.
This reaction was first reported in the seventies. At the beginning it was carried out thermally under relatively severe conditions, and led to transformations of generally low efficiency. Until the mid-nineties, dicobalt octacarbonyl was the only cluster used to mediate the reaction. With the exception of propynoic acid derivatives, all alkynes underwent the reaction. On the other hand, only strained olefins reacted efficiently under the original reaction conditions. With respect to regioselectivity, the bulkier substituent of the alkyne is placed adjacent to the carbonyl in the cyclopentenone product. Unsymmetrical olefins usually give mixtures of regioisomers (Scheme 2).2
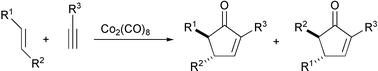 |
| | Scheme 2 Regiochemistry of the Pauson–Khand reaction. | |
In the early eighties Schore introduced the intramolecular version of this reaction. Until recently this version allowed the formation of 5.5- and 5.6-fused bicycles, (the former with generally greater levels of efficiency), and good conversions were achieved only with gem-disubstituted enynes (Scheme 3).3
 |
| | Scheme 3 Intramolecular PKR. | |
The main problem with the mechanism of the Pauson–Khand reaction is that beyond the cobalt-hexacarbonylalkyne complex, it is difficult to detect further intermediates. Nevertheless a generally accepted working mechanism has been proposed by Magnus (Scheme 4).4
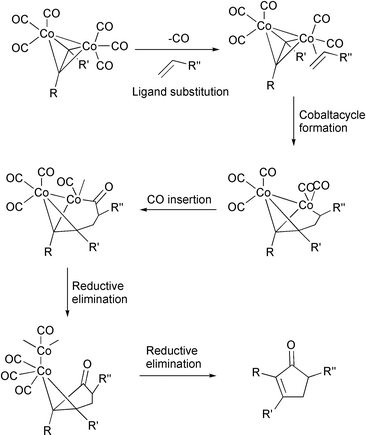 |
| | Scheme 4 | |
Several theoretical studies support this mechanism while it explains the regio- and stereochemical results of numerous examples. Starting from the initial hexacarbonyl complex, the first step would be the loss of one CO ligand. This step, strongly endothermic, is the rate-determining step and, consequently, acceleration of the process overall usually involves the use of promoters that act at this point, labilising one of the CO ligands. Some studies have shown, with the aid of IR spectroscopy, that this CO loss occurs after irradiation with shortwave UV, and pentacarbonyl dicobalt intermediates have been detected.5 Then, the olefin coordinates with the cobalt and is inserted into a Co–C bond forming a cobaltacycle. This is the other important stage in the reaction course as it determines its regiochemical and stereochemical outcome. Pericàs has studied these two important steps of the reaction using DFT calculations (Scheme 5).6 This author shows the importance of facilitating CO dissociation but points out this is not a sufficient condition for the reaction. Thus, the energy of the second step is also important and this is the reason why strained olefins react so well. Pericàs has established the relationship between the strain of the olefin and the energy of the cobaltacycle-forming step. In this step, the strain of cyclic olefins is liberated, favouring the process. Following this conclusion, they have carried out PKRs with cyclopropene, observing excellent yields under very mild conditions (Scheme 6). This group has also isolated sulfur ligated decarbonylated intermediates that support the dissociative decarbonylation step of the reaction course.
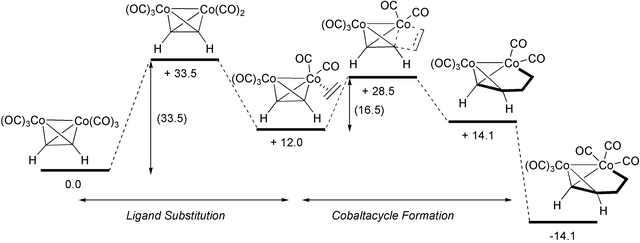 |
| | Scheme 5 Energy of the relevant steps of the PKR | |
 |
| | Scheme 6 | |
Other authors like Nakamura7 and Milet and Gimbert8 have performed high-level theoretical calculations on the cobaltacycle formation step. These authors show that the insertion of the olefin is the critical stereo- and regiochemical-determining step of the PKR. Nakamura points out that the bond-forming events occur only on one metal atom while the other acts as an anchor and exerts electronic influences through the Co–Co bond. Some further study is required with respect to the mechanism of this reaction, in particular on the reaction catalysed by other metals.
In addition to mechanistic studies, the main efforts in this chemistry at present are directed towards: (1) increasing the scope to new substrates, which involves the finding of new experimental protocols including the use of different metal clusters, and new promoters; (2) establishment of an efficient catalytic process; (3) development of the asymmetric version of the reaction; (4) the application of the PKR to the synthesis of natural products.
As we will show here, recent years have been excitingly fruitful and the scope of the PKR has widened remarkably due to the use of new promoters and reaction conditions. The reaction can be carried out catalytically and on solid support. There have been important advances in understanding what determines the stereochemical aspects of the reaction and several strategies have been developed to effect asymmetric processes. Although cobalt is still widely used, other metals are attaining great popularity, rhodium complexes being the most promising mediators. We can now say that this reaction is ready to be taken into account when planning a wide variety of syntheses of complex molecules.
2 Reaction conditions and scope
2.1 Accelerating the PKR
The main drawback of the PKR was originally its relatively low scope and, in many cases, poor conversions. Over the past years there has been an increase in the scope of this reaction, which has its origin in the finding of new reaction conditions. The first important report on the promotion of the PKR was by Smit and Caple, who effected the reaction with the reagents adsorbed on several solid supports (Dry State Adsorption Conditions, DSAC).9 This allowed the use of lower temperatures and shorter reaction times. Following that, the use of amine N-oxides, independently introduced by Jeong and Schreiber, has become the most popular way to promote the PKR.10 The N-oxides act by oxidising one CO ligand, which is transformed into CO2, thus forming a vacant site in the cobalt cluster (Scheme 7).
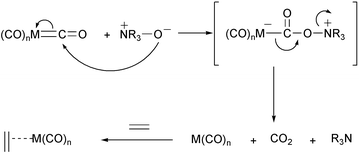 |
| | Scheme 7 | |
The amine N-oxides mostly used are trimethylamine N-oxide (TMANO) and N-methylmorpholine N-oxide (NMO). Recently the latter has been anchored to a solid support, facilitating the work up of the reaction and leading to excellent yields (Scheme 8).11
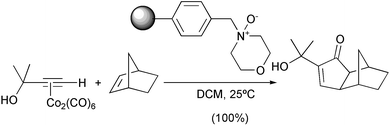 |
| | Scheme 8 | |
Another way to accelerate this reaction is by the addition of different chemical compounds. Several sulfides and sulfoxides have shown excellent results, especially in the intermolecular version of the reaction. One additive that has found widespread applicability is cyclohexylamine. This amine, introduced by Sugihara, has enhanced the yield of many intramolecular reactions. From the contributions of Sugihara we may say that sulfides are especially useful in the intermolecular version and with hydrolysable enynes, whereas amines are superior in the rest of the cases. These additives are added in 3.5 equivalent amounts with respect to the substrate and probably act by helping the displacement of a CO ligand and stabilising reaction intermediates (Scheme 9).12
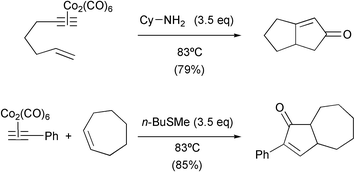 |
| | Scheme 9 | |
We have shown the positive effect of molecular sieves in both the catalytic and the stoichiometric version of the reaction. These zeolites probably retain CO molecules, increasing conversions even with unfavourable substrates, such as substituted olefins (Scheme 10).13
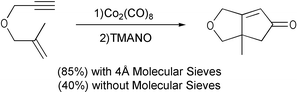 |
| | Scheme 10 | |
Other ways of labilising CO ligands consist of irradiating the complex. Ultraviolet light and ultrasound have shown a positive effect in the reaction. Most recently, several contributions have used microwaves as a way to accelerate the reaction. These latter conditions seem to shorten reaction times remarkably, although results are not spectacular with respect to conversions percentage.14
2.2 Other conditions for the PKR
Krafft has shown that modifications of the reaction conditions can lead to variations in the result of the intramolecular PKR. Thus, the use of a reductive atmosphere leads to the delivery of a cyclopentanone. The best results are obtained with Co4(CO)12 in alcohols under H2 or N2. Thermolysis of dicobalthexacarbonyl-alkyne complexes under H2 or O2 atmosphere in toluene forms monocyclic alkenes in moderate yields. These latter conditions are examples of so-called interrupted PKRs. Several groups have detected these and other types of products of interrupted processes that can become major under specially selected reaction conditions (Scheme 11).15
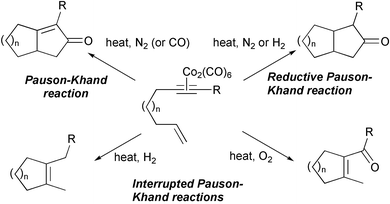 |
| | Scheme 11 | |
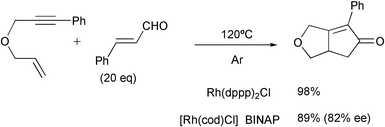
The PKR has been effected with reagents anchored on solid supports. The intermolecular version of this reaction, using one reagent anchored to a polymeric support, has been explored as a way to create libraries of compounds. In addition, when using these conditions with low weight starting compounds, the experimental work may be facilitated. After the early examples by Schore, who used alkynols linked to a solid support that were reacted with strained olefins, and the first intramolecular example by Bolton, Schreiber has recently used the PKR to carry out a split-pool synthesis of tricyclic and tetracyclic molecules. This diversity-oriented synthesis consisted of a Ferrier reaction followed by a PKR on a glycal template that yielded polycyclic scaffolds. They describe the synthesis of some individual products and a pilot library of 2500 compounds (Scheme 12).16
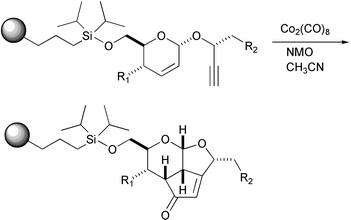 |
| | Scheme 12 | |
Other metals apart from cobalt can mediate the PKR. Although these have found best use in the catalytic version, as will be discussed later, the stoichiometric reaction has been performed with Zr, Ni, Fe, Ti, W and Mo. Many early contributions describe the use of these metals and have been reviewed previously.1 Christie and, more recently, Pericàs have studied the use of heterobimetallic Co–W and Co–Mo complexes, which are suitable precursors for the PKR and impart a high degree of selectivity in the process (Fig. 1).17
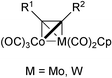 |
| | Fig. 1 | |
2.3 Scope of the reaction
Many functional groups such as ethers, alcohols, tertiary amines, acetals, esters, amides and heterocycles are compatible with the Pauson–Khand reaction. The intermolecular version is the most limited, as olefins without strain scarcely react. In the intramolecular version, relatively few carbon skeletons undergo the cyclisation. Most intramolecular PKRs use systems derived from hept-1-en-6-yne or propargyl allyl ethers or amines. Good conversions are generally achieved when a Thorpe–Ingold effect is helping. Substituted olefin fragments, in general, fail to react or lead to low yields. However, enynes connected through aromatic rings, allenes, enamines, ynamines and traceless tethers have recently joined the group of substrates used successfully in this reaction, involving the synthesis of some medium sized rings. In early times there was a common idea that alkenes possessing electron-withdrawing groups were not adequate substrates for Pauson–Khand reactions and underwent undesired CH insertion reactions. Despite this idea, Carretero has reported several examples of this reaction involving electron-deficient alkenes, such as α,β-unsaturated ketones, esters, nitriles, sulfoxides and sulfones (Scheme 13).18
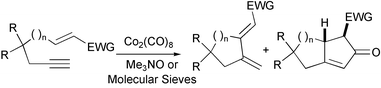 |
| | Scheme 13 | |
We and other groups have used aromatic substrates allowing the synthesis of aromatic polycycles. We have observed in some cases isomerisation of the emerging double bond.19 Lovely has reported an unusual regiochemistry in the PKR with similar substrates. In this case, due to the buttressing effect of the tert-butyl situated in the aromatic ring, a bridged compound is obtained involving the formation of seven and eight membered rings (Scheme 14).20
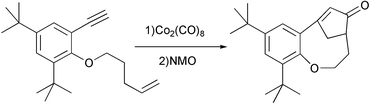 |
| | Scheme 14 | |
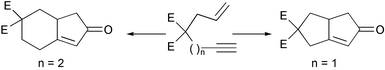
Some of the most promising new substrates for the PKR introduced in the past years are those that bear an allene that acts as the olefinic part of the reaction. Allenes have an interesting property: they can react with both double bonds depending on the substitution pattern and on the reaction conditions. The first studies were by Brummond, who established the substitution patterns for the reaction with either the external or the internal bond of the allenic fragment (Scheme 15). This group has applied these studies to the synthesis of hydroxymethylfulvalene, a potent anticancer agent, and has developed an asymmetric version of the reaction (vide infra).21
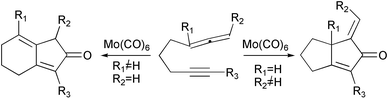 |
| | Scheme 15 | |
After these studies, several groups have used the intramolecular allenic PKR for the synthesis of medium sized rings fused to aromatic rings, cyclohexanes (vide infra) or β-lactams.22 The best results are obtained using rhodium catalysed reactions.
Cook has reported an impressive example of the allenic PKR this year. Dicyclopenta[a,e]pentalene was obtained via a molybdenum hexacarbonyl mediated tandem reaction (Scheme 16).23
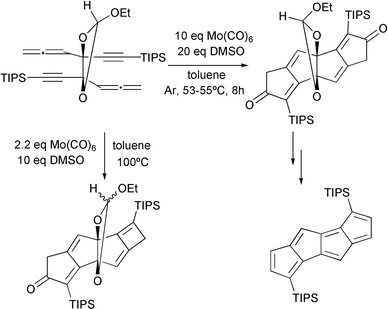 |
| | Scheme 16 | |
Exocyclic olefin fragments are among the few disubstituted double bonds that react efficiently in PKRs. Several examples have appeared in the literature and are covered by previous reviews. They include methylenecyclohexanes, methylenecyclopropanes and methylenepyranes (Fig. 2).
 |
| | Fig. 2 | |
Another group of interesting new substrates are those possessing traceless tethers. These compounds have the advantage of using the intramolecular version of the reaction, which is not limited to strained olefins and does not have regioselection problems. In a second step, which sometimes occurs in a domino fashion, they give a monocyclic compound upon cleavage of the tether. After the first tethers containing oxygen, used by Smit, and those by Pericàs, in which a sulfur atom was reductively eliminated after the PKR,24 the most recent reports use silicon as a traceless tether. Pagenkopf has succeeded in obtaining a monocyclic cyclopentenone in one step, starting from a vinylsilane. The mechanism of the reductive elimination of the silicon tether was investigated by this group and implies the participation of water in the process (Scheme 17).25
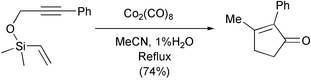 |
| | Scheme 17 | |
Several domino processes have also expanded the synthetic applications of the PKR. In addition to palladium allylic alkylations that will be commented on later, we have used a combined metathesis–PKR for the synthesis of tricyclic compounds in one step. The cobalt cluster acts first as a protecting group to avoid undesired enyne metathesis processes (Scheme 18).26
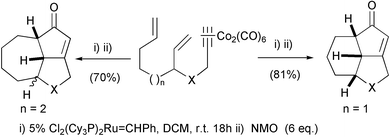 |
| | Scheme 18 | |
3 The catalytic Pauson–Khand reaction27
3.1 Catalysis by cobalt species
There has been great interest in developing efficient protocols for a catalytic version of the PKR. The synthetic applications that can be envisioned for this reaction make this aim an essential target in this research. After the first example of a real catalytic reaction reported by Rautenstrauch in 1990, during the nineties, several experimental conditions were reported.1 The common drawback of these protocols was the use of severe conditions involving high CO pressures and temperatures. The situation changed at the end of that decade when the groups of Livinghouse and Krafft reported new reaction conditions using only 1 atmosphere of CO. Livinghouse used either high intensity irradiation or heat to promote an efficient PKR using 5% of Co2(CO)8 under 1 atmosphere of CO. The thermal conditions gave best results when using a narrow gap of temperatures situated between 65 °C and 75 °C. Livinghouse put great emphasis on the use of high purity catalysts as an essential requirement for getting good results.28 On the other hand, Krafft showed that enyne–Co2(CO)6 complexes, previously formed and purified, or commercial Co2(CO)8 and Co4(CO)12 are effective enough in the catalytic PKR under 1 atmosphere of CO. The efficiency of the reaction is enhanced by the addition of Lewis bases, mainly 2–3 equivalents of cyclohexylamine, which act by stabilising reaction intermediates. The amine also facilitates the initial decarbonylation step (Scheme 19).29
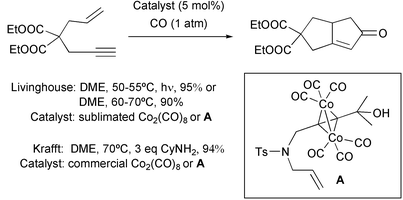 |
| | Scheme 19 | |
From the experience of our group, we can add to these considerations on the purity of Co2(CO)8 that recently opened bottles of this catalyst usually work well in catalytic PKR, but care must be taken with storage of this metal cluster, as it may lose activity in few days.
Krafft has described sub-stoichiometric reactions (35% of Co2(CO)8) under nitrogen atmosphere, in which cyclohexylamine was used as an iterative displacement agent of CO ligands (Scheme 20).29
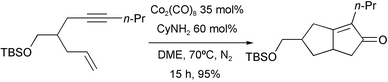 |
| | Scheme 20 | |
Jeong first made the addition of phosphines and phosphites to enhance the yields of the catalytic PKR. These additives may avoid the formation of inactive cobalt species, stabilising intermediate active clusters. These findings have recently been used in an enantioselective version of the reaction. When adding chiral phosphines such as BINAP, good enantiomeric excess is reached with moderate chemical yields. The best results are obtained with unsubstituted olefins and alkynes. Substitution patterns in the skeleton dramatically affect the stereochemical outcome of the reaction (Scheme 21).30
 |
| | Scheme 21 | |
Buchwald has recently used chiral phosphites derived from BINOL as additive in a cobalt catalysed reaction (Fig. 3). The enantiomeric excesses reached with these additives are moderate and far behind those obtained with these ligands in other transformations such as asymmetric hydrogenation. Only aryl substituted 1,6-enynes maintained a good degree of enantioselectivity.31
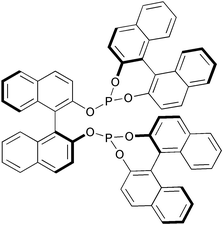 |
| | Fig. 3 | |
Other cobalt sources have been used recently and show high efficiency in the catalysis of the PKR. Chung has reported several ways to generate active cobalt species.32 the most recent contributions from this group deal with the description of several reusable catalysts. Thus, cobalt supported on mesoporous silica was presented as the first heterogeneous catalyst for the PKR. This catalyst is obtained by decomposition of Co2(CO)8 in the presence of silica (SBA-16) under refluxing toluene. Good yields are obtained using this system, even when reutilising the same catalyst two or three times (Scheme 22).
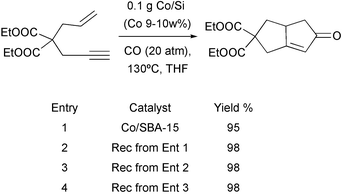 |
| | Scheme 22 | |
Another similar system described by this group consists of supporting cobalt on charcoal. In this case, the amount of cobalt supported by the solid is slightly superior. The main problem with these systems is that they use severe reaction conditions, in particular, high CO pressures. This drawback has been partially solved with the obtention of colloidal cobalt nanoparticles, which react at lower pressures. These particles have been used in the first PKR carried out in aqueous media.32
The combination of the PKR with other processes in one step increases the synthetic utility of this reaction. Chung has recently been very active in using palladium catalysed asymmetric allylic alkylations to obtain enynes that are cyclised in the same reaction. In their latest contribution, they use a combination of palladium and cobalt nanoparticles immobilised on silica (PCNS) to form bicyclic enones after sequential allylic alkylation–PKR (Scheme 23).32
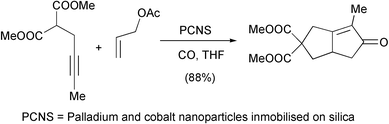 |
| | Scheme 23 | |
Carbonylcobalt complexes have also been modified by the interchange of several CO by phosphines. The resulting clusters are more resistant against high temperatures and thus are interesting alternatives for catalytic reactions performed under severe conditions. The phosphines can also be immobilised in resins thus giving anchored cobalt complexes. These catalysts, used at 5% give moderate to good yields in a series of substrates. When using dendron-functionalised polystyrene as the immobilising agent, the results are improved remarkably (Scheme 24).33
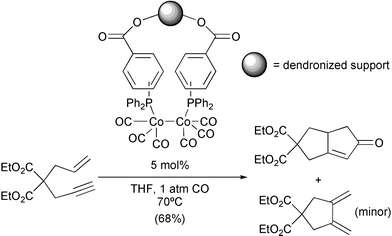 |
| | Scheme 24 | |
Other cobalt metal clusters apart from Co2(CO)8 have exhibited high reactivity in the catalytic PKR. In addition to Co4(CO)12, which can be activated by Lewis bases, Sugihara has introduced methylidynetricobalt nonacarbonyl as an efficient cluster, even in the absence of activators. The reactivity of this cluster may be due to the facile coordination of the substrate to the metal trough dissociative and associative mechanisms and/or cleavage of one Co–Co bond. Standard conditions with this cluster use 7 atmospheres of CO in toluene at 120 °C and lead to good results both in the inter- and the intramolecular versions. These kinds of clusters are easily prepared by reacting dicobalt octacarbonyl with trihaloalkanes. They are more stable against auto oxidation than dicobalt octacarbonyl itself (Fig. 4).34
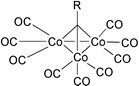 |
| | Fig. 4 | |
3.2 Catalysis by other metals
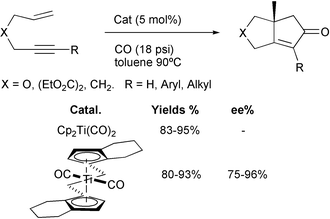
Several metals have been used to catalyse PKR besides cobalt. In addition to W, Zr and some recent reports with Ir, there has been much attention to the use of Ti, Ru and Rh. We would dare to say that most promising results are being reported with several Rh complexes and we can envision that this metal will overtake cobalt in the coming years. With respect to Ti, Buchwald has developed several titanocenes that act as efficient catalysts in the PKR and in PK-like reactions with cyanides. In particular, titanocene dicarbonyl is able to catalyse the reaction of different 1,6 and 1,7-enynes with excellent functional group tolerance, but fails to react with sterically hindered olefins and alkynes. In a very recent contribution, this group has prepared a series of aryloxide complexes. These new catalysts are able to promote cyclisation with some sterically hindered enynes. The efficiency of the cyclisation reactions is dependent upon the substitution of the aryloxide ligand (Scheme 25).35
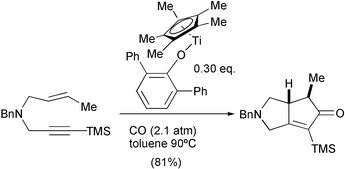 |
| | Scheme 25 | |
In an enantioselective version of this methodology, Buchwald has used chiral titanocenes to effect the PKR. These catalysts have achieved good chemical yields with moderate enantiomeric excesses (72–96%). In Scheme 26 a comparison of the racemic and asymmetric reactions is shown.35
 |
| | Scheme 26 | |
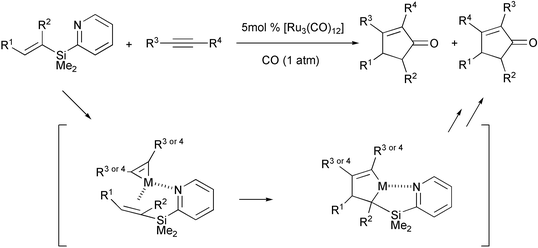
Late transition metals were introduced more recently. After the first reports by Murai and Mitsudo, both in 1997, ruthenium complexes have been used by Itami and Yoshida recently. In their work, they have contributed remarkably to the expansion of the scope of the intermolecular PKR. The strategy consists of using olefins bearing a pyridisilyl group which is readily removed after the reaction. These α or β substituted vinylsilanes are easily obtained from alkynes. The pyridyl group directs the PKR by a possible coordination of the nitrogen with the metal, which accelerates the process and gives complete regioselectivity. The directing group is eliminated in the reaction, giving rise directly to 4 and/or 5 substituted cyclopentenones. This avoids the use of ethylene or strained olefins in the intermolecular PKR and is a way to solve the problem of the regioselectivity with unsymmetrical olefins (Scheme 27).36
 |
| | Scheme 27 | |
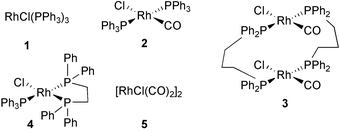
Rhodium complexes are effective catalysts for the PKR and are receiving increasing interest. Jeong has been especially active in this field and has shown that several complexes are able to promote the process. The scope and efficiency of the reaction depends on the catalyst used. Those depicted in Fig. 5 are the most active.37
 |
| | Fig. 5 | |
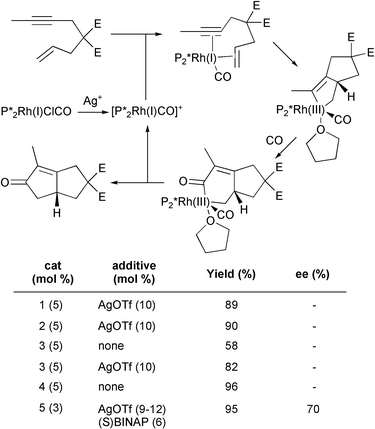
Some of these rhodium complexes need activation with AgOTf. Modification of complex 5 produces a chiral catalyst that can give rise to enantioselective processes. However, a careful choice of conditions, including CO pressure, activation, solvent and ligands, is essential, to obtain good enantioselectivity. In Scheme 28, some reactions with different catalysts are compared, including the best conditions found for the asymmetric process. Jeong has proposed a reaction course for the rhodium catalysed PKR which is also summarised in this Scheme. The face-selectivity in the first coordinating step would determine the stereochemical outcome of the reaction, and explains the influence of the solvent and the electronic character of the ligand used.37
 |
| | Scheme 28 | |
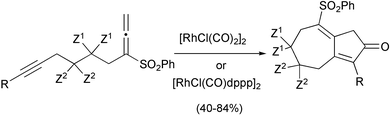
Two important advances using rhodium catalysts have recently appeared. The first is the efficient synthesis of seven membered rings via allenic PKRs. Mukai has synthesised 19 examples of bicyclo[5,3,0]decenones, using several rhodium catalysts. Different catalysts work best with different allenynes. The process tolerates hydroxy and silyloxy groups and reaches good yields in general (Scheme 29).38
 |
| | Scheme 29 | |
The second is an elegant CO-transfer carbonylation reaction in which aldehydes serve as a source of CO for a PKR catalysed by rhodium, ruthenium or iridium complexes under argon atmosphere. This approach begins with decarbonylation of the aldehyde and transfer of the CO to the enyne. The authors show there is no free CO in the reaction medium that is used to form the PK product, as they perform reactions in 13CO atmosphere without observing incorporation of 13C in the final product. The enantiomeric excess reached when using a chiral ligand is higher with this methodology than when using CO atmosphere in the absence of the aldehyde (Scheme 30).39
 |
| | Scheme 30 | |
4 Inducing asymmetry in the Pauson–Khand reaction
Previous reviews have covered the different efforts made by many groups regarding asymmetric PKR. However there have been important advances after 1999 in the different approaches that can be used to introduce asymmetry in the process. These are: (1) The chiral substrate approach; (2) the chiral auxiliary approach; (3) the chiral metal complex approach and (4) the chiral promoter approach. The best results have been reported using chiral substrates or chiral auxiliaries. The chiral metal complex approach is still in its beginnings and will probably show excellent results in the future.
4.1 Transfering chirality from the substrate
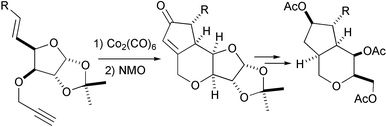
This approach uses chiral precursors that transfer their chirality to the final cyclopentenone. This implies the synthesis of chiral substrates, which have generally been made from classic chiral pools. While amino acids have received scarce attention as starting materials, carbohydrates have been used widely for the synthesis of chiral enynes. The most recent contribution has used an isopropylidenedioxyfuranoside as a scaffold to obtain a series of enynes that are transformed into cyclopenta[c]pyranes (Scheme 31).40
 |
| | Scheme 31 | |
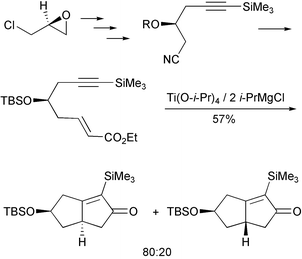
One recent contribution in this area uses epichlorhydrin to develop a new chiral block that gives enantiomerically pure 4-substituted bicyclooctanes (Scheme 32).41
 |
| | Scheme 32 | |
Brummond has reported the successful transfer of chirality from chiral allenes. The PKR was effected using molybdenum and zirconium complexes, and with the internal bond of the allene giving α-alkylidene and α-silylidene cyclopentenones. The enantioselectivity of these reactions was good, although partial isomerisation of the exocyclic double bond formed in the cyclisation process occurred. A typical example is depicted in Scheme 33.42
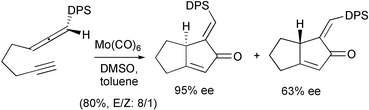 |
| | Scheme 33 | |
4.2 Using chiral auxiliaries
Major contributions in this area are due to Pericàs' group. Many of these have been reviewed previously. This group has worked with chiral sulfur moieties like 10-methylthioisoborneol, which coordinate with the metal, giving complexes that have sometimes been isolated. Some of the complexes studied by this group are shown in Fig. 6. Complexes like 6 give good stereochemical results, although they do not work well when sulfur is substituted by oxygen. Other auxiliaries like Oppolzer's camphorsultam 7 or chiral oxazolidinones 8 gave excellent results in stereocontrol and yields. On the other hand, chiral sulfoxides like 9 only reacted when bearing certain moieties at the alkyne, and in those cases gave mixtures of regioisomers with partial loss of the enantiomeric excess (Fig. 6).43
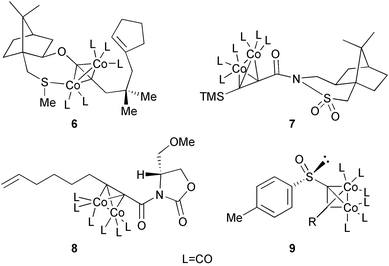 |
| | Fig. 6 | |
These chiral auxiliaries act by transmitting their chirality to the metal cluster due to the chelation with a cobalt atom, which upon diastereoselective complexation of the olefin, controls the absolute configuration of the final cyclopentenone (Fig. 7).
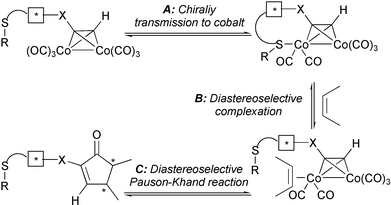 |
| | Fig. 7 | |
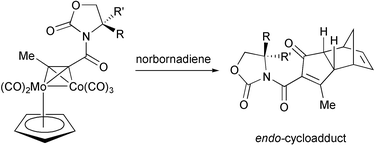
The most recent contribution from this group describes the use of chiral alkynylthiols instead of the parent ethers as so-called chelating auxiliaries. These new auxiliaries have higher stability against hydrolysis, are more efficiently prepared and exhibit excellent diastereoselectivities in PKR with norbornene and norbornadiene. The chiral auxiliary is eliminated using samarium iodide, and a retro-Diels–Alder reaction can finally afford highly enantiomerically enriched 4-substituted cyclopentenones (Scheme 34).43
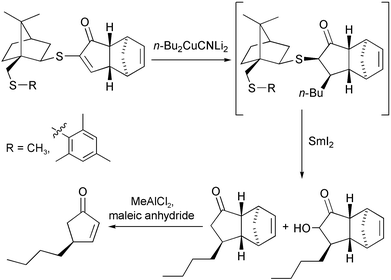 |
| | Scheme 34 | |
Carretero has demonstrated the feasibility of carrying out the PKR with electron deficient olefins. Among the different electronic withdrawing groups used by this group, chiral sulfoxides attached at the olefin are efficient auxiliaries and reach high enantiomeric excess. The proximity of the chiral sulfur to the reaction centre explains these good results. In addition, mixtures of Z and E vinylsulfoxides gave only one isomer after the cyclisation. Reductive cleavage of the sulfoxide is carried out by treatment with activated zinc (Scheme 35).44
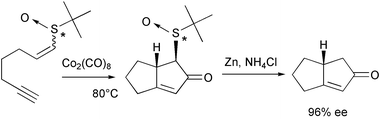 |
| | Scheme 35 | |
4.3 Using chiral metal complexes
This elegant approach actually groups several different methodologies. The most experimentally simple possibility has been shown above and consists of the addition of chiral ligands to metal complexes that form in situ chiral aggregates. In general the ligands used are well known for their success in other chemical transformations, but in the PKR only give good enantioselectivities in certain examples. As we have seen, this methodology has been carried out with cobalt, titanium, ruthenium and rhodium catalysts. Another possibility is to prepare desymmetrised metal clusters by interchanging one ligand for a chiral one. After the first report by Greene, Pericàs has recently obtained different chiral complexes using bidentate (P,N) and (P,S) ligands. The most effective result was achieved using a ligand called PuPHOS, which is readily obtained from natural product (+)-pulegone. This ligand reacted with cobalthexacarbonyl-alkyne complexes derived from terminal alkynes giving diastereomerically-enriched mixtures of chiral complexes that were separated. The major isomers, upon reaction with norbornadiene, gave excellent results in yields and stereoselectivities (Scheme 36).45
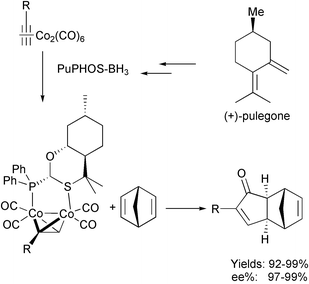 |
| | Scheme 36 | |
Finally metal clusters can be desymmetrised substituting one of the metal atoms for another metal. Pericàs has recently prepared heterobimetallic (Mo–Co) complexes. The chirality of the tetrahedral core formed by the alkyne and the metals is used to induce high levels of stereocontrol. The complexes obtained are enantiomerically pure with the aid of several traditional chiral auxiliaries attached to the alkyne. The complexation of the chiral alkyne with the two metals gives a mixture of diastereomeric bimetallic complexes that are separated. The major isomer reacted with norbornadiene efficiently and with total regioselectivity. Interestingly, the face-stereoselectivity is reversed with these complexes and the endo-fused norbornadiene adducts are obtained instead of the expected exo compounds (Scheme 37).46
 |
| | Scheme 37 | |
4.4 Using chiral promoters
Several groups have used chiral promoters, generally amine N-oxide derived from natural alkaloids. The chiral promoter should be able to make a selective decarbonylation of one carbonyl of the cobalt cluster, generating an enantiomerically enriched cyclopentenone after the cyclisation. To date, the enantiomeric excesses reached with this approach have been very low. In a recent report Kerr has modified this methodology using the chiral N-oxide to prepare a desymmetrised cobalt complex by selective substitution of one carbonyl by a phosphine. This leads to sterically and electronically differentiated cobalts. The resulting asymmetric complexes are isolated, with moderate to good enantiomeric excess. In the presence of norbornadiene and an achiral amine N-oxide, they lead to enantiomerically enriched PK adducts (Scheme 38).47
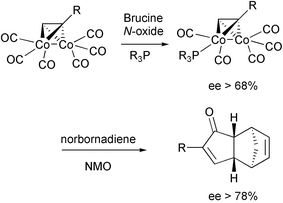 |
| | Scheme 38 | |
5 Natural product synthesis using the PKR
There has been a permanent increase in the use of the PKR for the synthesis of natural products. The cyclopentane ring is quite common in nature and the PK adducts are easily functionalised. Applications up to 1999 have been covered by previous reviews.1 These include the synthesis of prostaglandins, different triquinanes and polyquinanes like ceratopicanol, furanether B, kainic acid, hirsutene, fenestranes, brefeldine A, epoxidictimene, loganine, xestobergsterol, spatane, daphane, iridomirmecin, dendrobin, kalmanol and β-cuparenone. We will present herein some of the syntheses that have appeared in the last 3 years.
Krafft has reported the total synthesis of asteriscanolide, a sesquiterpene structurally related to epoxidictimene. The synthesis is based on a regioselective intermolecular PKR with propene. The formation of the cyclooctane ring is accomplished in the final stages by means of a ring closing metathesis reaction (Scheme 39).48
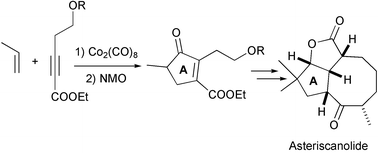 |
| | Scheme 39 | |
An intermolecular PKR has been used as a key step for the synthesis of the cedrene skeleton. The starting material was a monocyclic enyne with an exocyclic olefin fragment. A bridged adduct is obtained in high yield which is further manipulated to provide a total synthesis of α- and β-cedrene (Scheme 40).49
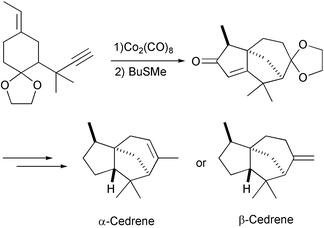 |
| | Scheme 40 | |
Zard has recently reported a total synthesis of 13-deoxyserratine, a lycopodium alkaloid with high structural complexity, in only 10 steps and 12% overall yield. The key steps in this synthesis were a highly diastereoselective PKR and a radical cyclisation cascade (Scheme 41).50
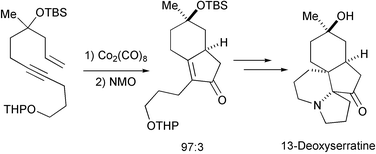 |
| | Scheme 41 | |
Mukai has effected a total synthesis of 8α-hydroxystreptazolone in which the key step is an intramolecular PKR carried out on a 2-oxazolone derivative. This implies the use of an enamine as the olefinic part of the reaction. The PKR is accomplished in a highly stereoselective manner. This compound is a natural product possessing antifungal and antibiotic activities (Scheme 42).51
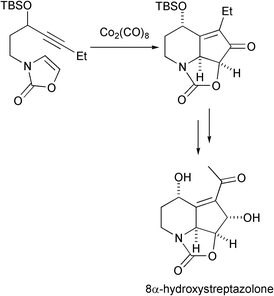 |
| | Scheme 42 | |
6 Concluding remarks
There are really few organic transformations that add so much molecular complexity in one step as the Pauson–Khand reaction. The process yields cyclopentenones, which can be easily transformed into different functionalised cyclopentanes, present in the structure of many natural products. This reaction had its main drawback in the limited scope, low yields and lack of efficient catalytic procedures. But we have tried to demonstrate that these problems have been, to a great extent, solved. There are several efficient ways to accelerate the reaction. Using these new procedures, the intramolecular PKR is now successfully used to obtain 5, 6 and sometimes 7 membered rings. Disubstituted and electronically poor olefin fragments are frequently reactive and allenes act efficiently as the olefinic part. The intermolecular version is still quite limited, as unstrained olefins scarcely react, but the use of traceless tethers or directing groups like pyridisilyl have circumvented this problem, also avoiding the formation of regioisomeric mixtures. There are a wide variety of catalytic protocols available to effect this reaction, some of them under mild conditions. Nevertheless the catalytic PKR is not a completely solved problem. Many protocols only give good results with favourable substrates. Others use interesting reusable catalysts but under severe reaction conditions. The most promising advances come from the use of late transition metal complexes (rhodium, ruthenium). Although there is not one single catalyst that works well with every substrate, with a careful selection of the metal complex, one can reasonably expect to achieve good results. Asymmetric PKR can be carried out using different strategies. Important findings will probably come from the use of chiral ligands and chiral metal complexes. Advances in the comprehension of the reaction mechanism will allow accurate prediction of the stereochemical outcome of the reaction. Finally, all this knowledge is producing impressive synthetic applications that will encourage synthetic chemists to rely on this reaction when planning their syntheses.
7 Acknowledgements
The authors are grateful to Spanish MCYT (grant BQU2002-00137) and the Fundación San Pablo (grant 2/01) for financial support. We wish to thank Linda Hamalainen for linguistic help.
References
- For a comprehensive review, see: K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263 Search PubMed
 .
.
- For a recent study on the regiochemistry of an intermolecular PKR, see: W. J. Kerr, M. McLaughlin, P. L. Pauson and S. M. Robertson, J. Organomet. Chem., 2001, 630, 104 Search PubMed .
- N. E. Schore and M. C. Croudace, J. Org. Chem., 1981, 46, 5436 CrossRef CAS ; C. Exon and P. Magnus, J. Am. Chem. Soc., 1983, 105, 2477 CrossRef CAS .
- P. Magnus and L.-M. Príncipe, Tetrahedron Lett., 1985, 26, 4851 CrossRef CAS .
- C. M. Gordon, M. Kiszka, I. R. Dunkin, W. J. Kerr, J. S. Scott and J. Gebicki, J. Organomet. Chem., 1998, 554, 147 CrossRef CAS .
- M. A. Pericàs, J. Balsells, J. Castro, I. Marchueta, A. Moyano, A. Riera, J. Vázquez and X. Verdaguer, Pure Appl. Chem., 2002, 74, 167 Search PubMed and references cited therein.
- M. Yamanaka and E. Nakamura, J. Am. Chem. Soc., 2001, 123, 1703 CrossRef CAS .
- T. J. M. de Bruin, A. Milet, F. Robert, Y. Gimbert and A. E. Green, J. Am. Chem. Soc., 2001, 123, 7184 CrossRef CAS .
- W. A. Smit, A. S. Gybin, A. S. Shashkov, Y. T. Struchkov, L. G. Kyzmina, G. S. Mikaelian, R. Caple and E. D. Swanson, Tetrahedron Lett., 1986, 27, 1241 CrossRef CAS .
- S. Shambayati, W. E. Crowe and S. L. Schreiber, Tetrahedron Lett., 1990, 31, 5289 CrossRef CAS ; N. Jeong, Y. K. Chung, B. Y. Lee, S. H. Lee and S.-E. Yoo, Synlett, 1991, 204 CrossRef CAS .
- D. S. Brown, E. Campbell, W. J. Kerr, D. M. Lindsay, A. J. Morrison, K. G. Pike and S. P. Watson, Synlett, 2000, 1573 CAS .
- T. Sugihara, M. Yamada, M. Yamaguchi and M. Nishizawa, Synlett, 1999, 771 CAS ; T. Sugihara, M. Yamada, H. Ban, M. Yamaguchi and C. Kaneko, Angew. Chem. Int. Ed. Engl., 1997, 36, 2801 CrossRef CAS .
- L. Pérez-Serrano, L. Casarrubios, G. Domínguez and J. Pérez-Castells, Org. Lett., 1999, 1, 1187 CrossRef CAS ; J. Blanco-Urgoiti, L. Casarrubios, G. Domínguez and J. Pérez-Castells, Tetrahedron Lett., 2002, 43, 5763 CrossRef CAS .
- S. Fischer, U. Groth, M. Jung and A. Schneider, Synlett, 2002, 2023 CAS ; M. Iqbal, N. Vyse, J. Dauvergne and P. Evans, Tetrahedron Lett., 2002, 43, 7859 CrossRef CAS .
- M. E. Krafft, L. V. R. Boñaga and C. Hirosawa, J. Org. Chem., 2002, 67, 1233 CrossRef CAS .
- H. Kubota, J. Lim, K. M. Depew and S. L. Schreiber, Chem. Biol., 2002, 9, 265 CrossRef CAS .
- R. Rios, R. M. A. Pericàs and A. Moyano, Tetrahedron Lett., 2002, 43, 4903 CrossRef CAS ; A. J. Fletcher, D. T. Rutherford and S. D. R. Christie, Synlett, 2000, 1040 CAS .
- M. R. Rivero and J. C. Carretero, J. Org. Chem., 2003, 68, 2975 CrossRef .
- L. Pérez-Serrano, J. Blanco-Urgoiti, L. Casarrubios, G. Domínguez and J. Pérez-Castells, J. Org. Chem., 2000, 65, 3513 CrossRef CAS .
- C. J. Lovely, H. Seshadri, B. R. Wayland and A. W. Cordes, Org. Lett., 2001, 3, 2607 CrossRef CAS .
- K. M. Brummond and J. Lu, J. Am. Chem. Soc., 1999, 121, 5087 CrossRef CAS ; K. M. Brummond, J. Lu and J. Petersen, J. Am. Chem. Soc., 2000, 122, 4915 CrossRef CAS .
- B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Eur. J., 2002, 8, 1719 CrossRef CAS .
- H. Cao, J. Flippen-Anderson and J. M. Cook, J. Am. Chem. Soc., 2003, 125, 3230 CrossRef CAS .
- J. Castro, A. Moyano, M. A. Pericás and A. J. Riera, J. Org. Chem., 1998, 63, 3546 .
- J. F. Reichwein, S. T. Iacono and B. L. Pagenkopf, Tetrahedron, 2002, 58, 3813 CrossRef CAS .
- M. Rosillo, L. Casarrubios, G. Domínguez and J. Pérez-Castells, Org. Biomol. Chem., 2003, 1, 1450 RSC .
- A review on the catalytic PKR has just appeared: S. E. Gibson and A. Stevenazzi, Angew. Chem. Int. Ed., 2003, 42, 1800 Search PubMed .
- B. L. Pagenkopf and T. Livinghouse, J. Am. Chem. Soc., 1996, 118, 2285 CrossRef CAS ; D. B. Belanger and T. Livinghouse, Tetrahedron Lett., 1998, 39, 7641 CrossRef CAS .
- M. E. Krafft, L. V. R. Boñaga and C. Hirosawa, J. Org. Chem., 2001, 66, 3004 CrossRef and references cited therein..
- K. Hiroi, T. Watanabe, R. Kawagishi and I. Abe, Tetrahedron: Asymmetry, 2000, 11, 797 CrossRef CAS .
- S. J. Sturla and S. L. Buchwald, J. Org. Chem., 2002, 67, 3398 CrossRef CAS .
- K. H. Park, S. U. Son, Y. K. Chung and T. Hyeon, Org. Lett., 2002, 4, 4361 CrossRef CAS and references cited therein.
- A. Dahan and M. Portnoy, Chem. Commun., 2002, 2700 RSC and references cited therein.
- T. Sugihara, M. Yamaguchi and M. Nishizawa, Chem. Eur. J., 2001, 7, 1589 CrossRef CAS .
- S. J. Sturla and S. L. Buchwald, Organometallics, 2002, 21, 739 CrossRef CAS and references cited therein.
- K. Itami, K. Mitsudo and J. Yoshida, Angew. Chem. Int. Ed., 2002, 41, 3481 CrossRef CAS .
- N. Jeong, B. K. Sung, J. S. Kim, S. B. Park, S. D. Seo, J. Y. Shin, K. Y. In and Y. K. Choi, Pure Appl. Chem., 2002, 74, 85 Search PubMed .
- C. Mukai, I. Nomura and S. Kitagaki, J. Org. Chem., 2003, 68, 1376 CrossRef CAS ; K. M. Brummond, H. Chen, K. D. Fischer, A. D. Kerekes, B. Rickards, P. C. Sill and S. J. Geib, Org. Lett., 2002, 4, 1931 CrossRef CAS .
- T. Morimoto, K. Fuji, K. Tsutsumi and K. Kakiuchi, J. Am. Chem. Soc., 2002, 124, 3806 CrossRef CAS ; T. Shibata, N. Toshida and K. Takagi, J. Org. Chem., 2002, 67, 7446 CrossRef CAS .
- A. Pal and A. Bhattacharjya, J. Org. Chem., 2001, 66, 9071 CrossRef CAS .
- K. Subburaj, S. Okamoto and F. Sato, J. Org. Chem., 2002, 67, 1024 CrossRef CAS .
- K. M. Brummond, A. D. Kerekes and H. Wan, J. Org. Chem., 2002, 67, 5156 CrossRef CAS and references cited therein.
- I. Marchueta, E. Montenegro, D. Panov, M. Poch, X. Verdaguer, A. Moyano, M. A. Pericàs and A. Riera, J. Org. Chem., 2001, 66, 6400 CrossRef CAS .
- M. R. Rivero, J. Adrio and J. C. Carretero, Eur. J. Org. Chem., 2002, 2881 CAS .
- X. Verdaguer, A. Moyano, M. A. Pericàs, A. Riera, M. A. Maestro and J. Mahía, J. Am. Chem. Soc., 2000, 122, 10242 CrossRef CAS .
- R. Rios, M. A. Pericàs, A. Moyano, M. A. Maestro and J. Mahía, Org. Lett., 2002, 4, 1205 CrossRef CAS .
- D. R. Carbery, W. J. Kerr, D. M. Lindsay, J. S. Scott and S. P. Watson, Tetrahedron Lett., 2000, 41, 3235 CrossRef CAS .
- M. E. Krafft, Y. Y. Cheung and K. A. Abboud, J. Org. Chem., 2001, 66, 7443 CrossRef CAS .
- W. J. Kerr, M. McLaughlin, A. J. Morrison and P. L. Pauson, Org. Lett., 2001, 3, 2945 CrossRef CAS .
- J. Cassayre, F. Gagosz and S. Z. Zard, Angew. Chem., Int. Ed., 2002, 41, 1783 CrossRef CAS .
- I. Nomura and C. Mukai, Org. Lett., 2002, 4, 4301 CrossRef CAS .
Footnote |
| † Present address: Centro de investigación Lilly S. A. Avda. de la industria 30. 28108 Alcobendas, Madrid, SPAIN. |
|
| This journal is © The Royal Society of Chemistry 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.