Novel and efficient synthesis of difficult sequence-containing peptides through O–N intramolecular acyl migration reaction of O-acyl isopeptides†
Youhei
Sohma
,
Masato
Sasaki
,
Yoshio
Hayashi
*,
Tooru
Kimura
and
Yoshiaki
Kiso
*
Department of Medicinal Chemistry, Center for Frontier Research in Medicinal Science, Kyoto Pharmaceutical University, Yamashina-Ku, Kyoto 607-8412, Japan. E-mail: kiso@mb.kyoto-phu.ac.jp; Fax: +81 75 591 9900; Tel: +81 75 595 4635
First published on 21st November 2003
Abstract
A novel and efficient method for the synthesis of difficult sequence-containing peptides has been developed based on the synthesis of O-acyl isopeptides followed by an O–N intramolecular acyl migration reaction, resulting in a remarkable improvement of the yields.
The synthesis of “difficult sequences”-containing peptides is one of the most problematic areas in peptide chemistry, and the peptides often have low yields and purity in solid-phase peptide synthesis (SPPS).1–3 It is known that the difficult sequences are generally hydrophobic and promote aggregation in solvents during synthesis and purification. This aggregation is attributed to intermolecular hydrophobic interactions and a hydrogen bond network among resin-bound peptide chains resulting in the formation of extended secondary structures such as β-sheets.1
To solve this problem, Mutter et al. developed building blocks, so-called pseudo-prolines, which are dipeptide derivatives, including Ser/Thr-derived oxazolidines or Cys-derived thiazolidine.2 Sheppard and Johnson et al. also reported a building block, 2-hydroxy-4-methoxybenzyl (Hmb), which is a protecting group for the backbone amide nitrogen.3 These special building blocks were designed to disrupt the secondary structure formed by interchain hydrogen bonds. However, in these approaches, prior modification of Fmoc-amino acids by 2–6 steps of solution phase synthesis is required, and strong acids are also required to remove the building blocks. Therefore, the development of novel methods using conventional amino acid derivatives are of great significance in the synthesis of difficult sequence-containing peptides.
We previously developed new water-soluble prodrugs of HIV-1 protease inhibitors4 and the anti-tumor agent, paclitaxel.5 These prodrugs are O-acyl isoforms of parent drugs that have α-hydroxy-β-amino acids, and the parent drugs are easily formed viaO–N intramolecular acyl migration, a well-known reaction seen in Ser/Thr-containing peptides.6 These prodrugs increased water solubility with a newly formed and ionized amino group, and migration to the N-acyl parent drugs could be controlled accurately by pH, proceeding in a short time with no side reaction under physiological conditions (pH 7.4).
Through these studies, we conceived the idea that the O–N intramolecular acyl migration could be applied to the synthesis of difficult sequence-containing peptides (Fig. 1). Namely, more hydrophilic “O-acyl isopeptides” derived from difficult sequence-containing peptides would overcome the solubility problem in HPLC purification. To demonstrate this hypothesis, a model of a difficult sequence-containing peptide, Ac-Val-Val-Pns-Val-Val-NH2 (1, Pns: phenylnorstatine, (2R,3S)-3-amino-2-hydroxy-4-phenylbutanoic acid),7 was selected. Pns, which has the hydroxymethylcarbonyl (HMC) isostere required for inhibition of aspartyl proteases,8 is an α-hydroxy-β-amino acid with O–N intramolecular acyl migration capability. Peptide 1 was synthesized by both the standard Fmoc-based SPPS method (Route A) and a new method through its O-acyl isopeptide 5 followed by migration (Route B, Scheme 1).
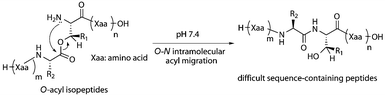 | ||
| Fig. 1 The synthetic strategy for difficult sequence-containing peptides via the O–N intramolecular acyl migration reaction of O-acyl isopeptide. | ||
 | ||
| Scheme 1 Reagents and conditions: i, 20% piperidine–DMF, 20 min; ii, Fmoc-Val-OH, DIPCDI (1,3-diisopropylcarbodiimide), HOBt, DMF, 2 h; iii, Boc-Pns-OH, DIPCDI, HOBt, DMF, 2 h; iv, Fmoc-Val-OH, DIPCDI, DMAP, CH2Cl2, 16 h × 2; v, Ac2O, TEA, DMF, 2 h; vi, TFA–m-cresol–thioanisole–H2O (92.5 ∶ 2.5 ∶ 2.5 ∶ 2.5), 90 min; vii, preparative HPLC (a linear gradient of CH3CN in 0.1% aqueous TFA); viii, phosphate buffered saline, pH 7.4, 25 °C. | ||
In Route A, Rink amide aminomethyl (AM) resin9 was employed and the Fmoc-protected amino acids were sequentially coupled using the DIPCDI–HOBt method (2 h)10 after removal of each Fmoc group with 20% piperidine–DMF (20 min). The resulting peptide resin was cleaved with TFA–m-cresol–thioanisole–H2O (92.5 ∶ 2.5 ∶ 2.5 ∶ 2.5)11 for 90 min. An undesired peptide, Fmoc-Val-Val-Pns-Val-Val-NH2, was obtained at a similar rate to peptide 1, indicating that the Fmoc group of the pentapeptide-resin was not deprotected during SPPS [see Fig. 3A-a in the ESI]. This suggests that the highly hydrophobic nature of the Fmoc-peptide-resin prevented the base from accessing the Fmoc group, probably by forming insoluble micro-aggregates on the resin. In addition, another by-product, H-Val-Val-Pns-Val-Val-NH2, was also detected (12% of 1), indicating that the amino group of the peptide-resin reacted incompletely with Ac2O. These results are well supported by the report that undesired aggregation can occur from as early as coupling of the fifth residue.12 Further purification of 1 by preparative scale HPLC was laborious due to the extremely low solubility of the products, the solubility of 1 in H2O, MeOH and DMSO being 0.008, 0.065 and 0.67 mg mL−1, respectively [see Table 1 in ESI]. When the DMSO solution of 1 was used for HPLC purification, the overall yield of 1 in Route A was only 6.9% [see Table 1 in ESI].
On the other hand, in Route B, shown in Scheme 1, Boc-Pns-OH was coupled to the H-Val-Val-NH-resin to obtain 2, and subsequent coupling with Fmoc-Val-OH to the α-hydroxy group of this β-amino acid was performed using the DIPCDI–DMAP method in CH2Cl2 to obtain ester 3. O-Acyl isopeptide 5·TFA was obtained as a major product through the coupling of another Val residue, its N-acetylation and TFA treatment [see Fig. 3A-b in ESI]. This result indicates that the protected peptide resin 4 is efficiently synthesized with no interference from the difficult sequences. Namely, the branched ester structure could modify the property of the “difficult sequence” as well as improving its solubility. In addition, since H-Pns-Val-Val-NH2 was not formed as a by-product, 1) the esterification of the secondary hydroxy group of Pns was successfully completed on the solid support, 2) the formed ester bond was stable in both piperidine and TFA treatments, and 3) diketopiperazine was not formed when the last Fmoc group was removed, corresponding to a report that diketopiperazine formation did not occur in a similar elongation of the peptide chain from the secondary hydroxy group.13 Although slight racemization (3.2%) of the esterified Val residue occurred in the DIPCDI–DMAP method, the racemized product could be removed by HPLC purification.
The solubility of 5·TFA in H2O and MeOH was 59.4 and 277.3 mg mL−1, respectively, 7500- and 4300-fold higher than that of N-acyl peptide 1 [see Table 1 in ESI]. Accordingly, a solution of 5·TFA in MeOH could easily be applied to preparative HPLC and 5·TFA was purified using 0.1% aqueous TFA–CH3CN as the eluant. Compound 5·TFA was stable at 4 °C for at least 30 days. Finally, 5·TFA was dissolved in phosphate buffered saline (PBS, pH 7.4) and completely converted to the corresponding parent peptide 1viaO–N intramolecular acyl migration at room temperature with no side reaction [see Fig. 4A in ESI]. This migration was very rapid with a half-life of <1 min [see Table 1 in ESI]. The resultant precipitate was centrifuged and washed with water and methanol to give highly pure 1. Consequently, the overall yield of 1 in Route B was 54% [see Table 1 in ESI]. These results suggest that synthesis via the O-acyl isopeptide is a powerful and efficient method for synthesizing difficult sequence-containing peptides.
To adapt this migration strategy for the synthesis of peptides with β-hydroxy-α-amino acid residues, 6 (Ac-Val-Val-Ser-Val-Val-NH2, Fig. 2) in which Pns in 1 was substituted with Ser, was synthesized in the same manner, i.e. by Routes A and B, as described for 1. By Route A, Fmoc-Val-Val-Ser-Val-Val-NH2, a similar by-product to that found in the synthesis of 1, was observed by HPLC analysis in an amount 1.1-fold higher than that of the desired peptide 6, while by Route B, O-acyl isopeptide 7 was obtained as the major product [see Fig. 3B in ESI]. This result also supported our hypothesis that the modification of 6 to the branched ester structure 7 changed the secondary structure to that favorable for Fmoc-deprotection. In this case, non-esterified H-Ser-Val-Val-NH2 was not detected in crude 7, although a low rate of racemization (0.8%) during esterification was detected. HPLC purification of 7·TFA was successfully achieved based on its excellent solubility in water and MeOH (59.6 and 126.6 mg mL−1, respectively), while purification of 6 by preparative scale HPLC was problematic due to its low solubility in H2O, MeOH and DMSO, with values of 0.013, 0.051, and 0.64 mg mL−1, respectively [see Table 1 in ESI]. The TFA salt of 7 was stable at 4 °C for at least 30 days. Moreover, this purified O-acyl form could be completely converted to 6 by O–N migration in PBS at pH 7.4 (25 °C) with no side reaction [see Fig. 4B and 5 in ESI]. As depicted in Fig. 5B in the ESI, 6 was clearly formed periodically as a white precipitate from 7 and migration was completed after 16 h. Interestingly, 7 exhibited more than 120-fold slower migration with a half-life of 2 h than that observed for 5 containing the α-hydroxy-β-amino acid (half-life <1 min) [see Table 1 and Fig. 4 in ESI], despite the formation of a similar five-membered ring transition state and a primary ester bond in 7. The faster migration in 5 may be attributed to a unique interlocking effect of the phenyl group in Pns, which has severe conformational restrictions, such as a gem-effect by geminal methyl substitution.14 Consequently, the overall yield of 6 by Route B was 41%, higher than that of Route A (6.0%).
 | ||
| Fig. 2 Conversion of O-acyl isopeptide 7 to 6viaO–N intramolecular acyl migration in PBS (pH 7.4, 25 °C). | ||
In conclusion, a novel method via the O–N intramolecular acyl migration reaction of O-acyl isopeptides is a powerful strategy for the synthesis of difficult sequence-containing peptides, since O-acyl isopeptides could improve not only the solubility in various media but also the nature of the difficult sequence during SPPS. This convenient method which requires no special building blocks and strong acidic deprotection agents would be advantageous in decreasing side reactions during synthesis and in increasing the solubility of peptides in HPLC purification, leading to higher yields of difficult sequence-containing peptides. Further studies using other model peptides, conformational analysis of O-acyl isopeptides and the synthesis of physiologically significant peptides with difficult sequences such as the Alzheimer's disease-related peptide, Aβ 1–4215 are under investigation.
This research was supported in part by the Frontier Research Program of the Ministry of Education, Science and Culture of Japan, and grants from the Ministry of Education, Science and Culture of Japan. We thank Dr Z. Ziora for useful discussions, Dr S. N. Rajesh for his help in preparing the manuscript, and Ms Y. Fukusako, Ms A. Shimizu and Ms N. Takahashi for technical assistance. We are grateful to Ms K. Oda for mass spectral measurements.
References
- S. B. H. Kent, Annu. Rev. Biochem., 1988, 57, 957 CrossRef CAS; J. P. Tam and Y. A. Lu, J. Am. Chem. Soc., 1995, 117, 12058 CrossRef CAS.
- J.-F. Guichou, L. Patiny and M. Mutter, Tetrahedron Lett., 2002, 43, 4389 CrossRef CAS.
- For a review, see: R. Sheppard, J. Peptide Sci., 2003, 9, 545 Search PubMed.
- Y. Hamada, H. Matsumoto, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2003, 13, 2727 CrossRef CAS.
- Y. Hayashi, M. Skwarczynski, Y. Hamada, Y. Sohma, T. Kimura and Y. Kiso, J. Med. Chem., 2003, 46, 3782 CrossRef CAS.
- J. M. Stewart, in The Peptide, eds. E. Gross and J. Meienhofer, Academic Press, New York, 1981, vol. 3, p. 170 Search PubMed.
- R. Nishizawa, T. Saino, T. Takita, H. Suda, T. Aoyagi and H. Umezawa, J. Med. Chem., 1977, 20, 510 CrossRef CAS.
- T. Mimoto, J. Imai, S. Tanaka, N. Hattori, O. Takahashi, S. Kisanuki, Y. Nagano, M. Shintani, H. Hayashi, H. Sakikawa, K. Akaji and Y. Kiso, Chem. Pharm. Bull., 1991, 39, 2465 CAS.
- H. Rink, Tetrahedron Lett., 1987, 28, 3787 CrossRef CAS.
- D. Sarantakis, J. Teichman, E. L. Lien and R. L. Fenichel, Biochem. Biophys. Res. Commun., 1976, 73, 336 CAS.
- Y. Kiso, K. Ukawa and T. Akita, J. Chem. Soc., Chem. Commun., 1980, 101 RSC; F. Albericio, N. Kneib-Cordonier, S. Biancalana, L. Gera, R. I. Masada, D. Hudson and G. Barany, J. Org. Chem., 1990, 55, 3730 CrossRef CAS.
- J. Bedford, C. Hyde, T. Johnson, W. Jun, D. Owen, M. Quibell and R. C. Sheppard, Int. J. Peptide Protein Res., 1992, 40, 300 CAS.
- H. Tamamura, T. Kato, A. Otaka and N. Fujii, Org. Biomol. Chem., 2003, 1, 2468 RSC.
- T. C. Bruce and W. C. Bradbury, J. Am. Chem. Soc., 1965, 87, 4838 CrossRef; H. Matsumoto, Y. Sohma, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2001, 11, 605 CrossRef CAS.
- C. Haass and D. J. Selkoe, Cell, 1993, 75, 1039 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: HPLC profiles of crude state, migration studies, and spectral data for 1, 5, 6, and 7. See http://www.rsc.org/suppdata/cc/b3/b312129a/ |
| This journal is © The Royal Society of Chemistry 2004 |