Resolving Mn framework sites in large cage aluminophosphate zeotypes by high field EPR and ENDOR spectroscopy
D.
Arieli
*a,
T. F.
Prisner
b,
M.
Hertel
b and
D.
Goldfarb
*a
aDepartment of Chemical Physics, Weizmann Institute of Science, Rehovot, Israel 76100. E-mail: daniella.goldfarb@weizmann.ac.il
bDepartment of Physical and Theoretical Chemistry, University of Frankfurt/Main, Germany
First published on 19th November 2003
Abstract
The structural features of Mn(II) incorporated into two large cage zeotypes, Mn-UCSB-10Mg and Mn-UCSB-6Mg, were explored by combining multifrequency CW-EPR with W-band ENDOR spectroscopy. As-synthesized samples, dried both at room temperature and 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C, were examined and the results were compared with the reference samples Mn-AlPO4-20 and Mn-AlPO4-5. High-frequency CW EPR experiments (at W- and G- bands) resolved two main types of Mn(II) framework sites with significantly different 55Mn hyperfine couplings and slightly different g values. These results were further corroborated by 55Mn ENDOR spectra, which enabled a more accurate determination of the two hyperfine coupling values and revealed the presence of a third species. ENDOR experiments carried out at magnetic fields away from the central, |−1/2,mI〉→|+1/2,mI〉 EPR transitions, established a negative sign for Aiso(55Mn). By comparison with as-synthesized samples that were mildly dehydrated the various species were assigned to framework sites with different degrees of water coordination. While one species is similar to the distorted (pseudo) tetrahedral sites found in the reference Mn-AlPO4-20,5 samples, the other two experience interaction with weakly bound water ligands. The transformation between the three types upon dehydration and rehydration is reversible. In an attempt to improve the spectral resolution, W-band EPR and ENDOR measurements were carried out on single-crystals of Mn-UCSB-10Mg (typical size of ∼0.02 mm3). Similar to the polycrystalline sample, two main Aiso(55Mn) components were resolved in the EPR spectra, their relative populations, however, differed from that of the polycrystalline material. This difference is attributed to variations in the water content originating from a crystal size effect. Surprisingly, the single crystal spectra did not show better resolution, and moreover, they did not exhibit significant orientation dependence in most of the experiments. These findings are ascribed to the presence of several chemically distinguishable sites combined with the multiplicity of symmetry related tetrahedral sites in one zeotype unit cell. Such a situation leads to an effective ‘powder like’ spectrum due to the small anisotropy of the magnetic interactions involved.
°C, were examined and the results were compared with the reference samples Mn-AlPO4-20 and Mn-AlPO4-5. High-frequency CW EPR experiments (at W- and G- bands) resolved two main types of Mn(II) framework sites with significantly different 55Mn hyperfine couplings and slightly different g values. These results were further corroborated by 55Mn ENDOR spectra, which enabled a more accurate determination of the two hyperfine coupling values and revealed the presence of a third species. ENDOR experiments carried out at magnetic fields away from the central, |−1/2,mI〉→|+1/2,mI〉 EPR transitions, established a negative sign for Aiso(55Mn). By comparison with as-synthesized samples that were mildly dehydrated the various species were assigned to framework sites with different degrees of water coordination. While one species is similar to the distorted (pseudo) tetrahedral sites found in the reference Mn-AlPO4-20,5 samples, the other two experience interaction with weakly bound water ligands. The transformation between the three types upon dehydration and rehydration is reversible. In an attempt to improve the spectral resolution, W-band EPR and ENDOR measurements were carried out on single-crystals of Mn-UCSB-10Mg (typical size of ∼0.02 mm3). Similar to the polycrystalline sample, two main Aiso(55Mn) components were resolved in the EPR spectra, their relative populations, however, differed from that of the polycrystalline material. This difference is attributed to variations in the water content originating from a crystal size effect. Surprisingly, the single crystal spectra did not show better resolution, and moreover, they did not exhibit significant orientation dependence in most of the experiments. These findings are ascribed to the presence of several chemically distinguishable sites combined with the multiplicity of symmetry related tetrahedral sites in one zeotype unit cell. Such a situation leads to an effective ‘powder like’ spectrum due to the small anisotropy of the magnetic interactions involved.
1. Introduction
Recently, extensive research has led to the synthesis of aluminophosphate zeotypes (AlPOs) with previously unseen compositions and framework topologies including large pore structures with multidimensional 12-ring channels.1 An example is the UCSB-n zeotype family1 (Fig. 1), which may have a potential advantage over large cage unidimensional zeotypes in case of diffusion limited reactions. While zeolite and zeotype micro-crystals (≈1–10 μm) are highly desired for catalysis, large crystals, on the order of 1 mm, are favored for applications as templates for the formation of arrayed nanoclusters as well as investigations of adsorption and diffusion in zeolites.2,3 For example, large crystals of AlPO4-5 hosting oriented dye molecules were reported to form microlasers4 and recently also used in monitoring the dynamics and orientational behavior of a single molecule in the micropores.5 | ||
| Fig. 1 The structures of UCSB-6-SBS (a), and UCSB-10-SBT (b). Fragments showing 2×2×1 unit cells are presented. | ||
Modifications of the zeotype framework with heteroatoms, particularly transition metal (TM) ions, is a common way to introduce new catalytic properties; substitution of divalent ions such as Mn(II) for Al(III) will impart charge imbalance, which when compensated by a proton results in a solid acid catalyst. Furthermore, Mn(II) and other TM ions have been found to introduce redox properties to these materials through their ability to change their oxidation state upon heating with oxygen or hydrogen.6,7 Mapping the precise atomic architecture of catalytically active sites in metal incorporated zeotypes (MAPO's) is, therefore, highly relevant, and a full panoply of spectroscopic tools is often deployed depending on the nature and valence state of the transition metal ion.8 Among these techniques, EPR is widely used for low concentrations of paramagnetic TM sites, which usually escape detection by X-ray diffraction. Nonetheless, the EPR spectrum alone is often not sufficient for a detailed structural characterization, particularly in polycrystalline (disordered) materials, where low spectral resolution is often encountered due to the anisotropic character of the various magnetic interactions involved. A better mapping of the close environment of paramagnetic metal sites can be provided by the electron-nuclear-double-resonance (ENDOR) technique.9,10
Recent developments in high-field EPR (ν0≳70 GHz) offer new opportunities for the investigation of high-spin TM ion sites (S>½) in polycrystalline materials.11–13 The advantages of high-fields over conventional X-band (ν0∼9 GHz) experiments stem from the increased sensitivity and resolution due to the reduced effect of high-order terms arising from the zero field splitting (ZFS) in combination with the high thermal polarization that can be achieved at low temperatures.11,14 ENDOR applications benefit further from the appreciably larger nuclear Zeeman interaction, which helps in resolving signals of nuclei with close gyromagnetic ratios (γ), and improves the detection of low γ nuclei with small hyperfine couplings. As the sample size is inversely related to the applied frequency, high field EPR is most useful for size-limited samples, such as small single crystals. This has been nicely demonstrated on metalloproteins containing Cu(II)15 and Mn(II),16 as well as on fullerenes.17 All these samples were measured at W-band (ν0∼95 GHz) and showed highly resolved EPR and ENDOR spectra. These, in turn, enabled the determination of various hyperfine tensors, namely, magnitude and orientation with respect to the crystal frame. To the best of our knowledge, no EPR experiments were so far carried out on a zeolite (or zeotype) single crystal, predominantly due to the fact that conventional zeolite preparation procedures result in crystals that are often too small for X- and even for W-band measurements. An alternative sample preparation technique designed to overcome this difficulty was recently reported by Belford and co-workers, who succeeded to observe single crystal EPR spectra at X-band from monolayers of copper-exchanged zeolite-A crystals assembled on glass plates.18
Natural zeolites that are found as large crystals are often impure or rare and require growth conditions that cannot be reproduced in the laboratory. In conventional hydrothermal synthesis, crystal formation is controlled by the solubility of the reagent particles, the rate of generation of nucleation centers and the solubility of the resulting products. Usually, the low solubility of the crystalline products leads to a high supersaturation which results in many small crystals and there is no routine method to obtain large zeolite single crystals.19,20 Yet, crystals of AlPO4-5 and the two UCSB-nMg type of materials, investigated in this work, have been successfully grown to a maximum dimension of 1 mm.1,21
Recently, we have characterized the isomorphous substitution of Mn(II) for Al/Mg in a series of Mn incorporated zeotypes, including the two large open Mn-UCSB-nMg structures by W-band 31P ENDOR.13 A substantial 31P hyperfine couplings, in the range of 5–8 MHz, was found for Mn(II) framework sites, where the two large open structures exhibited the lower hyperfine values. Interestingly, the EPR spectra of these two samples gave an indication for the presence of more than one Mn(II) framework site, but the origin of the difference has not been determined. This is reported in the present work. Here we combined multi-frequency CW EPR (9–180 GHz) with W-band pulsed 55Mn and 1H ENDOR measurements on as-synthesized and dried polycrystalline materials, as well as single crystals. The current study allowed us to provide evidence for the presence of three types of Mn(II) framework sites, rather than the two reported earlier,13 which differ in their 55Mn hyperfine coupling. While two sites were resolved at W- and G-band CW-EPR, 55Mn ENDOR spectra revealed a third species. The difference between the three was found to be associated with different degrees of hydration.
2.1 Experimental
×500×200 μm3 were found as well. An example for such a crystal is shown in Fig. 2b. The phase purity of the powder samples was verified by powder X-Ray Diffraction (XRD) using a RIGAKU D diffractometer. Single crystals of Mn-UCSB-10Mg were subjected to XRD measurements using a Nonius Kappa CCD diffractometer with MoKα radiation. The parameters obtained for a typical crystal (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group) are: a=17.191, b=17.191, c=41.030 Å; α=β=90°, γ=120°. All polycrystalline samples were ground prior to introduction into the W-band tubes (0.84 mm OD). The single crystal (C1) was mounted with its b axis (010 plane) parallel to the tube employing a thin paper film, as shown in Fig. 2c.
space group) are: a=17.191, b=17.191, c=41.030 Å; α=β=90°, γ=120°. All polycrystalline samples were ground prior to introduction into the W-band tubes (0.84 mm OD). The single crystal (C1) was mounted with its b axis (010 plane) parallel to the tube employing a thin paper film, as shown in Fig. 2c.
 | ||
| Fig. 2 Mn-UCSB-nMg crystals. (a) optical micrograph of Mn-UCSB-6Mg crystals, (b) SEM micrograph of a typical large Mn-UCSB-10Mg single crystal, and (c) optical micrograph of the W-band tube containing a single Mn-UCSB-10Mg crystal attached to a thin paper film. | ||
| Sample | Type | P/Al | Mg/Al | Mn/Al | Number of T sitesa |
|---|---|---|---|---|---|
| a Crystallographic tetrahedral (T) sites. b Polycrystalline material. c Single crystals. | |||||
| Mn-AlPO4-20 | PCb, as-synthesized | 0.99 | 0.0014 | 1 | |
| Mn-AlPO4-5 | PC, as-synthesized | 1.00 | 0.0015 | 1 | |
| Mn-UCSB-6Mg | PC, as-synthesized | 1.75 | 0.71 | 0.0012 | 4 |
| Mn-UCSB-6Mg | PC, 150°C dehydrated |
||||
| Mn-UCSB-10Mg | PC, as-synthesized | 1.86 | 0.84 | 0.0016 | 4 |
| Mn-UCSB-10Mg | PC, 150°C dehydrated |
||||
| Mn-UCSB-10Mg-C1 | SCc, as-synthesized | ||||
| Mn-UCSB-10Mg-C2 | SC, as-synthesized | ||||
Dehydration was carried out on a vacuum line using an apparatus constructed of an X-band quartz tube (3 mm OD) to which two W-band tubes (∼0.6 mm OD) were connected. The samples were initially placed in the X-band section and transferred to the thinner W-band part only after dehydration and sealing the X-band tube. The samples were heated to 150°C at a vacuum of 5×10−4 mbar for 20 h. Rehydration was done by exposing the dehydrated samples to air at room temperature for 48 h. These procedures were carried out for the as-synthesized samples reported in Table 1.
=15 μs, and a typical τ value of 0.4 μs. 55Mn Davies ENDOR spectra were recorded with MW pulses of 70 and 35 ns, respectively, tRF=15 μs, and a τ value of 0.4 μs. All 55Mn ENDOR spectra were recorded using the random acquisition method26 to eliminate base-line distortions. The sign of the hyperfine coupling was determined by measuring the spectra at magnetic fields away from the central |−1/2,mI〉→|+1/2,mI〉
55Mn hyperfine components.16 Determination of the field values in the ENDOR experiments was done by measuring the corresponding 1H Larmor frequencies (ν1H).
The average ENDOR frequency, ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) =[ν(+|MS|)+ν(−|MS|)]/2, and the frequency splitting, Δν=ν(+|MS|)−ν(−|MS|), are:29
=[ν(+|MS|)+ν(−|MS|)]/2, and the frequency splitting, Δν=ν(+|MS|)−ν(−|MS|), are:29
 | (1) |
 | (2) |
→mI+1;−5/2≤mI≤3/2) or ‘down’
(mI→mI−1;−3/2≤mI≤5/2) ENDOR transitions. The above equations generate a total of 30 different ENDOR frequencies for an S=5/2, I=5/2 system. When ν0≫A only six frequencies are observed, which can be approximated by: | νMS=|νI−MSA|. | (3) |
≅Aiso). Note that the ZFS interaction was neglected as well. Using eqn. (8) of ref. 27 we have verified that the contribution of the ZFS to the peak position of the ENDOR frequencies is indeed very small.31
3. Results and discussion
3.1. CW EPR and 1H ENDOR experiments
The W-band CW EPR spectra of the different samples in the region of the central |−1/2,mI〉→|+1/2,mI〉 EPR transitions are shown in Fig. 3a. The room temperature spectra of Mn–AlPO4-20 and Mn–AlPO4-5 are very similar and are both dominated by one well resolved sextet, typical for 55Mn at high-field. The line splitting, determined by Aiso(55Mn) is provided in Table 2. The spectra of the two as-synthesized Mn–UCSB-n structures show a splitting in the two outermost hyperfine components, as already found in low temperature FS-ED studies.13 The main Mn(II) sites of all materials have already been shown, through their 31P hyperfine coupling, to be framework sites.13,32 The splitting may be a result of an overlap of two sextets due to the existence of two chemically distinguishable Mn(II) sites with different Aiso(55Mn) values. These, for example, can be associated with different crystallographic tetrahedral (T) sites (see Table 1), or with different Mn(II) environments as a consequence of water coordination. Alternatively, it could be assigned to powder pattern features originating from second order effects of a relatively large ZFS interaction.30,33 The appearance of the splitting only in the high and low field 55Mn hyperfine components suggests that the latter option is less likely.
 | ||
| Fig. 3 (a) W-band CW EPR spectra of the various zeotypes investigated. All spectra of the as-synthesized polycrystalline samples were measured at room temperature. Dehydrated samples were measured at 9 K and the single crystal spectrum was recorded at 5 K. The asterisks mark the signal of a standard with g=2.0026. (b) 5 K W-band Davies 1H ENDOR spectra of the various Mn-UCSB-nMg samples measured at the center of the EPR spectrum (g=2.0). | ||
| EPR simulation | New simulationa | ENDOR | |||||
|---|---|---|---|---|---|---|---|
| Sample (species) | |Aiso|/MHz | w b | D c/MHz | Δ ±1/2 d/G | w b | Δ ±1/2/G | A iso/MHz |
|
a Parameters corresponding to the thin line spectrum in Fig. 4c. See Section 3.2 for more explanation.
b Relative amount.
c Estimated error ±20%.
d The EPR line width of the central, |−1/2,mI〉→|+1/2,mI〉 transition at Q, W and G-bands, respectively. The line widths of all other transitions were set to 300 G.
e Determined from the line splitting in the CW EPR spectra.
|
|||||||
| Mn-AlPO4-20 | 241±5e |
−251.2±0.7 |
|||||
| Mn-AlPO4-5 | 251±5e |
−251.3±0.9 |
|||||
| Mn-UCSB-6Mg, no 1 | 244±4 |
0.66 | 750 | 25,9,10, | 0.63 | 10 | −247±1.3 |
| Mn-UCSB-6Mg, no 2 | 261±4 |
0.33 | 600 | 30,15,15 | 0.21 | 8 | −267±2.3 |
| Mn-UCSB-6Mg, no 3 | 0.16 | 9 | −257±2 |
||||
| Mn-UCSB-10Mg, no 1 | 248±4 |
0.47 | 750 | 22,18,15 | −248±3 |
||
| Mn-UCSB-10Mg, no 2 | 263±4 |
0.53 | 600 | 18,9,8 | −264.5±1 |
||
| Mn-UCSB-6Mg-D | 244±4 |
1000 | 18 | −247±1.5 |
|||
| Mn-UCSB-10Mg-D, no 1 | 246±4 |
0.7 | 700 | 9 | −247±1.4 |
||
| Mn-UCSB-10Mg-D, no 2 | 261±4 |
0.3 | 700 | 17 | −266±3 |
||
| Mn-UCSB-10Mg-D, no 3 | −256±1 |
||||||
| Mn-UCSB-10Mg-SC#1, no 1 | 248±4 |
0.75 | 750 | 8 | |||
| Mn-UCSB-10Mg-SC#1, no 2 | 263±4 |
0.25 | 600 | 17 | |||
To unambiguously distinguish between the above two possibilities, CW EPR spectra were measured at four different frequencies, and the results are presented in Fig. 4. The effect of the frequency is similar for the two structures; at 9 GHz (X-band, Fig. 4a) the condition D<gβeB0 holds, and the spectrum is dominated by the central transitions centered around g∼2, superimposed on a background signal due to the other |MS, mI〉→|MS±1, mI〉 transitions.30 Higher order effects of the ZFS are well evident by the increased splitting and line width from low to high field and the low resolution due to the appearance of forbidden transitions, |−1/2,mI〉→|+1/2,mI±1〉.28 This reduced resolution prevents the distinction of the two Mn(II) species. The 35 GHz (Q-band, Fig. 4b) is somewhat better resolved, yet, the six hyperfine lines are still broad. At 180 GHz (G-band, Fig. 4d), the resolution increases and the line splitting, observed at W-band, can be also detected in the fifth hyperfine line. If the splitting is a consequence of the ZFS powder broadening, then it should be reduced as the frequency increases from 95 to 180 GHz. Therefore, we conclude that the splitting is due to the presence of two Mn(II) sites with different Aiso(55Mn) values. Furthermore, the G-band spectra show a clear asymmetry due to a slight g-shift, (g1−g2)/2∼30 ppm, between the two Mn(II) species.
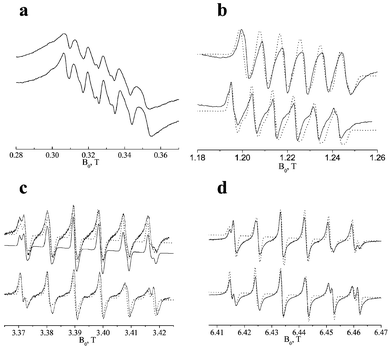 | ||
| Fig. 4 Variable frequency CW EPR of as-synthesized Mn-UCSB-nMg. All upper spectra correspond to Mn-UCSB-6Mg and the lower ones to Mn-UCSB-10Mg. (a) X-band, 160 K, ν0=9.2 GHz, (b) Q-band, 298 K, ν0=35 GHz, (c) W-band, 298 K, ν0=95 GHz, and (d) G-band, 298 K, ν0=180 GHz. Simulated Q-, W- and G-band spectra are represented by dotted lines and the simulation parameters are given in Table 2. The thin line in (c) represents a simulation carried with the hyperfine parameters obtained from the ENDOR measurements (Section 3.2). | ||
Spectral simulations using third order perturbation theory for the ZFS term28 were carried out in order to determine the two Aiso(55Mn) values and to estimate the size of the ZFS parameter. The X-band spectra were not simulated because the condition D≪gβeB0 required for the perturbation treatment does not hold. The simulations are represented by the dotted traces in Fig. 4, and the corresponding parameters are summarized in Table 2. In all simulations g1=1.9995 and g2=1.9996, based on the W- and G-band experiments. The relative amounts of the species is denoted in Table 2 by w. The disagreement between the simulated and experimental spectra is attributed to the presence of a distribution in the two ZFS parameters, D and E as well as in Aiso, all of which were not taken into account in the simulations. This strain also prevents an unambiguous determination of η
(E/D), which was set to 0.33 in all cases. This was done based on an earlier study of Fe-AlPO4-2034 and it corresponds to the maximum E value where ZFS broadening effects the largest. The line width used in the simulations of the Q-band EPR spectra is larger than that used for the W- and G- band spectra. This accounts for the larger effect of distribution in D and E at lower frequencies.
To clarify whether the two Mn(II) species are due to preferential occupation of two crystallographic T sites (alternatively, to the separation of the four T sites to two ‘groups’), or to another source of variance in the chemical environment, such as coordination to water, the effect of dehydration was examined. Attempts to calcine the samples at 600–800°C resulted either in incomplete template removal and/or the collapse of the structure, which is attributed to the high framework charge density associated with substantial amount of framework Mg.1,35 We have, therefore, performed only mild dehydration of the as-synthesized samples by heating them under vacuum to 150°C. Due to technical limitations in introducing dehydrated samples into the W-band tubes under vacuum, only a small amount could be measured, thus requiring low temperatures to improve the sensitivity in the CW EPR measurements. Variable temperature (5–298 K) CW experiments on Mn-UCSB-10 showed practically no temperature dependence. The comparison of the corresponding spectra before and after mild dehydration reveals changes for both UCSB-n samples (Fig. 3a). In Mn-UCSB-10D, two Mn(II) species are still present after dehydration, yet, the two Aiso(55Mn) values, determined by simulation (Table 2), are slightly different than those obtained before dehydration. Furthermore, the ratio between the two species has changed considerably. This indicates that the site with the larger |Aiso(55Mn)| value is associated with a hydrated sphere, whereas the site with the smaller |Aiso(55Mn)| has fewer water molecule in its close environment. The small hyperfine value is still too large to fall in the range of the typical values reported in the literature for tetrahedral Mn(II),30,39 hence, we assign it to Mn(II) in a highly distorted tetrahedral environment. This is summarized in Table 2. The spectral changes occurring upon dehydration are completely reversible and the original EPR spectra of the two Mn–UCSB-n samples were regenerated after rehydration.
The frameworks of the two UCSB-n structures (SBS and SBT) are structurally related (Fig. 1), such that both share a common type of cage and have similar framework densities. Hence, for a similar Mn(II) concentration (Table 1) one would not expect a large difference between the statistical distribution of the two species. The main difference between the two samples is in the crystal size; the average size of the Mn-UCSB-10 crystal is ∼1 μm since most of the material consists of very small crystals, while in the case of Mn-UCSB-6 the average crystal size is ∼70 μm. Therefore, we suggest that the variation in the ratio between the two species is related to the crystal size. As the crystals become smaller, their relative water content increases, which directly leads to a larger amount of Mn(II) sites with a higher degree of weakly bound water molecules (larger |Aiso(55Mn)|). This suggestion will be later substantiated by measurements of a single crystal of Mn-UCSB-10.
The effect of mild dehydration on the Mn(II) species was also monitored by 1H ENDOR measurements, the results of which are depicted in Fig. 3b. The spectra displayed were measured at a field corresponding to the center of the spectrum (g=2.0). Measurements of the spectrum at the highest field 55Mn hyperfine component gave the same spectrum. The spectra of both as-synthesized samples are dominated by an intense matrix peak at ν1H, overlapping with weak, resolved, singularities. The matrix line arises from distant water ligands and template protons, hence, it should be reduced upon dehydration. This is indeed observed in the spectra of both Mn-UCSB-nD samples, where additional singularities, attributed to the template protons, become clear. The spectral changes may have also contributions from template molecules which can undergo a reversible re distribution within the pores at moderate temperature, similar to the phenomenon known for organic guest molecules in zeolites.36 Note that since only partial dehydration was carried out at 150°C, the signal at ν1H is still relatively strong.
3.2. 55Mn ENDOR measurements
To confirm the existence of the two Mn(II) sites resolved in the EPR spectra, and determine more accurately their Aiso(55Mn) values, 55Mn ENDOR measurements were carried out as well. All spectra, presented in Fig. 5, except for the top ones, were collected at the |−1/2,+1/2〉→|+1/2,+1/2〉 EPR transition, marked as “a” on the inset figure. The top spectra were measured at a field position away from the central transitions (“b”) in order to determine the sign of Aiso(55Mn)
(see below). The general expressions for the ENDOR frequencies, given in eqns. 1–2 predict four ENDOR transitions for each Mn(II) species, when the |−1/2,+1/2〉→|+1/2,+1/2〉 EPR transition is excited,29 as schematically shown in Fig. 6. The spectra, however, resolved only the two major components belonging to the two MS manifolds for each site. The low resolution is attributed to both A-strain and ZFS broadening effects.29 Note that the relatively weak signal at ∼80 MHz observed in the two reference samples, Mn-AlPO4-20(5), is an instrumental artifact.37
 | ||
| Fig. 5 5 K W-band 55Mn ENDOR spectra of the various zeotypes investigated. The top spectra were measured at a field position ‘b’, marked with a dashed arrow on the inset figure. All other spectra were recorded at a field set to the |−1/2,+1/2〉→|+1/2,+1/2〉 EPR transition, marked as ‘a’. The signals marked with * are instrumental artifacts. The arrows mark transitions due to the third species (see text). The inset figure shows the central part of the FS-ED spectrum of Mn-AlPO4-20 at 5 K. | ||
 | ||
| Fig. 6 A schematic diagram showing the EPR transition selected in all except the top ENDOR experiments in Fig. 5 (dotted line), along with the four allowed NMR transitions (solid lines). | ||
The sign of Aiso(55Mn) was determined by recording the ENDOR spectrum of Mn-AlPO4-20 at a field position where only the |−5/2,mI〉→|−3/2,mI〉 and |−3/2,mI〉→|−1/2,mI〉 transitions contribute to the spectrum at 5 K.38 Under these conditions, only one of the doublet components, ν−, should be detected, the position of which is dictated by the sign of the hyperfine coupling.11 Comparison of the upper two spectra in Fig. 5 reveals a clear reduction in the intensity of the high frequency component, thus substituting ν=87 MHz and MS=−1/2 in eqn. (3) results in a negative sign for Aiso(55Mn)
(note that the first order ENDOR expression is actually sufficient for sign determination). This is in agreement with the sign reported in the literature,39,40 and also with theoretical DFT calculations performed on tetrahedral/octahedral cluster models representing Mn(II) framework sites.13 Based on the structural similarity between the various framework systems, it is reasonable to assume a common sign for all Aiso(55Mn) measured in this work.
Considering the results of the two reference samples, the 55Mn ENDOR spectra of the two Mn-UCSB-n samples can be easily interpreted. Two transitions are detected in the low frequency range (ν−, MS=−1/2), where the peaks around 85 and 95 MHz correspond to the species with Aiso1 and Aiso2, respectively (see Table 2). This assignment was done by fitting the Aiso values using eqns. (1) and (2). From this fitting the positions of the other two transitions (ν+, MS=+1/2) are predicted at ∼162 and ∼172 MHz, in good correlation with the experimental observation. However, the spectrum of Mn-UCSB-6 has an additional peak at an intermediate frequency (ν+=167 MHz), implying the presence of a third Mn(II) species. Qualitatively, the ratio between the two integrated signals in the low frequency range of the two UCSB-n samples nicely correlates with the ratio determined by spectral simulation of the EPR spectrum (see Table 2). Moreover, the spectral changes that occur upon mild dehydration also support our earlier interpretation of the CW EPR and 1H ENDOR data. The ENDOR signals corresponding to Aiso2=−265 MHz either disappear (Mn-UCSB-6D) or show reduced intensities (Mn-UCSB-10D), in full agreement with the CW EPR observations.
Interestingly, the high-frequency spectrum of Mn-UCSB-10D also shows the additional line at 166.5 MHz. A close look at the corresponding high-frequency ENDOR spectra reveals that this species is also present in as-synthesized Mn-UCSB-10 (marked by an arrow on Fig. 5). Furthermore, the low frequency range of the spectra of Mn-UCSB-6 and Mn-UCSB-10D show a weak hump around 90 MHz, where the corresponding ν− should appear. The latter are not clearly resolved because of the large line width of the two major ENDOR components in this frequency regime. The hyperfine interaction of the third species is, therefore, associated with an intermediate value between Aiso1 and Aiso2. Comparison of the spectra of the as-synthesized and partially dehydrated samples lead to the assignment of this species to framework Mn(II) with weakly coordinated water molecules. Since its Aiso value is intermediate between the other two it is possible that it has only one weakly coordinated water molecule, whereas the Mn(II) with Aiso2 has two such water molecules. We specify weak coordination because the 1H ENDOR spectra exhibit maximum couplings that are significantly smaller than those appearing for water ligands in Mn(H2O)62+ (∼8 MHz).41
The extraction of the various Aiso values from the ENDOR spectra was done by numerical fitting of eqns. (1) and (2). Since the two ENDOR frequencies originating from the same MS value (see Fig. 6) are not resolved in the spectra, the fitting was done for their averaged frequency and the results are summarized in the last column of Table 2. In all cases, except the third Mn(II) species, for which only one ENDOR transition was clearly resolved, both ν+ and ν− were used for the fitting procedure. A comparison between the hyperfine parameters extracted from the EPR simulation and those determined by fitting the ENDOR frequencies shows a difference of 0–6 MHz between the two, except for the third species, which is not resolved at all in the EPR spectra. Furthermore, whenever different, the EPR simulation parameters are smaller than the ENDOR values. This difference is attributed to the limited resolution of the EPR spectra, that shows only two rather than three distinguishable Mn(II) species, and the error in determining the magnitude of the external field. It is also possible that errors inherent to the application of perturbation theory in both type of theoretical analyses contribute as well.27,29 Due to the higher resolution and better accuracy of the RF frequencies the hyperfine values extracted from the ENDOR analysis are, of course, favored. Once the presence of the third Mn(II) type was established, we have simulated again the W-band CW EPR spectrum of Mn-UCSB-6 using the Aiso(55Mn) values extracted from the ENDOR data, and the results are shown in Fig. 4c. In this simulation we kept the same g and ZFS parameters of the original simulation (g1,2 and D1,2) and set g3=1.9996, D3=650 MHz for the new species (other parameters are listed Table 2). The relative amounts of the three types were estimated from the relative intensities of the corresponding high frequency ENDOR lines in Fig. 5. In agreement with the experimental spectrum, this new simulation did not resolve the presence of the third species, yet, a slight improvement in the fit between the experimental and simulated spectra was attained.
Once assigning the various ENDOR signals and establishing the sign of Aiso(55Mn), we have selected two samples, containing either a single or multiple Mn(II) species, and carried out ENDOR measurements on all six |−1/2,mI〉→|+1/2,mI〉 EPR transitions. This is useful for further understanding of the mechanisms contributing to the broadening of the ENDOR lines, and confirming the presence of the third species in Mn-UCSB-n. Figs. 7–8 present the results obtained for Mn-AlPO4-20 and Mn-UCSB-6, respectively, where the third order predictions are represented by sticks superimposed on the experimental spectra. The predictions were calculated using eqns. (1) and (2) by substituting the Aiso values determined by fitting (Table 2). Whenever one of the four inner EPR transitions were excited, the average of two corresponding ENDOR transitions was selected. While the calculations performed on Mn-AlPO4-20 and Mn-UCSB-6 predict a monotonous change in the peak position with increasing mI
(see markers on individual lines on top of the upper ENDOR spectra in Figs 7 and 8), the spectra show a symmetric pattern, in which the peak position and overall line shape is a function of |mI| rather than mI. This small but significant |mI| dependence may be associated with the increased contribution of the |−3/2,mI〉→|−1/2,mI〉 transitions as the field position is shifted away from the central |−1/2, ±1/2〉→|+1/2, ±1/2〉 transitions. Since the ENDOR frequencies calculated for the MS=±3/2 manifolds are far beyond our spectral range ( is about 400 MHz), the ENDOR frequencies must come from the MS=±1/2 manifolds, yet, the EPR transitions from which they originate are now different and may include some orientation selection. In the above analysis the Mn nuclear quadrupole interaction has been neglected because it is expected to be small.29 Moreover, the quadrupolar shifts, in the case of a dominant Zeeman and hyperfine interaction should depend on mI and not |mI| and therefore it is unlikely that it accounts for the experimentally observed |mI| dependence.
 | ||
| Fig. 7 Bottom: 5 K 55Mn W-band ENDOR spectra measured at fields set to the six |−1/2,mI〉→|+1/2,mI〉 EPR transitions of Mn-AlPO4-20. The transitions are marked on the top FS-ED spectrum. Theoretical calculations based on third order perturbation theory are presented as sticks, where the arrows indicate their mI origin. | ||
 | ||
| Fig. 8 5 K 55Mn W-band ENDOR spectra measured at fields set to the six |−1/2,mI〉→|+1/2,mI〉 EPR transitions of Mn-UCSB-6Mg. Calculations of transition frequencies based on third order perturbation theory are presented as sticks, where the arrows indicate their mI origin. The peak marked with an asterisk was not assigned in this work. | ||
The spectra of Mn-UCSB-6 indeed confirm the presence of the third species, whose ENDOR lines appear at all field positions, and are significantly enhanced when measuring at the two outermost field positions (see high frequency range in Fig. 8). This enhancement is due to the site selectivity that is achieved at these fields (Fig. 4) arising from the limited excitation band width of the MW pulses. Since the field was set such as to maximize the echo signal, the third species with an intermediate hyperfine value (|Aiso1|<|Aiso3|<|Aiso2|) is very likely the dominant one at this field. Finally, we note that the origin of the peak at 154 MHz, marked with an asterisk in Fig. 8, is unclear at this stage. ENDOR experiments at other radiofrequency regimes may be needed in order to explain this additional feature, and in particular to provide a full account for the spectral variation which appears in Figs. 7 and 8.
3.3. Single crystal EPR and ENDOR experiments
Single crystals of Mn-UCSB-10Mg (Mn-UCSB-10C#1,2) were grown to about 0.02 mm3 and their EPR/ENDOR spectra gave a fairly good signal to noise ratio at low temperatures. Unfortunately, most of the spectra did not reveal any noticeable orientation dependence, nor an improvement in resolution with respect to the polycrystalline samples.An example of a CW-EPR spectrum of a typical crystal, Mn-UCSB-10C1, mounted with its b axis parallel to the tube, is depicted in the bottom spectrum of Fig. 3a. The EPR spectrum again resolves the two species, yet, their relative intensity is different, and is closer to that of Mn-UCSB-6 (Table 2). As mentioned earlier, this difference is attributed to the different crystal size where larger crystals contains relatively less of the hydrated Mn(II) species, characterized by the larger |Aiso(55Mn)|. Other crystals oriented differently with respect to the tube axis, as well as crystals prepared with higher Mn/Al concentrations in the range of 0.001–0.005 exhibited similar EPR spectra and, likewise, no orientation dependence. We, therefore, conclude that this powder-like behavior of the |−1/2,mI〉→|+1/2,mI〉 transition is inherent to the structure of the zeotype and the distribution of Mn(II) sites in it.
Measurements of the EPR spectra by echo detection at low temperature (FS-ED) showed, however, that the anisotropy of the ZFS could be indirectly tracked (Fig. 10). In this experiment we measured the FS-ED spectra of Mn-UCSB-10C2 for a full rotation at an arbitrary plane and representative spectra for three selected rotation angles, θ, are shown in Fig. 10a. The variation in the width of the |−5/2,mI〉→|−3/2,mI〉 and |−3/2,mI〉→|−1/2,mI〉 EPR transitions is manifested as a change in the contribution of the broad background caused by these transitions with respect to the sharp sextet due to the central transitions. All spectra were normalized and the relative intensity of the central transitions, denoted by R, was measured at a fixed field. The full orientation dependence of R is depicted in Fig. 10b, which shows a clear periodic behavior. This periodic pattern is explained by the large anisotropy of the |−5/2,mI〉→|−3/2,mI〉 and |−3/2,mI〉→|−1/2,mI〉 EPR transitions that have significant contribution to the FS-ED spectra at low temperatures. Unfortunately, the low resolution does not allow a quantitative analysis of these spectra, which in principal could provide the orientation of the ZFS components with respect to the molecular frame.
We have also carried out ENDOR experiments on Mn-UCSB-10C1, which showed only a subtle line narrowing compared with respect to the polycrystalline material, as presented in the bottom spectrum of Fig. 3b and in Fig. 9. The broad 1H ENDOR spectrum shows additional singularities around ±1 MHz, not detected for polycrystalline Mn-UCSB-10, and is very similar to the spectrum of the mildly dehydrated polycrystalline sample, Mn-UCSB-10D. As the CW EPR spectra of these two samples (Mn-UCSB-10D and Mn-UCSB-10C1) are also very similar, we believe that this slight change in the ENDOR spectrum is not a direct consequence of measuring a single crystal, but rather due to the decreased intensity of the matrix line at ν1H as a consequence of the lower water content in the single crystal. 31P Davies ENDOR spectra show a doublet due to the central EPR transitions,32 and the Mims spectra reveal the expected blind spot pattern with minima at n/τ
(n=1,2,…),32,42 accompanied by a strong matrix line at ν31P.42 Some line narrowing of the broad doublet components with respect to the polycrystalline sample is detected, yet, this change is very small. Considering the CW EPR results described above, the ENDOR spectra are not surprising since the 31P doublet is also associated with the central EPR transitions, and the line width of the 31P hyperfine components are determined primarily by a distribution in Aiso rather than the anisotropic hyperfine coupling.32
 | ||
| Fig. 9 5 K 31P W-band ENDOR spectra. (a) Davies spectrum measured for polycrystalline Mn-UCSB-10Mg, (b) same as (a) for the single crystal, Mn-UCSB-10Mg-C1, and (c) Mims spectrum recorded for Mn-UCSB-10Mg-C1 with τ=700 ns. | ||
 | ||
| Fig. 10 Top: 5 K FS-ED normalized spectra for Mn-UCSB-10Mg-C2 at three different goniometer orientations. Bottom: The full orientation dependence of R (defined at the top of the figure). | ||
In order to account for the overall spectral behavior of the single crystals, we have analyzed the structure of UCSB-10 (SBT) in terms of the possible Mn(II) occupation sites. The SBT unit cell is composed of 432 atoms, out of which 72 are Al/Mg sites that can be substituted by Mn(II). These sites are schematically shown in Fig. 11a. Although only four out of these 72 sites are crystallographically distinguishable, the orientation of the ZFS main component with respect to the external field can generally assume up to 72 different angles. An illustration for such a distribution is given in Fig. 11b, where twelve framework sites, derived from the structure shown in Fig. 11a, are shown. If we assume, for the purpose of the illustration only, that the main principal axis system of the ZFS, z, is along one of the ‘bonds’ connecting two neighboring T sites, then we can easily draw the angle between zi and the external field, ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) . Each T site will, generally, have a different value of θi, so that the total EPR spectrum is a superposition of 72 individual sub-spectra (some may coincide, though) and the total line width can eventually converge to an inhomogeneous Gaussian line shape, as schematically depicted in the inset figure. If one takes into account the non-uniform distribution of weakly coupled water ligands around the metal site, possible distortions created by framework substitution,13 as well as the presence of defects and vacancies in the zeotype lattice, additional broadening is expected as a result of D strain. All the above, combined with the small anisotropy of the |−1/2,mI〉→|+1/2,mI〉 transitions at high fields, lead to the powder-like behavior of the Mn incorporated UCSB-10 crystals investigated in this work.
. Each T site will, generally, have a different value of θi, so that the total EPR spectrum is a superposition of 72 individual sub-spectra (some may coincide, though) and the total line width can eventually converge to an inhomogeneous Gaussian line shape, as schematically depicted in the inset figure. If one takes into account the non-uniform distribution of weakly coupled water ligands around the metal site, possible distortions created by framework substitution,13 as well as the presence of defects and vacancies in the zeotype lattice, additional broadening is expected as a result of D strain. All the above, combined with the small anisotropy of the |−1/2,mI〉→|+1/2,mI〉 transitions at high fields, lead to the powder-like behavior of the Mn incorporated UCSB-10 crystals investigated in this work.
 | ||
| Fig. 11 (a) The 72 sites of Al/Mg in one unit cell of the SBT (UCSB-10) structure, and (b) two possible orientations of the ZFS principal axis, zi, with respect to a fixed magnetic field vector, , drawn for the 12 central Al/Mg sites cut from scheme (a). A schematic explanation for the powder-like single crystal behavior is given in the inset figure. | ||
4. Summary
Multifrequency EPR in combination with W-band 55Mn ENDOR experiments showed that three different Mn(II) framework sites are present in the large open structures USCB-nMg (n=6, 10). The three types differ in their water interactions, as determined by comparing as-synthesized materials with mildly as-synthesized dehydrated samples. While one of the species, characterized by the smallest hyperfine coupling, Aiso1=−247 MHz, is assigned to a distorted tetrahedral site, also found in Mn-AlPO4-20(5), the other two are associated with extra water ligands. One of the hydrated species with Aiso2=−265 MHz is attributed to a framework Mn(II) with two weakly coordinated water molecules, whereas the second one, with Aiso3=−256 MHz, has probably only one weakly coordinated water molecule. As opposed to the other two, this third species was resolved only in the 55Mn ENDOR and not in the EPR spectra. Hydration and dehydration result in a reversible transformation between the three Mn(II) types.
Single crystal EPR/ENDOR measurements were carried out for the first time. The spectra, however, did not exhibit better resolution than those of the polycrystalline samples and practically no orientation dependence was observed. These findings are ascribed to the multiplicity of symmetry related tetrahedral sites in the zeotype unit cell, effectively leading to a ‘powder like’ spectrum due to the small anisotropy of the magnetic interactions involved.
Acknowledgements
This research was supported by a grant from the United States-Israel Binational Science Foundation (BSF), Jerusalem, Israel and by the DFG Schwerpunkt program “High field EPR in Physics, Chemistry and Biology”. We thank Prof. Lev Weiner for performing the Q-band experiments, and Dr Linda Shimon is acknowledged for her help with the single crystals analyses using X-ray diffraction.References
- X. Bu, P. Feng and G. D. Stucky, Science, 1997, 278, 2080–2085 CrossRef CAS.
- E. Lehmann, S. Vasenkov, J. Karger, G. Zadrozna and J. Kornatowski, J. Chem. Phys., 2003, 118, 6129–6132 CrossRef CAS.
- G. D. Li, Z. K. Tang, N. Wang and J. S. Chen, Carbon, 2002, 40, 917–921 CrossRef CAS.
- U. Vietze, O. Krauß, F. Laeri, G. Ihlein, F. Schüth, B. Limburg and M. Abraham, Phys. Rev. Lett., 1998, 81, 4628–4631 CrossRef CAS.
- C. Seebacher, C. Hellriegel and C. Bräuchle, J. Phys. Chem. B, 2003, 107, 5445–5452 CrossRef CAS.
- M. Hartmann and L. Kevan, Chem. Rev., 1999, 99, 635–663 CrossRef CAS.
- B. M. Weckhuysen, R. R. Ramachandra, J. A. Martens and R. A. Schoonheydt, Eur. J. Inorg. Chem., 1999, 565–577 CrossRef CAS.
- J. M. Thomas, Ang. Chem. Int. Ed., 1999, 38, 3588–3628 Search PubMed.
- H. Kurreck, B. Kirste and W. Lubitz, Electron Nuclear Double Resonance Spectroscopy of Radicals in Solution, VCH Publishers, New York, 1988 Search PubMed.
- C. Gemperle and A. Schweiger, Chem. Rev., 1991, 91, 1491–1505.
- D. Goldfarb, M. Bernardo, K. G. Strohmaier, D. E. W. Vaughan, H. Thomann, O. G. Poluektov and J. Schmidt, J. Am. Chem. Soc., 1996, 118, 4665–4671 CrossRef.
- J. Zhang and D. Goldfarb, J. Am. Chem. Soc., 2000, 122, 7034–7041 CrossRef CAS.
- D. Arieli, K. G. Strohmaier, D. E. W. Vaughan and D. Goldfarb, J. Phys. Chem. B, 2002, 106, 7509–7519 CrossRef CAS.
- B. F. Bellew, C. J. Halkides, J. G. Gerfen, R. G. Griffin and D. J. Singel, Biochemistry, 1996, 35, 12186–12193 CrossRef CAS.
-
(a) W. A. Coremans, O. G. Poluektov, E. J. J. Groenen, G. W. Canters, H. J. Nar and A. Messerschmidt, J. Am. Chem. Soc., 1996, 118, 12141–12153 CrossRef CAS;
(b) W. A. Coremans, O. G. Poluektov, E. J. J. Groenen, G. W. Canters, H. J. Nar and A. Messerschmidt, J. Am. Chem. Soc., 1997, 119, 4726–4731 CrossRef CAS.
- (a) P. Manikandan, R. Carmieli, T. Shane, A. J. Kalb (Gilboa) and D. Goldfarb, J. Am. Chem. Soc., 2000, 122, 3488–3494 CrossRef CAS; (b) R. Carmieli, P. Manikandan, A. J. Kalb (Gilboa) and D. Goldfarb, J. Am. Chem. Soc., 2001, 123, 8378–8386 CrossRef CAS.
- E. J. J. Groenen, O. G. Poluektov, M. Matsushita, J. Schmidt and J. H. van der Waals, Chem. Phys. Lett., 1992, 197, 314–318 CrossRef CAS.
- S. Hyunsoo, H. Kwang, L. Yun-Jo, Y. Kyung Byung and L. Belford, J. Phys. Chem. B, 2003, 107, 8281–8284 CrossRef.
- A. Kuperman, S. Nadimi, S. Oliver, G. A. Ozin, J. M. Garces and M. M. Olken, Nature, 1993, 365, 239–242 CrossRef CAS.
- S. Qiu, J. Yu, G. Zhu, O. Terasaki, Y. Nozue, W. Pang and R. Xu, Microporous Mesoporous Mater., 1998, 21, 245–251 CrossRef CAS.
- O. Weif, G. Ihlein and F. Schuth, Microporous Mesoporous Mater., 2000, 35–36, 617–620.
- I. Gromov, V. Krymov, P. Manikandan, D. Arieli and D. Goldfarb, J. Magn. Reson., 1999, 139, 8–17 CrossRef CAS.
- M. Rohrer, O. Brügmann and T. F. Prisner, Appl. Magn. Reson., 2001, 21, 257–274 CAS.
- W. B. Mims, Proc. R. Soc. London, Ser. A, 1965, 283, 452–457 CAS.
- E. R. Davies, Phys. Lett. A, 1974, 47, 1–2 CrossRef CAS.
- B. Epel, D. Arieli, D. Baute and D. Goldfarb, J. Magn. Reson., 2003, 164, 78–83 CrossRef CAS.
- R. Vardi, M. Bernardo, H. Thomann, K. G. Strohmaier, D. E. W. Vaughan and D. Goldfarb, J. Magn. Reson., 1997, 126, 229–241 CrossRef CAS.
- E. Meirovitch and R. Poupko, J. Chem. Phys., 1978, 82, 1920–1925 CAS.
- B. E. Sturgeon, J. A. Ball, D. W. Randall and R. D. Britt, J. Phys. Chem., 1994, 98, 12871–12883 CAS.
- G. H. Reed and D. Markham, in Biological Magnetic Resonance, eds. L. J. Berliner and J. Reuben, Plenum Press, New York, 1984, vol. 6, p. 73–142 Search PubMed.
- The largest contribution of the ZFS term to the ENDOR energies for D=700 MHz and η=0.3 is calculated smaller than 1%.
- D. Arieli, D. E. W. Vaughan, K. G. Strohmaier and D. Goldfarb, J. Am. Chem. Soc., 1999, 121, 6028–6032 CrossRef CAS.
- B. J. Gaffney and H. Silverstone, in Biological Magnetic Resonance, eds. L. J. Berliner and J. Reuben, Plenum Press, New York, 1993, vol. 13, p. 1–57 Search PubMed.
- D. Arieli, D. E. W. Vaughan, K. G. Strohmaier, H. Thomann, M. Bernardo and D. Goldfarb, Magn. Reson. Chem., 1999, 37, S43–S54 CrossRef CAS.
- J. M. Thomas, R. H. Jones, R. Xu, J. Chen, A. M. Chippindale, S. Natarajan and A. K. Cheetham, J. Chem. Soc. Chem. Commum., 1992, 929–931 RSC.
- S. B. Hong, H. M. Cho and M. E. Davis, J. Phys. Chem., 1993, 97, 1622–1628 CrossRef CAS.
- These peaks are due to the second harmonics of the strong signals at ∼160 MHz, and are eliminated by lowering the RF power.
- The relative populations at 4.3 K of a S=5/2 with a dominant electron Zeeman interaction at 94.9 GHz are 1: 0.35: 0.12: 0.04: 0.01: 0.005 for |−5/2〉:|−3/2〉:|−1/2〉: |+1/2〉: |+3/2〉: |+5/2〉.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Calderon Press, Oxford, 1970, p. 436–442 Search PubMed.
- J. L. Manson, W. E. Buschmann and J. S. Miller, Ang. Chem. Int. Ed., 1998, 37, 783–784 Search PubMed.
- X. Tan, M. Bernardo, H. Thomann and C. P. Scholes, J. Chem. Phys., 1992, 98, 5147–5157 CrossRef.
- A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press, Oxford, 2001 Search PubMed.
| This journal is © the Owner Societies 2004 |