Integration of combined heteroduplex/restriction fragment length polymorphism analysis on an electrophoresis microchip for the detection of hereditary haemochromatosis
Tim
Footz
a,
Martin J.
Somerville
b,
Robert
Tomaszewski
b,
Basil
Elyas
b and
Christopher J.
Backhouse
*a
aDepartment of Electrical and Computer Engineering, 2nd Floor, ECERF Building (9107 - 116St.), University of Alberta, Edmonton, Alberta, Canada T6G 2V4
bDepartment of Medical Genetics, University of Alberta, Edmonton, Alberta, Canada
First published on 26th November 2003
Abstract
This work describes an integrated method of enzymatic digestion, heteroduplex analysis (HA) and electrophoretic sizing on a microfluidic chip. HA techniques based on microchip electrophoresis are capable of the high sensitivity detection of subtle mutations such as single nucleotide polymorphisms (SNPs) but are not readily able to detect homozygous mutant genotypes. Such homozygous conditions are commonly encountered with the gene implicated in hereditary haemochromatosis, HFE. We employed the restriction fragment length polymorphism (RFLP) method of mutation detection to complement the HA method in a rapid novel on-chip procedure that separated digested PCR fragments to reliably determine the presence or absence of the most important mutations associated with haemochromatosis. This method was able to distinguish the homozygous mutant, heterozygous and homozygous wildtype genotypes. The mutations investigated here (C282Y, H63D and S65C) are often the mutation targets used in the genetic testing for haemochromatosis. This method provides the extremely specific digestion methods needed for the analysis of the known and relatively common mutations that have a significant probability of occurring in a homozygous form. However, the high sensitivity of the HA method is useful in detecting other mutations of lesser likelihood which, by virtue of their rarity, are likely to be present only in a heterozygous form. Although the conventional methods of analysing these mutations require as much as a day to perform, this microchip method, even without robotics or multiplexed operation, can be performed in about 10 min per sample.
Introduction
Considerable effort has been applied to develop improved mutation analysis methods for integration with capillary electrophoresis (CE) microchips (e.g.refs. 1–5). Such microfluidic devices offer many advantages over conventional capillary or gel-based methods in terms of speed, reagent usage and integration of sample preparation techniques to the extent that they are referred to as “micro total analysis systems” or “lab-on-a-chip” devices.6 Perhaps the most important application of microchips will be the accurate detection of genetic variation in clinical research, the genetic testing of individuals, identifying disease genes and in population screening.7,8 A common situation is the desire to detect specific mutations that are known to be the most significant indicators of disease risk while trying to detect as many other mutations as possible (both known and unknown).The mutation detection methods that are candidates for integration into high-throughput microfabricated devices each have significant advantages and disadvantages.1–3 Some techniques7 are more suitable for the purpose of “scanning” a diverse population of molecules for highly polymorphic genes (i.e. many different mutation sites) or for researching candidate genes (i.e. the polymorphic sites are unknown). Some techniques however are only effective in the role of “genotyping”, and discern only the presence or absence of a well-characterized mutation. Rapid low-cost scanning techniques would facilitate fundamental genomics research and high-volume medical interrogations of large genes whereas genotyping may find its niche in point-of-care medical examination and high-accuracy population screening.
If no mutations are present in a given genetic sequence then that sequence is referred to as being wildtype. Given that humans typically have two copies of each gene, the genetic information carried by the two copies may be identical (homozygous) or may differ (heterozygous). The mutation status of a specific region of DNA may therefore be homozygous mutant (both copies are mutants), homozygous wildtype (neither copy is a mutant) or heterozygous (one copy is a mutant). DNA is normally double stranded but in many procedures (e.g. genetic amplification methods) the DNA is separated into single strands. When these single strands are able to recombine they each may recombine with a perfectly complementary sequence (forming a homoduplex) or a nearly perfectly complementary sequence (forming a heteroduplex, i.e. a mutant sequence paired with a wildtype sequence). Heteroduplexes contain a “bulge” or “bubble” in any regions containing an imperfect fit and as a result in electrophoresis these duplexes typically migrate more slowly than the homoduplexes. The DNA from a heterozygous sample will generate four different duplexes, two homoduplexes and two heteroduplexes, but often the molecules co-migrate such that separate peaks are not resolved. Electrophoresis under conditions that enhance mobility differences can be used to determine the presence of a heterozygous state, and this powerful technique for detecting mutations is known as heteroduplex analysis (HA).
Hereditary haemochromatosis, one of the most common autosomal recessive diseases, is characterized by iron overload of major organs, especially the liver, and is linked to defects in the gene HFE.9 The majority of HFE mutations represent single nucleotide sequence polymorphisms (SNPs) and there are few affected loci.10–12 The predominant mutation is “C282Y” (denoting the 845G>A transition in codon 282 of exon 4), but also of significance are “H63D” (the 187C>G transversion in codon 63 of exon 2) and “S65C” (the 193A>T transversion in codon 65 of exon 2). These mutations are important enough that clinically relevant testing often focuses on them alone. The accurate detection of SNPs requires technology that exhibits superior sensitivity, referring to the ability to identify a sample as containing mutant DNA (i.e. high sensitivity entails a low rate of false negatives), and is also discriminatory, easily distinguishing one genotype from another (e.g. H63D vs. S65C). It is generally regarded that direct sequencing can resolve mutations with 100% sensitivity and provide discrimination to the nucleotide level and as such is the most accurate mutation detection technology. However, the main drawback of sequencing is that it is the most costly in regards to time, reagents and commercial equipment.13 The desire to fully integrate the sequencing technology on micro- or nanoscale devices has been hampered by the need for highly purified samples.14
The commonly used macroscopic mutation detection techniques applied to analysis of HFE have recently been reviewed by Pointon et al.15 The analysis of HFE is often the benchmark for developing new SNP detection techniques and most reports (below) focus on the detection of C282Y, H63D and S65C. Restriction Fragment Length Polymorphism analysis (RFLP16,17), Allele-Specific Polymerase Chain Reaction (ASPCR5) and other PCR-based methods that rely on sequence-specific reagents,18 are appropriate for genotyping but not for mutation scanning. These methods rely on a significant degree of prior characterization of the region of interest in order to develop sequence-specific restriction enzymes (also known as endonucleases) or nested primers, but the methods are well suited to identifying mutations in a homozygous state and can provide 100% sensitivity. Previously, macroscopic RFLP analysis was a time consuming approach due to the need for lengthy post-PCR treatment and conventional electrophoretic separation.19
The most familiar mutation scanning techniques that do not require sequence-specific reagents other than the PCR primers are Denaturing High Performance Liquid Chromatography (DHPLC20), HA11 and Single Strand Conformation Polymorphism analysis (SSCP21–23). DHPLC and HA are inherently able to identify only heterozygous genotypes (unless post-PCR samples are reannealed in the presence of a heteroduplex generator24) and even SSCP requires high-precision controls to convincingly detect a homozygous mutant condition. DHPLC is accepted to be the most sensitive of these procedures in general (ca. 96% sensitivity25) and also provides considerable discrimination.13 The equipment and consumables for DHPLC are expensive (the columns are of limited lifetime) but the process is fully automated. HA and SSCP are relatively inexpensive but their sensitivities are typically no more than 90%.26
The speed of the electrophoresis-based analysis of HFE was greatly increased by utilizing capillary systems.27–29 Recent work has demonstrated microfluidic devices with large arrays of separation channels that can significantly improve the throughput of electrophoretic separations; this was demonstrated for RFLP (with off-chip digestion)30 and ASPCR-based methods5 as well as for sequencing.14,31 Although microchip HA and SSCP have been reported,4,32–34 their mutation detection sensitivities in a scanning mode were not evaluated. However, recent analogous work in a capillary array electrophoresis system demonstrated SNP-detection sensitivities of 75% and 93% (for the p53 gene), for HA and SSCP respectively, but combining the results in tandem afforded 100% sensitivity35 with similar results reported by others.11,36
To our knowledge only one report has been published concerning the integration of on-chip restriction enzyme digestion with CE separation.37 In that report the restriction enzyme and its buffer were placed in two wells and the sieving matrix between the wells was equilibrated with them, taking up enzyme and both positive and negative ions from the enzyme buffer. Although this is a potentially powerful method of implementing restriction enzyme digestions on-chip, it is unclear how applicable it is in general, considering that pioneering CE and microchip CE researchers are still investigating a variety of reagents for efficient mutation detection, including highly-denaturing polymers.
We have integrated fragment sizing with an extremely fast HA method and a simple on-chip RFLP analysis on a single microfluidic chip. Without prior conditioning of its channels, the microchip was loaded directly with commercially available PDMA-based sieving matrix38,39 and a sample previously subjected only to the PCR. The primers were not designed specially for this application but were chosen simply to bracket the desired mutation, not to amplify homologues, and to have a length of ca. 200–300 bp. The microchip was run at room temperature with a single automated protocol. We estimate that each run utilized 1/4000th of the volume of loaded PCR sample, about 75 pL. The peak arrival times are reproducible to within ca. 2%. An initial separation was established to conveniently identify heteroduplex-containing samples with an estimated sensitivity of 80%. Next, enzymatic digestion, using Bcl I or Sna BI restriction enzymes (for H63D or C282Y, respectively), allowed the use of electrophoretic sizing to provide the supplementary RFLP analysis. HA alone was sufficient to uniquely characterize heterozygosity for H63D, S65C and H63D/S65C (compound mutation), but the RFLP analysis was necessary to correctly identify the homozygous samples and also to detect heterozygosity for C282Y. Although Jackson et al.27 developed a powerful method of HA that was tailored to a specific mutation, such a non-general method is unsuited for mutation scanning. In the present work we seek to integrate a general mutation scanning HA method and to our knowledge there have been no reports of the successful application of such a general HA method to the detection of the C282Y mutation. Moreover, in macroscopic methods the “gold standard” appears to be provided by restriction enzyme methods. In the present work, the combined use of the RFLP and HA techniques allows screening for the most significant mutations associated with HFE while simultaneously scanning for other rare mutations.
Materials and methods
Reagents
Genomic DNA was extracted from lymphocytes of individuals (obtained with informed consent), purified using either phenol–chloroform–isoamyl alcohol extraction40 or with the QIAmp DNA Blood Kit (QIAGEN, Mississauga, ON, Canada) and solubilized in TE (pH 8.0). Sequencing reactions were performed with AmpliTaq® DNA Polymerase and the ABI PRISM® BigDye® Terminator v3.0 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Streetsville, ON, Canada) and genotypes confirmed on the ABI PRISM® 377 Slab Gel Sequencer (Applied Biosystems). PCR reagents (polymerases, buffers and primers) were from Invitrogen (Burlington, ON, Canada). Performance Optimized Polymer-6 (POP-6™, part #402837) and Genetic Analyzer Buffer with EDTA (GABE, part #402824) were from Applied Biosystems. Bcl I and Sna BI restriction enzymes and SuRE/Cut incubation Buffer M were from Roche Diagnostics (Laval, QB, Canada).PCR
HFE exons 2 (234 bp) and 4 (223 bp) were PCR-amplified, separately, for a test panel of clinically-relevant DNA templates. The samples were wildtype (denoted −/−) as well as homozygous (denoted +/+) and heterozygous (denoted +/−) for C282Y, H63D and S65C. “Hot-start” PCR was performed in 25 or 50 µL reactions with 1.2–6 ng µL−1 of genomic template DNA (described above), 200 µM of each dNTP, 1.5 mM MgCl2, 1 × reaction buffer, 1.25 Units of Taq DNA Polymerase and with the primers as detailed in Table 1.| Amplicon - primer | Sequence | 5′ Label | Final concentration |
|---|---|---|---|
| HFE exon 2 - forward | 5′-TCA GAG CAG GAC CTT GGT CTT TCC-3′ | HEX | 0.4 µM |
| HFE exon 2 - reverse | 5′-CAT ACC CTT GCT GTG GTT GTG ATT-3′ | n/a | 0.4 µM |
| HFE exon 4 - forward | 5′-GTG TCG GGC CTT GAA CTA CT-3′ | HEX | 0.4 µM |
| HFE exon 4 - reverse | 5′-ACC CCA GAT CAC AAT GAG GG-3′ | n/a | 0.4 µM |
Thermal cycling was performed as follows: 2 min at 94 °C, 10 “touch-down” cycles of (30 s at 94 °C, 30 s at 66 °C with −0.5 °C per cycle, 30 s at 72 °C), 30 cycles of (30 s at 94 °C, 30 s at 61 °C, 30 s at 72 °C) and 10 min at 72 °C.
Microchip methods
The microfluidic devices used here consist of 4 reservoirs (or wells) linked by two microchannels nominally 50 µm wide and 20 µm deep (Fig. 1). These “simple cross” glass microchips were supplied by Micralyne (Edmonton, AB, Canada). As described previously,41 the Microfluidic Tool Kit (µTK, also from Micralyne) was used to manipulate the reagents and DNA upon these microchips. For the present work we used a laser induced fluorescence (LIF) system that provides excitation at a wavelength of 532 nm and detection at 578 nm. A compiled LabVIEW interface supplied by Micralyne was used to record the LIF signal at 200 Hz. The LIF detection took place at 30 or 76 mm downstream from the intersection. | ||
| Fig. 1 Diagram of the “simple cross” glass microchip, from Micralyne. The circles represent reservoirs 2 mm in diameter and 1.1 mm deep, each holding ca. 3 µL. The lines linking the circles represent microchannels nominally 50 µm wide, 20 µm deep and having an approximately semi-circular cross section. The X symbols represent the locations of optical detection at 30 and 76 mm from the intersection. | ||
The untreated channels of microchips were filled with POP-6™ as described previously41 with 1 µL of POP-6™ + 2 µL of 1 × GABE in the sample and buffer waste reservoirs, 3 µL of 1 × GABE in the buffer supply reservoir and 2.7 µL of 0.1 × GABE + 0.3 µL of unpurified, non-denatured PCR sample in the sample supply reservoir (Fig. 1). Aliquots of POP-6™ were prepared fresh daily by heating at 65 °C for 10 min, which we found necessary to dissolve precipitates (we attribute these to urea). For each run, samples were injected from the sample well to the sample waste well at 500 V cm−1 for 60 s (Fig. 1). After this injection step the portion of the sample DNA within the intersection of the channels (a volume of ca. 50 pL) was separated by applying a field of 706 V cm−1 between the buffer and buffer waste wells for 200 or 250 s. Subsequent runs of a sample were done without replacing the polymer matrix, but the polymer was replenished when changing samples.
A C++ program was used to analyse the fluorescence data, first removing spurious, isolated spikes due to electrical noise (i.e. peaks consisting of a single point) and then applying a low-pass filter to the data by performing a running average with a neighbourhood of 30 points. Because of the high rate of data acquisition (200 Hz), this low-pass filtering had no discernable effect upon the HA peaks although it was very effective in removing the high levels of noise encountered in sizing the restriction digests (discussed below). The analysis program wrote postscript files for all of the analysed data. More details can be found in previous work.41
HA and sizing
Each raw PCR sample was diluted with electrophoresis buffer on the microfluidic chip and electrophoretically injected into the microchip (Fig. 1). The first analysis performed on each sample was an electrophoretic separation wherein the arrival time of the homoduplex peak provides a size determination (to within several percent, data not shown) and the presence of homoduplex and heteroduplex peaks indicates the presence of a mutation. Moreover, the identity of mutations can be distinguished on the basis of their characteristic peak patterns (described below). A typical electropherogram of this first run is shown in Fig. 2 in which the primer DNA arrives first, along with additional high-mobility species that represent either impurities in the primer preparation (a common artefact) or nonspecifically-amplified products. This is then followed by the specifically-amplified double-stranded product DNA showing duplex peaks. | ||
| Fig. 2 Microchip electrophoresis of raw PCR sample. Sample is injected for 60 s with the LIF detector turned off (included in the analysis time), followed by separation to 76 mm. HEX-labelled primers and PCR products are detected as they pass by the LIF system, represented by peaks in the electropherogram. RFU = relative fluorescence units. | ||
Digestion and sizing
Immediately following the HA step, a restriction enzyme solution diluted with incubation buffer was added to the sample well of the microchip and sizing was performed in subsequent electrophoretic runs. After 1 min of digestion at room temperature, an electrophoretic run sampled the partially-digested DNA. After 6 min of digestion another electrophoretic run sampled the DNA (by which time the digestions were complete). The diluted mixture of Bcl I was comprised of incubation buffer M at a concentration of 7 × and 2 Unit µL−1 of enzyme, of which 0.3 µL was added to the sample. The diluted mixture of Sna BI contained 1 × buffer M and 1 Unit µL−1 of enzyme, of which 0.2 µL was added to the sample.For HFE exon 2, the Bcl I enzyme cleaves the 234 bp fragment (recognition sequence: TGA TCA) only when the H63D mutation is not present in the sequence. This results in fragments of 72 bp (retaining the fluorescently-tagged primer) and 162 bp (unlabelled, and so undetectable). Therefore, the wildtype, H63D heterozygous, S65C heterozygous and the H63D/S65C compound heterozygous samples will produce digestion and thus a new higher-mobility peak representing the 72 bp fragment. Duplexes having one or more strands with the H63D mutation (TGA) are not affected (i.e. in the H63D homo- and heterozygous samples).
For HFE exon 4, the enzyme Sna BI was used to cleave the fragments (recognition sequence: TAC GTA) only when the C282Y mutant sequence (underlined) is present (wildtype is GTG>), resulting in fragments of 171 bp (labelled) and 52 bp (unlabelled, undetected). In this case only the C282Y heterozygous and the C282Y homozygous samples will contain copies of the mutation and so only these samples will produce electropherograms that will show digestion products.
Peak velocities are reproducible to within ca. 2% and this allows accurate fragment size determinations by comparing the times-of-arrival of peaks, although for the purposes of the present work such quantitation was not required.
Results and discussion
As predicted, each homozygous sample was confirmed to possess a single peak in heteroduplex analysis (Fig. 3a, c, f and h), corresponding to the presence of the entirely homoduplex DNA (wildtype or mutant) generated during PCR. However, for samples heterozygous for the H63D and S65C mutations (both in exon 2), the profiles are altered (Fig. 3b, d and e) with additional peaks detected, thereby confirming the heterozygous nature of these samples. | ||
| Fig. 3 Heteroduplex analysis of HFE by microchip electrophoresis. Primer peaks were omitted from the electropherograms to show fine detail of the PCR product peaks. Peak velocity is ca. 98% reproducible in this polymer buffer system (i.e. run-to-run variation of ca. 1–3 s) and peak arrival times can be offset by several seconds when fresh polymer is prepared. (a–e) Exon 2, 234 bp products. (f–h) Exon 4, 223 bp products. 60 s injections are included in the analysis times. For genotype nomenclature: “+” = mutation present; “−” = mutation absent. RFU = relative fluorescence units. | ||
As expected from the lack of any report in the literature that used a general HA method to detect C282Y, our HA method was unable to distinguish the heterozygous C282Y sample (in exon 4) from the wildtype and mutant homozygous samples (Fig. 3f–h). In an ideal PCR, the amount of DNA is doubled in each thermal cycle of the so-called exponential regime, as each strand of denatured DNA is used as a template to construct an exact match—i.e. only homoduplexes are formed. With our PCR procedure we have found that there is no need to reanneal PCR product to ensure that heteroduplexes are formed—this finding is a consequence of cycling the PCR past the exponential regime (after many thermal cycles, much of the DNA does not participate in any given cycle of replication). To corroborate this, a heterozygous (C282Y+/−) sample was re-examined after applying a standard reannealing protocol (95 °C for 3 min, ramp down to 65 °C at 1 °C min−1)42 to an aliquot of the PCR to promote the formation of heteroduplexes, but there were no differences in the electrophoretic profiles (data not shown).
In HA methods the mobility shift of the heteroduplexes is a monotonic function of the size of the mutation.43,44 This mechanism tends to provide a diversity of electropherograms, suggesting that in general the HA method will provide efficient discrimination. It can be seen that the peak profiles for the H63D heterozygote (Fig. 3b) and the S65C heterozygote (Fig. 3d) are dissimilar. Moreover, the profile of the H63D/S65C compound mutation (Fig. 3e) appears to be a composite of the constituent heteroduplex profiles. This suggests that in many cases an electropherogram may be used to identify or categorize the combinations of mutations in a sequence. Although we are still optimizing our HA method for maximum sensitivity and ease of integration, we expect to achieve a performance comparable to that reported by others in macroscopic HA work, about 80% sensitivity. Other (non-microchip) HA-based electrophoretic mutation detection methods have obtained higher sensitivities (nearing 100%) by combining HA and SSCP.35,36 Since the RFLP approach is incapable of detecting unexpected polymorphisms, a profound benefit would be realised if the RFLP method could be applied in tandem with such highly sensitive HA methods.
After digesting each sample for 1 min, an electrophoretic run sampled the DNA at a point when digestion was incomplete (Figs. 4b and 5b). For the Bcl I digestions of HFE exon 2 samples, the restriction enzyme-containing sample produced very weak signals when separation was allowed to proceed to 76 mm (data not shown). We believe that these weak signals are a result of contamination from the Bcl I enzyme and its buffer. Such weak signals could arise from the tendency of excess salts or proteins to result in low or noisy signals within this polymer-buffer system (such phenomena are described in the ABI PRISM® 3100 Genetic Analyzer User's Manual). These phenomena were avoided by placing the LIF detector at the 30 mm position—this provided suitable separation of digested from undigested fragments and still produced an adequate signal intensity. Fig. 4b shows the patterns of incomplete Bcl I digestion for the different samples. When compared to the pre-digestion electropherograms (Fig. 4a) it is evident that the lowest mobility peaks (on the right) represent undigested molecules and that samples that contain copies of the gene without the H63D mutation show the effects of digestion. Such samples (the wildtype, H63D heterozygous, S65C heterozygous and the H63D/S65C compound heterozygous samples) show the emergence of a new higher mobility peak (on the left) after only 1 min of digestion, this new peak representing the 72 bp fragment. Any sequences containing the H63D mutation (i.e. with a mutated codon 63 (TGA)) are left undigested by the enzyme.
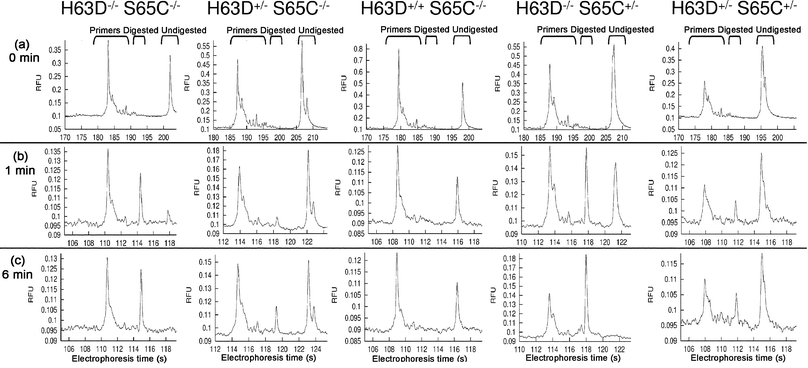 | ||
| Fig. 4 RFLP analysis of HFE exon 2 by microchip electrophoresis with on-chip digestion. (a) 76 mm separation of PCR samples before adding restriction enzyme. (b) 30 mm separation of samples after incubation with Bcl I for 1 min. (c) 30 mm separation of samples after incubation with Bcl I for 6 min. 60 s injections are included in the analysis times. The electropherograms were aligned by the invariant primer peak for easier interpretation. For genotype nomenclature: “+” = mutation present; “−” = mutation absent. RFU = relative fluorescence units. | ||
 | ||
| Fig. 5 RFLP analysis of HFE exon 4 by microchip electrophoresis with on-chip digestion. (a) 76 mm separation of PCR samples before adding restriction enzyme. (b) 76 mm separation of samples after incubation with Sna BI for 1 min. (c) 76 mm separation of samples after incubation with Sna BI for 6 min. 60 s injections are included in the analysis times. The electropherograms were aligned by the invariant primer peak for easier interpretation. For genotype nomenclature: “+” = mutation present; “−” = mutation absent. RFU = relative fluorescence units. | ||
After 6 min of digestion with restriction enzymes, another electrophoretic run sampled the DNA after the site-specific digestion was completed (based on the observation of when the cleavable fragments of the appropriate homozygous sample were no longer detected). This 6 min period of digestion gave enough time for the sample injection, the separation stage and note taking before starting the next run. Fig. 4c clearly indicates that samples containing the H63D mutation on both copies of the gene are left undigested, those samples without any copies of the H63D mutation are completely digested and those samples containing one copy of the mutation are partially digested. It can be seen then that the Bcl I digestion readily allows the wildtype (for H63D), H63D homozygous and H63D heterozygous conditions to be distinguished.
The restriction enzyme digestion of HFE exon 4 is similar, except that all separation lengths were 76 mm and that the enzyme Sna BI specifically cleaves the C282Y mutant sequence. The measures required for the analysis of Bcl I fragments (i.e. shorter separation distances) were not required here, presumably because this enzyme (and associated reagents) was more compatible with the microchip environment. As shown in Fig. 5, the electropherograms for the C282Y heterozygous and the C282Y homozygous samples show the emergence of a new peak (for the 171 bp fragment) after 1 min of digestion. Furthermore, only the C282Y homozygous sample shows complete digestion after 6 min of incubation with Sna BI (Fig. 5c). The Sna BI digestion readily allows the wildtype for C282Y (no digestion), C282Y homozygous (complete digestion) and C282Y heterozygous (partial digestion) conditions to be distinguished. In light of the inability of the HA separation technology to determine the presence or absence of the C282Y mutation, the RFLP method was necessary for identifying these genotypes.
By sizing the products of the Bcl I digestion alone (i.e. without HA), it is impossible to distinguish between the H63D heterozygous and the H63C/S65C compound heterozgyous samples or between the wildtype and S65C heterozygous samples. However, the shape of the HA electropherogram for heterozygous samples clearly allows one to distinguish the three heterozygous samples (Fig. 3b, d and e). Moreover, the shape of the HA electropherogram of the compound heterozgyous sample appears to be a composite of the individual heterozygote electropherograms. This resemblance was sufficient to enable us to surmise the identity of the H63C/S65C compound heterozgyous sample before subsequent confirmation by direct sequencing.
Conclusions
This work is intended to demonstrate the importance of attaining higher levels of integration upon microfluidic systems. Although effective in scanning for both known and unknown mutations, a failing of the HA method is that with its sensitivity of 80% it is not as reliable as the RFLP method. On the other hand the RFLP method is restricted to detecting a very limited number of known mutations. In this way these two methods complement each other well and neither would be effective alone. The present method is readily able to detect the mutations associated with haemochromatosis that are of greatest interest (C282Y, H63D and S65C) while being able to scan for other less common mutations (both known and unknown) with high sensitivity. Once detected, these less common mutations may be identified by the appearance of their characteristic electropherograms or by follow-up sequencing.We are in the process of improving the sensitivity of the HA method with improved polymer formulations and have made some preliminary tests of this method with varied temperatures. We expect to improve the sensitivity of the HA method slightly (by perhaps 5 to 10%). A drawback of the HA method is that in order to detect homozygous mutations it would be necessary to reanneal samples with known wildtype sequences prior to performing the HA analysis. Although this could have been done manually here, we have developed a means of reannealing on-chip for future applications. Most importantly, we are integrating SSCP with HA. The SSCP method is capable of detecting both heterozygous and homozygous mutations and it has already been demonstrated by others that this combination of HA and SSCP can approach 100% sensitivity.35,36 We are now attempting to combine on-chip denaturation, reannealing, RFLP, HA and SSCP methods. We do not expect that this further integration will result in significantly longer analysis times.
Electrophoresis is particularly suited for microfabricated structures, is readily extended to higher levels of integration and permits the use of a readily replaceable sieving matrix. The recent advances in integrating sub-microlitre volume PCR reactions with microchip capillary arrays will permit the PCR and mutation analysis of hundreds of samples in a matter of minutes in a completely automated system. The present work is not fully automated, but the present approach is well suited to the decision-based genotyping process usually applied to analysis of the HFE gene.
In future it may be possible to optimise the reaction and electrophoresis conditions of this work to provide stronger signals and faster analysis. Our technique may also be applicable to the microchip integration of procedures employing restriction enzymes to fingerprint large gene fragments prior to applying a mutation scanning technique.45,46 By comparison, the macroscopic methods for RFLP-based detection of HFE genotypes may involve the purification of the PCR product, digestion for up to several hours at elevated temperature and subsequent electrophoretic separation of the digested products (e.g. ref. 47) with a total elapsed time of a day or more. Our novel combined HA/RFLP approach provides a very rapid method of determining the HFE genotype with an analysis time of about 10 min per sample in a method that tests for the most important known mutations (i.e. genotyping) and that has a high (ca. 80%) probability of detecting unknown mutations (i.e. scanning).
Acknowledgements
We gratefully acknowledge the support of the Natural Sciences and Engineering Research Council of Canada and Micralyne.References
- L. J. Jin, J. Ferrance and J. P. Landers, Biotechniques, 2001, 31, 1332 Search PubMed.
- J. Khandurina and A. Guttman, J. Chromatogr., A, 2002, 943, 159–183 CrossRef CAS.
- E. Verpoorte, Electrophoresis, 2002, 23, 677–712 CrossRef CAS.
- H. J. Tian, L. C. Brody and J. P. Landers, Genome Res., 2000, 10, 1403–1413 CrossRef CAS.
- I. Medintz, W. W. Wong, L. Berti, L. Shiow, J. Tom, J. Scherer, G. Sensabaugh and R. A. Mathies, Genome Res., 2001, 11, 413–421 CrossRef CAS.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators B, 1990, 1, 244–248 CrossRef.
- A. Ahmadian and J. Lundeberg, Biotechniques, 2002, 32, 1122 Search PubMed.
- V. N. Kristensen, D. Kelefiotis, T. Kristensen and A. L. Borresen-Dale, Biotechniques, 2001, 30, 318 Search PubMed.
- J. N. Feder, A. Gnirke, W. Thomas, Z. Tsuchihashi, D. A. Ruddy, A. Basava, F. Dormishian, R. Domingo, M. C. Ellis, A. Fullan, L. M. Hinton, N. L. Jones, B. E. Kimmel, G. S. Kronmal, P. Lauer, V. K. Lee, D. B. Loeb, F. A. Mapa, E. McClelland, N. C. Meyer, G. A. Mintier, N. Moeller, T. Moore, E. Morikang, C. E. Prass, L. Quintana, S. M. Starnes, R. C. Schatzman, K. J. Brunke, D. T. Drayna, N. J. Risch, B. R. Bacon and R. K. Wolff, Nat. Genet., 1996, 13, 399–408 CAS.
- A. Asberg, K. Thorstensen, K. Hveem and K. S. Bjerve, Genet. Test., 2002, 6, 59–62 CrossRef CAS.
- J. N. P. de Villiers, R. Hillermann, L. Loubser and M. J. Kotze, Hum. Mol. Genet., 1999, 8, 1517–1522 CrossRef CAS.
- J. J. Pointon, D. Wallace, A. T. Merryweather-Clarke and K. J. Robson, Genet. Test., 2000, 4, 151–161 CrossRef CAS.
- E. Gross, N. Arnold, J. Goette, U. Schwarz-Boeger and M. Kiechle, Hum. Genet., 1999, 105, 72–78 CrossRef CAS.
- B. M. Paegel, S. H. I. Yeung and R. A. Mathies, Anal. Chem., 2002, 74, 5092–5098 CrossRef CAS.
- J. J. Pointon, A. T. Merryweather-Clarke, M. Carella and K. J. H. Robson, Genet. Test., 2000, 4, 115–120 CrossRef CAS.
- A. Koeken, C. Cobbaert, W. Quint and L. J. van Doorn, Clin. Chem. Lab. Med., 2002, 40, 122–125 Search PubMed.
- Q. Liang, P. A. Davis, B. H. Thompson and J. T. Simpson, J. Chromatogr., B, 2001, 754, 265–270 CrossRef CAS.
- J. M. Devaney, E. L. Pettit, S. G. Kaler, P. M. Vallone, J. M. Butler and M. A. Marino, Anal. Chem., 2001, 73, 620–624 CrossRef CAS.
- A. Guttman, Z. Ronai, C. Barta, Y. M. Hou, M. Sasvari-Szekely, X. Wang and S. P. Briggs, Electrophoresis, 2002, 23, 1524–1530 CrossRef CAS.
- G. Le Gac, C. Mura and C. Ferec, Clin. Chem., 2001, 47, 1633–1640 CAS.
- C. Hellerbrand, A. K. Bosserhoff, S. Seegers, G. Lingner, C. Wrede, G. Lock, J. Scholmerich and R. Buttner, Scand. J. Gastroenterol., 2001, 36, 1211–1216 CrossRef CAS.
- M. S. Hertzberg, D. McDonald and O. Mirochnik, Am. J. Hematol., 1998, 57, 260–261 CrossRef CAS.
- K. Simonsen, J. Dissing, L. Rudbeck and M. Schwartz, Ann. Hum. Genet., 1999, 63, 193–197 CrossRef.
- S. Fruchon, M. Bensaid, N. Borot, M. P. Roth and H. Coppin, Clin. Chem., 2003, 49, 822–824 CAS.
- W. Z. Xiao and P. J. Oefner, Hum. Mutat., 2001, 17, 439–474 CrossRef CAS.
- N. Arnold, E. Gross, U. Schwarz-Boeger, J. Pfisterer, W. Jonat and M. Kiechle, Hum. Mutat., 1999, 14, 333–339 CrossRef CAS.
- H. A. Jackson, D. J. Bowen and M. Worwood, Br. J. Haematol., 1997, 98, 856–859 CrossRef CAS.
- I. M. Lubin, N. A. Yamada, R. M. Stansel, R. G. Pace, E. M. Rohlfs and L. M. Silverman, Arch. Pathol. Lab. Med., 1999, 123, 1177–1181 Search PubMed.
- H. M. Wenz, S. Baumhueter, S. Ramachandra and M. Worwood, Hum. Genet., 1999, 104, 29–35 CrossRef CAS.
- C. A. Emrich, H. J. Tian, I. L. Medintz and R. A. Mathies, Anal. Chem., 2002, 74, 5076–5083 CrossRef CAS.
- C. Backhouse, M. Caamano, F. Oaks, E. Nordman, A. Carrillo, B. Johnson and S. Bay, Electrophoresis, 2000, 21, 150–156 CrossRef CAS.
- H. J. Tian, L. C. Brody, S. J. Fan, Z. L. Huang and J. P. Landers, Clin. Chem., 2001, 47, 173–185 CAS.
- H. J. Tian and J. P. Landers, Anal. Biochem., 2002, 309, 212–223 CrossRef CAS.
- H. J. Tian, A. Jaquins-Gerstl, N. Munro, M. Trucco, L. C. Brody and J. P. Landers, Genomics, 2000, 63, 25–34 CrossRef CAS.
- I. V. Kourkine, C. N. Hestekin, B. A. Buchholz and A. E. Barron, Anal. Chem., 2002, 74, 2565–2572 CrossRef CAS.
- P. Kozlowski and W. J. Krzyzosiak, Nucleic Acids Res., 2001, 29(14), e71 CrossRef CAS.
- S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1996, 68, 720–723 CrossRef CAS.
- V. Barbier, B. A. Buchholz, A. E. Barron and J. L. Viovy, Electrophoresis, 2002, 23, 1441–1449 CrossRef CAS.
- R. S. Madabhushi, Electrophoresis, 1998, 19, 224–30 CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning : a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 3rd edn., 2001 Search PubMed.
- T. Footz, S. Wunsam, S. Kulak, H. J. Crabtree, D. M. Glerum and C. J. Backhouse, Electrophoresis, 2001, 22, 3868–3875 CrossRef CAS.
- T. Wagner, D. Stoppa-Lyonnet, E. Fleischmann, D. Muhr, S. Pages, T. Sandberg, V. Caux, R. Moeslinger, G. Langbauer, A. Borg and P. Oefner, Genomics, 1999, 62, 369–376 CrossRef CAS.
- J. Bowyer, N. Verrills, M. R. Gillings and A. J. Holmes, J. Microbiol Methods, 2000, 41, 155–60 CrossRef CAS.
- E. A. Oussatcheva, L. S. Shlyakhtenko, R. Glass, R. R. Sinden, Y. L. Lyubchenko and V. N. Potaman, J. Mol. Biol., 1999, 292, 75–86 CrossRef CAS.
- J. S. Herzog, E. M. Jancis, S. D. Liao, G. Somlo and J. N. Weitzel, Hum. Mutat., 2002, 19, 656–663 CrossRef CAS.
- P. Kringen, S. Egedal, J. C. Pedersen, T. B. Haribtz, K. M. Tveit, K. Berg, A. L. Borresen-Dale and T. I. Andersen, Electrophoresis, 2002, 23, 4085–4091 CrossRef CAS.
- C. Lynas, Blood, 1997, 90, 4235–4236 CAS.
| This journal is © The Royal Society of Chemistry 2004 |