In-situ generated three component ruthenium-based catalyst for ROMP
Ricardo
Castarlenas
a,
Inaam
Alaoui-Abdallaoui
b,
David
Sémeril
a,
Bouchaib
Mernari
b,
Salaheddine
Guesmi
b and
Pierre H.
Dixneuf
*a
aInstitut de Chimie de Rennes, UMR 6509 Université de Rennes-CNRS, Organométalliques et Catalyse, Campus de Beaulieu, 35042, Rennes, France. E-mail: pierre.dixneuf@univ-rennes1.fr
bLaboratoire de Chimie de Coordination et Analytique, Faculté des Sciences, Université Chouaib Doukali, BP 20, El-Jadida, Morocco
First published on 18th November 2002
Abstract
The in-situ prepared three component system [RuCl2(p-cymene)]2/1,3-bis(R)-imidazolinium chloride/base (0.5/1/2) catalyses quantitatively the ROMP of cyclooctene at 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C for 20 min. After the activation of this catalytic system by preliminary heating at 80°C in chlorobenzene for 1 h, the resulting catalyst was able to polymerise cyclooctene at room temperature. Other ruthenium sources have also been evaluated in similar in-situ prepared systems for ROMP.
°C for 20 min. After the activation of this catalytic system by preliminary heating at 80°C in chlorobenzene for 1 h, the resulting catalyst was able to polymerise cyclooctene at room temperature. Other ruthenium sources have also been evaluated in similar in-situ prepared systems for ROMP.
Ring opening metathesis polymerisation (ROMP) constitutes an excellent tool to transform cyclic olefins into a variety of linear macromolecules, with regularly displayed double bonds and specific architectures.1 This is due to the discovery of well-defined single-site alkene metathesis catalysts, such as alkylidene–molybdenum2 and especially alkylidene–ruthenium catalysts tolerating a large variety of functional groups.3–11 These ruthenium precatalysts contain a bulky electron-rich ligand and, linked in close proximity to the metal site, an alkylidene,3,6,7,11 vinylidene,8 allenylidene,9 or alkylidyne10 moiety, or an in-situ generated carbene from diazoalkane.4 The introduction of bulky electron-rich imidazolylidene or imidazolinylidene ligands in ruthenium(II) catalyst precursors has opened a new door toward alkene metathesis and especially has led to the creation of efficient catalysts for ROMP.6,12,13 These ligands attract interest as their specific geometry, well contrasting with that of bulky phosphines (e.g. PCy3), may create or increase activity of the resulting coordinatively unsaturated ruthenium catalytic species.14
The straightforward preparation of RuCl2(1,3-imidazolylidene)(arene) has recently been shown by simple reaction of the dinuclear complex [RuCl2(arene)]2
(1) with electron rich olefins, sources of non sterically hindered 1,3-imidazolylidene,15,16 or with a source of bulky imidazolylidene such as a 1,3-bis(mesityl)imidazolium salt in the presence of a base.17 By contrast, it was recently found that the corresponding reaction of 1 with a bulky non aromatic 1,3-imidazolinium salt in the presence of Cs2CO3 did not lead to the corresponding heterocyclic carbene–ruthenium complex but led to the discovery of a new in-situ prepared three component catalyst for alkene metathesis and fine chemistry.18–21 Indeed, the ruthenium complex [RuCl2(p-cymene)]2
(1a), the imidazolinium chloride 2a
(R=mesityl) and the base Cs2CO3 in the molar ratio 0.5/1/2, led to the in-situ prepared, as yet unidentified catalyst A
(Scheme 1), which promoted the selective transformation of enynes into conjugated alkenyl cycloalkenes,18,19 and the ring closing metathesis of dienes in the presence of acetylene.20,21 This in-situ prepared catalyst has not been used profitably in the related ROMP of cyclic olefins. We now wish to report that the combination of the ruthenium source 1a, an imidazolinium salt 2 and Cs2CO3 leads to an efficient catalyst for high yield ROMP of cyclooctene at 80°C and at room temperature after preliminary thermal activation of the catalytic system.
 | ||
| Scheme 1 Preparation of catalyst A. | ||
The polymerisation of cyclooctene was studied with several types of catalysts based on the same ruthenium source [RuCl2(p-cymene)]2
(1a), with three different sterically hindered imidazolinium chlorides 2a, 2b, and 2c
(Fig. 1) and three different bases Cs2CO3, tBuOK, and NEt3 in order to give evidence of the influence of the base and of the electron-rich carbene nature. The results are gathered in Table 1. All experiments were performed at 80°C for 20 min and repeated at least twice. With the 1,3-bis(mesityl)imidazolinium salt 2a, it was showed that NEt3 inhibits the polymerisation (Table 1, entry 1), tBuOK gives relatively good yields (80%) with the highest Mn value (35×103)
(entry 2), whereas Cs2CO3 leads to the best results in term of excellent polymer yield (92%), Mn value (28×103) and lower polydispersity (1.3)
(entry 3). The nature of caesium carbonate appears to play a specific role, with respect to tBuOK, even if the Mn value is slightly smaller. Thus, Cs2CO3 not only deprotonates 2a, but modifies the catalyst in a way that is not yet understood, possibly by substituting the chloride ligand(s) and/or favouring the displacement of p-cymene ligand. An experiment with the well established ruthenium ROMP catalyst RuCl2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh)(PCy3)23
(3) was carried out under the same conditions for comparison with our catalytic systems (entry 8). It shows that the system 1a/2a with Cs2CO3
(entry 3) gives better yield and polydispersity than 3 whereas the latter offers a higher Mn value (70×103)
CHPh)(PCy3)23
(3) was carried out under the same conditions for comparison with our catalytic systems (entry 8). It shows that the system 1a/2a with Cs2CO3
(entry 3) gives better yield and polydispersity than 3 whereas the latter offers a higher Mn value (70×103)
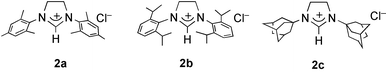 | ||
| Fig. 1 Imidazolinium chlorides. | ||
| Entry | Imidazolinium salt | Base | Yield (%) | 10−3×Mnb |
PDIc | % transd |
|---|---|---|---|---|---|---|
|
a General conditions: The catalyst was in-situ prepared from 7.5×10−6 mol of [RuCl2(p-cymene)]2, 1.5×10−5 mol of the imidazolinium chloride, and 3.0×10−5 mol of base in 2.5 mL of chlorobenzene. Polymerisation was performed by addition of 4.5×10−3 mol of cyclooctene ([monomer]/[Ru]=300).
b Determined by GPC in THF vs. polystyrene standards.
c Polydispersity index Mw/Mn.
d Determined by 13C NMR.
e 20 min at 80°C.
f Activation 1 h at 80°C and polymerisation 1 h at r. t.
|
||||||
| 1e | 2a | NEt3 | 0 | — | — | — |
| 2e | 2a | tBuOK | 80 | 35 | 1.5 | 75 |
| 3e | 2a | Cs2CO3 | 92 | 28 | 1.3 | 82 |
| 4e | 2b | Cs2CO3 | 65 | 9 | 1.5 | — |
| 5e | 2c | Cs2CO3 | 0 | — | — | — |
| 6f | 2a | Cs2CO3 | 85 | 69 | 1.4 | 81 |
| 7f | 2a | tBuOK | 50 | 41 | 1.3 | 78 |
| 8e | 3 | 65 | 70 | 1.5 | — | |
The influence of the imidazolinium chlorides with the bulky groups R=mesityl (2a), 2,6-bis(isopropyl)phenyl (2b), and adamantyl (2c) was evaluated using the most favourable base Cs2CO3 in the polymerisation of cyclooctene. It is surprising that 2c inhibits completely the polymerisation (Table 1, entry 5) whereas 2b leads to moderate yield and molecular weight (entry 4). Thus the combination of 2a and Cs2CO3 appears the best association for cyclooctene polymerisation when 1a is used as source of the ruthenium (entry 3).
Activation of the ruthenium catalyst
The performance of cyclooctene polymerisation at room temperature remains a challenge, especially for the transformation of functionalised cyclooctene and for application to fragile cyclic olefins. In order to make a step in this direction we considered that in the previous experiments (Table 1), the heating of the ruthenium source 1a with the in-situ generated bulky heterocyclic carbene might initially lead to the displacement of the arene, to generate a coordinatively unsaturated catalytic species of type RuX2(carbene)(solvent or cyclooctene)n. Indeed, the heating of benzimidazolinium salt with pendent aryl groups in the presence of 1a and Cs2CO3 leads to the coordination of the formed carbene and to the intramolecular displacement of p-cymene ligand by the pendent aryl group.22 Thus, in the absence of cyclooctene, the three component system [RuCl2(p-cymene)]2/2a/Cs2CO3 (0.5/1/2) was first heated at 80°C in 2.5 mL of PhCl for 1 h, during which time the solution colour changed from red to brown. After the solution was cooled to room temperature, the cyclooctene was added and the solution was stirred for 1 h at room temperature. Conversion was completed and good yield of polyoctenamer was obtained especially when Cs2CO3 was used as a base (Table 1, entry 6) with respect to tBuOK (entry 7). Under these conditions the catalyst gives access to high molecular weight Mn=69×103
(PDI=1.4) for a theoretical Mn=33×103 thus showing that an average of 50% of ruthenium sites were active. Several efficient ruthenium catalysts successfully performed the ROMP of cyclooctene at room temperature such as the Herrmann catalyst RuCl2(CHPh){1,3-bis(mesityl)imidazolylidene}2
[95% after 1.5 h, Mn=266×103, PDI=1.76],6 the Grubbs catalyst RuCl2(CHPh)(PCy3){1,3-bis(mesityl)imidazolinylidene}
[total conversion in 30 min],12 the binuclear Hofmann catalyst [RuCl(CHR){(tBu)2PCH2P(tBu)2}]2[OTf]2
[95% after 20 min],7 the Werner carbyne [RuHCl(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMe)(OEt2)(PCy3)2]BF4
[90% after 3 min],10 the RuCl2(imidazolylidene)(p-cymene) complex under visible light irradiation reported by Noels [99% after 2 h, Mn=537×103, PDI=1.33]13 and the thermally activated allenylidene complex [RuCl(CCCPh2)(PCy3)(p-cymene)][OTf]. [97% after 10 min, Mn=151×103, PDI=1.7].23 Although the nature of the catalytic species arising from thermally activated catalyst A is not known, this approach to generate a catalyst operating at room temperature has potential for fragile functional cyclic olefins polymerisation.
CMe)(OEt2)(PCy3)2]BF4
[90% after 3 min],10 the RuCl2(imidazolylidene)(p-cymene) complex under visible light irradiation reported by Noels [99% after 2 h, Mn=537×103, PDI=1.33]13 and the thermally activated allenylidene complex [RuCl(CCCPh2)(PCy3)(p-cymene)][OTf]. [97% after 10 min, Mn=151×103, PDI=1.7].23 Although the nature of the catalytic species arising from thermally activated catalyst A is not known, this approach to generate a catalyst operating at room temperature has potential for fragile functional cyclic olefins polymerisation.
Polymerisation based on various ruthenium sources
The present study shows the drastic influence of the nature of both N-heterocyclic carbene and the base. It can be predicted that another ruthenium source should modify significantly the catalyst activity. Thus, the same concept of in-situ prepared catalyst was applied to simple easily available ruthenium sources RuCl3·xH2O, [RuCl2(NBD)]n (NBD=norbornadiene), [RuCl2(COD)]n
(COD=1,5-cyclooctadiene), and RuCl2(dmso)4
(dmso=dimethylsulfoxide). The previously most active imidazolinium salt 2a and Cs2CO3 were added to the ruthenium precursor in the ratio [Ru]/2a/Cs2CO3
(1/1/2) to perform the polymerisation of norbornene and cyclooctene. The resulting in-situ prepared catalysts were directly used to polymerise 300 equivalents of norbornene (Table 2). The reaction performed at 60°C in 2.5 mL of PhCl for only 5 min gives in each case complete conversion and RuCl3·xH2O and especially RuCl2(dmso)4 lead to high molecular weight with controlled polydispersity (1.5–1.7). A blank test was carried out in the absence of the imidazolinium salt to rule out any erroneous activity and less than 5% of polymer was formed.
| Entry | Ruthenium source | Yield (%) | 10−3×Mnb |
PDIc |
|---|---|---|---|---|
|
a General conditions: The catalyst was in-situ prepared from 1.5×10−5 mol of [Ru], 1.5×10−5 mol of 2a, and 3.0×10−5 mol of Cs2CO3 in 2.5 mL of chlorobenzene. Polymerisation was performed by addition of 4.5×10−3 mol of norbornene ([monomer]/[Ru]=300) for 5 min at 60°C.
b Determined by GPC in THF vs. polystyrene standards.
c Polydispersity index Mw/Mn.
|
||||
| 1 | [RuCl2(NBD)]n | 99 | 12 | 1.7 |
| 2 | [RuCl2(COD)]n | 98 | 14 | 1.6 |
| 3 | RuCl3·xH2O | 96 | 46 | 1.5 |
| 4 | RuCl2(dmso)4 | 98 | 56 | 1.6 |
| 5 | [RuCl2(p-cymene)]2 | 97 | 13 | 1.5 |
The same in-situ prepared catalysts were evaluated for cyclooctene polymerisation which requires higher temperature (80°C). The results are displayed in Table 3. After 20 min at 80°C complete conversion was not reached. However, RuCl3·xH2O and RuCl2(dmso)4 gave materials with good molecular weights. These results revealed that for cyclooctene polymerisation the use of the ruthenium source [RuCl2(p-cymene)]2 is the best choice.
| Entry | Ruthenium source | Yield (%) | 10−3×Mnb |
PDIc |
|---|---|---|---|---|
|
a General conditions as in Table 2. Polymerisation was performed for 20 min at 80°C.
b Determined by GPC in THF vs. polystyrene standards.
c Polydispersity index Mw/Mn.
|
||||
| 1 | [RuCl2(NBD)]n | 20 | 12 | 2.7 |
| 2 | [RuCl2(COD)]n | 30 | 4 | 1.1 |
| 3 | RuCl3·xH2O | 48 | 39 | 1.5 |
| 4 | RuCl2(dmso)4 | 80 | 73 | 1.4 |
The above results show that for cyclooctene polymerisation at both 80°C or, after thermal activation, at room temperature, the best catalytic system is made from the stable easy available components: [RuCl2(p-cymene)]2/2a/Cs2CO3. These in-situ systems offer potential at the same time to raise hopes for the polymerisation of functional unstrained or fragile cyclic olefins and for the discovery of new catalytic systems by changing the ruthenium source.
Experimental
All reactions were carried out with rigorous exclusion of air using Schlenk-tube techniques. Chlorobenzene was dried under P2O5 and distilled under argon prior to use. The ruthenium complexes were purchased directly from available commercial sources except 3 which was synthesised according to the literature procedure.3 The imidazolinium salts were prepared as described in the literature.24 Norbornene was used as purchased from commercial sources and cyclooctene was distilled from powdered NaOH and stored under argon with 4 Å molecular sieves.Polymerisation of cycloolefins
×10−5 mol of [Ru] complex, 1.5×10−5 mol of the imidazolinium chloride, and 3.0×10−5 mol of the corresponding base were dissolved in 2.5 mL of dry chlorobenzene under argon atmosphere. Freshly distilled monomer (4.5×10−3 mol) was added immediately and the suspension was stirred for 20 min at 80°C. After reaction, the resulting viscous mixture was dissolved with 20 mL of CHCl3 containing 0.1% of 2,6-di-tert-butyl-4-methylphenol (BHT) and 0.3 mL of vinyl ether. Then the solution was poured in 200 mL of methanol to precipitate the polymer. The crude product was further purified by using silica gel column chromatography and reprecipitation in MeOH (200 mL, containing 0.1% BHT) to give the polymer as a white solid, which was collected by filtration, dried under vacuum, and characterised by 1H and 13C NMR and GPC calibrated from polystyrene standards.
×10−6 mol of [RuCl2(p-cymene)]2 complex, 1.5×10−5 mol of the imidazolinium chloride, and 3.0×10−5 mol of the corresponding base were dissolved in 2.5 mL of dry PhCl under argon atmosphere. The reaction was heated to 80°C for 1 h and when the solution had cooled to 20°C, 4.5×10−3 mol of cyclooctene was added and the solution was stirred for 1 h at r.t. The resulting polymer was purified as described in method A.
Acknowledgements
The authors are grateful for support to the European Union within the EU network POLYCAT (UPRN-CT-2000-00010), the COST-Chemistry Program D17 (0003/00), and the Region Bretagne for support within the Regional program PRIR (No. 169 AOC).References
- K. J. Ivin and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press, San Diego, CA, 1997 Search PubMed.
- R. R. Schrock, Acc. Chem. Res., 1990, 23, 158 CrossRef CAS.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100 CrossRef CAS.
- A. W. Stumpf, E. Saive, A. Demonceau and A. F. Noels, J. Chem. Soc., Chem. Commun., 1995, 1127 RSC.
- A. Hafner, A. Mühlebach and P. A. van der Schaaf, Angew. Chem., Int. Ed. Engl., 1997, 36, 2121 CrossRef CAS.
- T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, Angew. Chem., Int. Ed., 1998, 37, 2490 CrossRef CAS.
- S. M. Hansen, M. A. O. Volland, F. Rominger, F. Eisenträger and P. Hofmann, Angew. Chem., Int. Ed., 1999, 38, 1273 CrossRef CAS.
- H. Katayama, H. Urushima and F. Ozawa, J. Organomet. Chem., 2000, 606, 16 CrossRef CAS.
- A. Fürstner, M. Liebl, C. W. Lehmann, M. Picquet, R. Kunz, C. Bruneau, D. Touchard and P. H. Dixneuf, Chem. Eur. J., 2000, 6, 1847 CrossRef CAS.
- W. Stüer, J. Wolf, H. Werner, P. Schwab and M. Schulz, Angew. Chem., Int. Ed., 1998, 37, 3421 CrossRef CAS.
- J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus Jr. and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903 CrossRef CAS.
- L. Delaude, A. Demonceau and A. F. Noels, Chem. Commun., 2001, 986 RSC.
- B. Çetinkaya, S. Demir, I. Özdemir, L. Toupet, D. Sémeril, C. Bruneau and P. H. Dixneuf, New J. Chem., 2001, 25, 519 RSC.
- H. Kücükbay, B. Çetinkaya, S. Guesmi and P. H. Dixneuf, Organometallics, 1996, 15, 2434 CrossRef CAS.
- B. Çetinkaya, I. Özdemir and P. H. Dixneuf, J. Organomet. Chem., 1997, 534, 153 CrossRef CAS.
- L. Jafarpour, J. Huang, E. D. Stevens and P. S. Nolan, Organometallics, 1999, 18, 3760 CrossRef CAS.
- D. Sémeril, M. Cléran, C. Bruneau and P. H. Dixneuf, Adv. Synth. Catal., 2001, 343, 184 CrossRef CAS.
- L. Ackermann, C. Bruneau and P. H. Dixneuf, Synlett, 2001, 3, 397.
- D. Sémeril, C. Bruneau and P. H. Dixneuf, Helv. Chim. Acta, 2001, 84, 3335 CrossRef CAS.
- D. Sémeril, C. Bruneau and P. H. Dixneuf, Adv. Synth. Catal., 2002, 344, 585 Search PubMed.
- B. Çetinkaya and I. Özdemir, personal communication.
- R. Castarlenas, D. Sémeril, A. F. Noels, A. Demonceau and P. H. Dixneuf, J. Organomet. Chem. Search PubMed , in press.
- A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |