DOI:
10.1039/B208634D
(Communication)
Org. Biomol. Chem., 2003,
1, 24-26
Resolution of the non-steady-state kinetics of the elimination of HBr from 2-(p-nitrophenyl)ethyl bromide in alcohol/alkoxide media
Received 5th September 2002, Accepted 28th October 2002
First published on 21st November 2002
Abstract
Non-steady-state kinetic studies reveal that the elimination of HBr from 2-(p-nitrophenyl)ethyl bromide in alcohol/alkoxide media, the classical concerted E2 reaction, actually takes place by a two-step mechanism involving the intermediate formation of the carbanion.
Introduction
We find that kinetic data for the elimination of HBr from 2-(p-nitrophenyl)ethyl bromide (NPEB) in ethanol or methanol containing the corresponding alkoxide ions are inconsistent with the concerted E2 mechanism. Extent of reaction–time profiles for these reactions deviate significantly from the expected response for the E2 mechanism and extent-of-reaction-dependent apparent deuterium kinetic isotope effects (KIEapp) were observed. The kinetic data are consistent with the reversible consecutive second-order mechanism, eqn. (1). The formation of the carbanion is rate determining by virtue of the fact that loss of bromide ion (ke) is rapid compared to protonation of the carbanion (kb). The latter precludes D/H exchange during the elimination reactions of NPEB-2-d2.| | 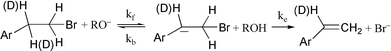 | (1) |
The elimination reactions of NPEB in ethanol containing ethoxide ions is a classic example1–3 of a reaction believed to take place by a concerted E2 mechanism. Recent studies published by the Thibblin group4–15 and others16–19 continue to provide significant new mechanistic detail. These studies have been concerned with the factors influencing elimination reactions in the border-line region between the E1CB and E2 mechanisms. The effect of structure on reactivity, kinetic isotope effects, and Brønsted parameters has been heavily relied upon in these discussions.
Our recent non-steady-state kinetic studies20–26 have revealed that many organic reactions previously believed to follow simple second-order kinetics follow the reversible consecutive second-order mechanism and do not reach a steady state before late in the first half-life. These include proton transfer reactions of methylarene radical cations,19,20,22 proton transfer between a nitroalkane and hydroxide ion,21 hydride transfer of an NADH model compound,25 and the SN2 reaction between the p-nitrophenoxide ion and methyl iodide in aqueous acetonitrile.26 We anticipated that our non-steady-state kinetic method would be applicable to the E2–E1CB manifold of reactions.
Results and discussion
The analysis of extent of reaction–time profiles is an effective means for distinguishing between the irreversible second-order mechanism (2) and the reversible consecutive second-order mechanism (3) providing that kinetic data in the pre-steady-state time period can be accessed.24 Once steady-state is reached, the two mechanisms| |  | (2) |
| | 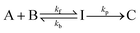 | (3) |
are kinetically indistinguishable. In our non-steady-state kinetic studies we determine two parameters, kinit/kpfo and t0.50/t0.05, which serve as mechanism probes. The first is defined as the ratio of the initial rate constant (kinit) during the extent of reaction ranging from 0 to 0.05 divided by the apparent pseudo first-order rate constant (kpfo) in the range from 0.05 to 0.50. The second is the time ratio, which corresponds to time at extent of reaction 0.50 (t0.50) divided by that at extent of reaction equal to 0.05 (t0.05). For mechanism (2)kinit/kpfo is equal to 1.00 and t0.50/t0.05 is equal to 13.51 under pseudo-first-order conditions with reactant B in large excess. In the early states of a reaction following mechanism (3), increase in [C] lags behind the decrease in [A]. When the increase in [C] is monitored under these conditions, kinit will be diminished and t0.05 will be increased giving rise to kinit/kpfo less than unity and t0.50/t0.05 less than 13.51. Just how large these deviations will be will depend upon the relative magnitudes of the rate constants kf, kb and kp and in the limit both values approach those for mechanism (2). For elimination reactions, eqn. (2) corresponds to the concerted E2 mechanism and eqn. (3) to the E1CB mechanism in which I is the carbanion.The data in Table 1 summarizes the mechanism probe parameters observed for the elimination of HBr from NPEB and NPEB-2-d2 in EtOH and in MeOH containing the corresponding alkoxide ions at 293 K. The last 2 columns in Table 1 show that both kinit/kpfo and t0.50/t0.05 deviate drastically from the theoretical values for mechanism (2), 1.00 and 13.51, respectively. The extent of reaction–time profiles for the reactions of NPEB and NPEB-d2 in ethanol are illustrated in Figs. 1 and 2, respectively. The experimental data are shown as solid squares and the solid lines represent the expected response for mechanism (2). The data in Table 1 and Figs. 1 and 2 force the conclusion that the elimination of HBr from NPEB in alcohols containing alkoxide ions does not follow the concerted E2 mechanism.
![Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and theoretical data for the E2 mechanism (solid lines). [EtO−] indicated on the figure.](/image/article/2003/OB/b208634d/b208634d-f1.gif) |
| | Fig. 1 Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and theoretical data for the E2 mechanism (solid lines). [EtO−] indicated on the figure. | |
![Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide-d2 in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and theoretical data for the E2 mechanism (solid lines). [EtO−] indicated on the figure.](/image/article/2003/OB/b208634d/b208634d-f2.gif) |
| | Fig. 2 Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide-d2 in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and theoretical data for the E2 mechanism (solid lines). [EtO−] indicated on the figure. | |
Table 1 Pseudo-first-order initial and apparent rate constants for the elimination reactions of 2-(p-nitrophenyl)ethyl bromide in alcohols containing alkoxide ions at 293 K
| Substratea | Solvent | [RO−]/mM | kinit/kpfobc | t0.50/t0.05bc |
|---|
| NPEB = 2-(p-nitrophenyl)ethyl bromide As defined in the text. Each value was derived from 40–60 extent of reaction–time profiles processed from digitally smoothed absorbance (335 nm)–time data (2000 points) generated with either a Hi-Tech model SF-61 or a SF-62 stopped flow instrument. Extent of reaction defined as [NS]/[NPEB]0 where NS is the product, p-nitrostyrene. |
|---|
| NPEB | EtOH | 300 | 0.810 | 11.12 |
| | | 150 | 0.763 | 10.52 |
| NPEB-d2 | EtOH | 300 | 0.881 | 12.03 |
| | | 150 | 0.929 | 12.61 |
| NPEB | MeOH | 300 | 0.840 | 11.58 |
| | | 150 | 0.783 | 11.08 |
| NPEB-d2 | MeOH | 300 | 0.714 | 8.72 |
| | | 150 | 0.750 | 9.04 |
The fit of experimental data to theoretical data for mechanism (3), carried out using procedures recently described,24 for the elimination reactions of NPEB and NPEB-d2 in ethanol are illustrated in Figs. 3 and 4. Once again the experimental data are shown as solid squares and the solid lines are theoretical data for mechanism (3). In all cases, reasonably close correspondence between experimental and theoretical data are observed. The fitting procedure which involved systematically changing the rate constants and minimizing the difference between experimental and theoretical data until a best fit was obtained invariably resulted in kf very nearly equal to kapp, defined by eqn. (4). Since kf can only equal kapp when kb is zero, the fitting results require that kb is very small.
It is likely that the
kb are non-zero but not large enough to influence the fitting of experimental to theoretical data. It should be pointed out that an additional constraint was applied during the fitting for the reactions of NPEB-d
2
(
Fig. 4) that consisted of assuming the value for
kp to be equal to that derived during the fitting of data for NPEB (
Fig. 3).
![Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and best-fit theoretical data for the reversible consecutive second-order mechanism (solid lines). [EtO−] indicated on the figure.](/image/article/2003/OB/b208634d/b208634d-f3.gif) |
| | Fig. 3 Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and best-fit theoretical data for the reversible consecutive second-order mechanism (solid lines). [EtO−] indicated on the figure. | |
![Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide-d2 in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and best-fit theoretical data for the reversible consecutive second-order mechanism (solid lines). [EtO−] indicated on the figure.](/image/article/2003/OB/b208634d/b208634d-f4.gif) |
| | Fig. 4 Extent of reaction–time profiles for the reaction of 2-(p-nitrophenyl)ethyl bromide-d2 in ethanol containing ethoxide at 293.2 K. Experimental data (solid squares) and best-fit theoretical data for the reversible consecutive second-order mechanism (solid lines). [EtO−] indicated on the figure. | |
The experimental (kapp) and best-fit (kf and kp) rate constants derived from extent of reaction–time profiles in both solvent systems are summarized in Table 2. The features of most interest are (a) that the relatively small kp values are about two orders of magnitude greater than the corresponding kf values and (b)
kf are about 15 times as great in ethanol as compared to methanol. Another feature of interest of the experimental data is that the apparent deuterium kinetic isotope effects (KIEapp) are extent-of-reaction dependent. This is illustrated by the plots in Fig. 5a (EtOH) and 5b (MeOH).
![Apparent deuterium kinetic isotope effects as a function of extent of reaction ([EtO−]
= 150 mM (a) and 300 mM (b)) for the elimination of HBr from 2-(p-nitrophenyl)ethyl bromide.](/image/article/2003/OB/b208634d/b208634d-f5.gif) |
| | Fig. 5 Apparent deuterium kinetic isotope effects as a function of extent of reaction ([EtO−]
= 150 mM (a) and 300 mM (b)) for the elimination of HBr from 2-(p-nitrophenyl)ethyl bromide. | |
Table 2 Rate constants for the elimination reactions of NPEB and NPEB-d2 in alcohols containing alkoxide ions at 293 K
| Substrate | Solvent | kapp/M−1 s−1a | kf/M−1 s−1b | kp/s−1c |
|---|
| Experimental value evaluated from t0.50 and refined in the fitting procedure. Best fit value obtained during the fitting procedure. Best fit value obtained during the fitting procedure. Constraint placed on the fitting that kp for NPEB-d2 be equal to that for NPEB. |
|---|
| NPEB | EtOH | 0.228 | 0.228 | 18.1 |
| NPEB-d2 | EtOH | 0.0288 | 0.0288 | 18.1d |
| NPEB | MeOH | 0.0150 | 0.0150 | 1.53 |
| NPEB-d2 | MeOH | 0.00218 | 0.00218 | 1.53d |
In order to provide further insight into how the relative values of kf and kp influence the magnitudes of the mechanistic probes, (kinit/kpfo and t0.50/t0.05), theoretical data were calculated for the hypothetical case where kapp
=
kf
(1 M−1s−1), kb
= 0, and kp was varied over a wide range. These results are summarized in Table 3. The most interesting feature of these data is that kpfo approaches kf when kp/kf is about 32 while kinit does not approach the kf value until the ratio is nearly 10,000. This provides a very wide range of kp values where t0.50/t0.05 deviates significantly from the limiting value of 13.51.
Table 3 The effect of the kp/kf ratio on the non-steady-state mechanism probes for the E2–E1CB mechanism manifolda
| kp/s−1 | kinit/s−1 | kpfo/s−1 | t0.50/t0.05 |
|---|
| Mechanism (3), [A]0
= 0.001 M, [B]0
= 1 M, kf
= 1 M−1 s−1, kb
= 0. |
|---|
| 1 | 0.144 | 0.478 | 4.72 |
| 2 | 0.230 | 0.650 | 4.85 |
| 4 | 0.280 | 0.814 | 5.28 |
| 8 | 0.380 | 0.930 | 6.12 |
| 16 | 0.502 | 0.983 | 7.41 |
| 32 | 0.638 | 0.997 | 9.01 |
| 64 | 0.768 | 1.000 | 10.01 |
| 128 | 0.867 | 1.000 | 11.85 |
| 256 | 0.929 | 1.000 | 12.63 |
| 512 | 0.963 | 1.000 | 13.06 |
| 1000 | 0.981 | 1.000 | 13.28 |
| 10000 | 0.998 | 1.000 | 13.49 |
Conclusions
Our primary conclusion from this preliminary study of the kinetics of elimination reactions is that the classic example of a concerted E2 mechanism, the elimination of HBr from NPEB, takes place by the E1CB mechanism accompanied by the formation of the carbanion which undergoes loss of bromide ion at a moderate rate. Our secondary conclusion, which is equally important, is that the non-steady-state mechanism probes are highly effective in the analysis of elimination reactions. Our recently developed data fitting procedure facilitates the resolution of the kinetics of the 2-step mechanism into the microscopic rate constants of the individual steps.Acknowledgements
We gratefully acknowledge the National Science Foundation (CHE-0074405) for support of this work.References
- R. F. Hudson and G. Klopman, J. Chem. Soc., 1964, 5–15 RSC.
- L. F. Blackwell, P. D. Buckley, J. W. Jolley and A. K. H. MacGibbon, J. Chem. Soc., Perkin Trans. 2, 1973, 169–173 RSC.
- J. R. Gandler and W. P. Jencks, J. Am. Chem. Soc., 1982, 104, 1937–1951 CrossRef CAS.
- X. Zeng and A. Thibblin, J. Chem. Soc., Perkin Trans. 2, 2002, 1352–1358 RSC.
- Z. S. Jia, J. Rudzinski, P. Paneth and A. Thibblin, J. Org. Chem., 2002, 67, 177–181 CrossRef CAS.
- Q. Meng and A. Thibblin, J. Chem. Soc., Perkin Trans. 2, 1999, 1397–1404 RSC.
- Q. Meng, B. Du and A. Thibblin, J. Phys. Org. Chem., 1999, 12, 116–122 CrossRef CAS.
- Q. Meng and A. Thibblin, J. Chem. Soc., Perkin Trans. 2, 1998, 583–590 RSC.
- Q. Meng and A. Thibblin, J. Am. Chem. Soc., 1997, 119, 1224–1229 CrossRef CAS.
- Q. Meng, A. Gogoll and A. Thibblin, J. Am. Chem. Soc., 1997, 119, 1217–1223 CrossRef CAS.
- A. Thibblin and Y. Saeki, J. Org. Chem., 1997, 62, 1079–1082 CrossRef CAS.
- Q. Meng and A. Thibblin, J. Chem. Soc., Chem. Commun., 1996, 345–346 RSC.
- Q. Meng and A. Thibblin, J. Am. Chem. Soc., 1995, 117, 9399–9407 CrossRef CAS.
- Q. Meng and A. Thibblin, J. Am. Chem. Soc., 1995, 117, 1839–1840 CrossRef CAS.
- A. Thibblin, Organic Reaction Mechanisms, 1992, 327–343 Search PubMed; A. Thibblin, Organic Reaction Mechanisms, 1991, 335–346 Search PubMed.
- P. Ryberg and O. Matsson, J. Am. Chem. Soc., 2001, 123, 2712–2718 CrossRef CAS.
- F. G. Larkin, R. A. More O'Ferrall and D. G. Murphy, Coll. Czech. Chem. Commun., 1999, 64, 1833–1848 CrossRef CAS.
- B. R. Cho, H. S. Chung and S. Y. Pyun, J. Org. Chem., 1999, 64, 8375–8378 CrossRef CAS.
- W. H. Saunders, Jr., J. Org. Chem., 1999, 64, 861–865 CrossRef CAS.
- V. D. Parker, Y. Zhao, Y. Lu and G. Zheng, J. Am. Chem. Soc., 1998, 120, 12720–12727 CrossRef CAS.
- Y. Zhao, Y. Lu and V. D. Parker, J. Am. Chem. Soc., 2001, 123, 1579–1586 CrossRef CAS.
- Y. Lu, Y. Zhao and V. D. Parker, J. Am. Chem. Soc., 2001, 123, 5900–5907 CrossRef CAS.
- Y. Zhao, Y. Lu and V. D. Parker, J. Chem. Soc., Perkin Trans. 2, 2001, 1481–1487 RSC.
- V. D. Parker and Y. Zhao, J. Phys. Org. Chem., 2001, 14, 604–611 CrossRef CAS.
- Y. Lu, Y. Zhao, K. L. Handoo and V. D. Parker, Org. Biomol. Chem., 2003, 1 Search PubMed (DOI: 10.1039/b208186e).
- Y. Lu, K. L. Handoo and V. D. Parker, Org. Biomol. Chem., 2003, 1 Search PubMed (DOI: 10.1039/b208635b).
|
| This journal is © The Royal Society of Chemistry 2003 |
Click here to see how this site uses Cookies. View our privacy policy here.