‘Latent’ ultraviolet light absorbers
Christoph
Kocher
*a,
Christoph
Weder
b and
Paul
Smith
a
aDepartment of Materials, Eidgenössische Technische Hochschule (ETH) Zürich, Universitätsstrasse 41, UNO C14, CH-8092 Zürich, Switzerland. E-mail: ckocher@ifp.mat.ethz.ch
bDepartment of Macromolecular Science and Engineering, Case Western Reserve University, 2100 Adelbert Road, Cleveland, OH 44106-7202, USA
First published on 15th November 2002
Abstract
We have synthesized and characterized photo-labile and acid-labile esters of 2-hydroxybenzophenone and 2-hydroxybenzotriazoles, which, upon appropriate treatment, release classical UV absorbers. Two concepts of ‘latent’ UV absorbers are presented, namely, benzoyl-caged species which release their parent UV absorber upon short-wavelength UV irradiation, and acid-labile species caged with the tert-butoxycarbonyl group which were employed in combination with a photoacid generator. The latent UV absorbers were melt processed into thermoplastic polymers such as linear low density polyethylene and absorbing–non-absorbing patterns were produced in the polymer blend films by spatially resolved conversion, illustrating their usefulness for optical data storage applications.
Introduction
Photochromic or, more generally, chromogenic molecules, in which a photochemically induced change in the structure causes a significant alteration of the absorption characteristics, have proven useful in many applications, including, for example, in optical filters,1 as tracer molecules for rheology studies,2 and in optical data storage media.3 Typically, the wavelength range in which such chromogenic dyes function is confined to the visible spectrum. In the design of new systems, the UV range has long been neglected, despite the fact that in applications such as optical data storage, performance can be improved by using UV-sensitive species. Moreover, photochromic dyes capable of developing a UV absorption band would allow for additional applications such as patterned UV filters, invisible markings, and selective UV screening in fluorescent tracer experiments. Here, we present systems which, upon appropriate treatment, release classical UV absorbers and, thus, represent a potentially useful extension of the available range of chromogenic dyes.Classical UV absorbers are benzophenones and benzotriazoles with ortho-hydroxy substituents.4,5 These molecules very efficiently absorb incident UV radiation, typically in the range 300–400 nm. They dissipate the absorbed energy by a non-radiative internal conversion process which involves the reversible formation of a six-membered hydrogen-bonded ring6—a process which is commonly known as excited-state intramolecular proton transfer (ESIPT).7,8 In the strategy presented herein, we derivatized a number of classical UV absorbers through esterification with benzoic acid and produced esters lacking the ability to dissipate incident UV radiation by internal conversion. Moreover, these caged UV absorbers are characterized by absorption spectra that are significantly shifted to shorter wavelengths when compared to those of their parent hydroxy compounds; long wavelength UV light is therefore transmitted.
The benzoyl group can be removed9 by exposure to short wavelength UV light, restoring the parent UV absorber. Additionally, we employed the acid-labile tert-butoxycarbonyl (t-Boc) group10 in combination with a so-called photoacid generator (PAG).11,12 Both concepts allow for photochemically induced, spacially resolved formation of the parent UV absorber.
Results and discussion
UV absorbers and derivatives
Amongst the vast array of available UV absorbers, we chose to derivatize 2-hydroxybenzophenone (5), 2-(2-hydroxy-5-methylphenyl)benzotriazole (6), and 2,4-di-tert-butyl-6-(5-chloro-2H-benzotriazol-2-yl)phenol (7) (Fig. 1). The first two compounds are structurally the most simple, commercially available representatives of their absorber classes (benzophenones and benzotriazoles, respectively), while the latter compound was selected to demonstrate the limitations of the photo-lability of the benzoyl caging group on the one hand, and the alternative approach of t-Boc protection on the other. This chlorine-substituted UV-absorber is characterized by a red-shifted absorption spectrum with respect to non-derivatized benzotriazoles and is therefore particularly suited for applications where UV light from the widely employed fluorescent tubes (also known as ‘blacklight’) is selectively screened out.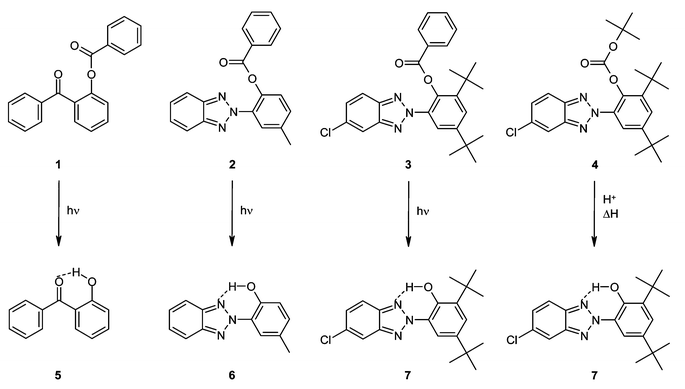 | ||
| Fig. 1 Structures of the photo-labile latent UV absorbers 1–3 and of the acid-labile latent UV absorber 4. Upon appropriate treatment (see text), the corresponding UV absorbers (5–7) are released. | ||
Benzoyl chloride was used to produce the photo-labile compounds 1–3, and di-tert-butyl dicarbonate to yield the acid-labile species 4 (Fig. 1). As will be shown in detail below, photocleavage of benzoic acid esters of the latent UV absorbers has certain limitations. In systems in which the benzoyl group represents the only substituent allowing for vibrational energy dissipation, such as in the benzoyl derivatives (1 and 2) of 2-hydroxybenzophenone (5) and 2-(2-hydroxy-5-methylphenyl)benzotriazole (6), fast and exclusive restoration of the parent UV absorber is observed upon irradiation with short wavelength UV light. In contrast, photolytic ester cleavage is strongly reduced in latent UV absorbers based on parents comprising additional substituents that allow for vibrational energy dissipation, such as 7. As a matter of fact, commercial UV absorbers of the benzotriazole class often contain tert-butyl, 1,1,2,2-tetramethylpropyl, and 1-methyl-1-phenylethyl groups to increase the photostability. These groups not only lock the molecule in its planar configuration,13 but also allow for vibrational energy dissipation.14 In hydroxyphenylbenzotriazenes, another important class of UV absorbers, the three phenyl groups have a similar effect. Consequently, photo-labile latent UV absorbers bearing such additional substituents are expected, and in fact were found, to have a reduced photosensitivity when compared to systems where the only substitiuent is the photocleavable benzoyl group. In order to also make use of such substituted UV absorbers in the context of this work, the hydroxyl functionality was protected with an acid-labile t-Boc group. Deprotection occurs in this case in the presence of acid at elevated temperatures. Since the acid can be photochemically released by employing a PAG, t-Boc-protected latent UV absorbers embedded in a polymer matrix offer similar opportunities for photolithographic patterning15,16 as their benzoyl-protected counterparts.9 Finally, the PAG can be sublimed off after selective activation, which ultimately renders patterned features insusceptible to unwanted conversion, e.g. in daylight.
Synthesis of latent UV absorbers
The latent UV absorbers 1–4 were synthesized by reacting the respective parent UV absorbers (ortho-hydroxy compounds) 5–7 with either benzoyl chloride in the case of the photo-labile systems 1–3, or with di-tert-butyl dicarbonate in the case of the acid-labile version 4, in anhydrous pyridine as solvent. The products were obtained in 77–96% yields after purification. All latent absorbers prepared were of high purity, as confirmed by 1H NMR spectroscopy and elemental analysis. It should be noted that some of the compounds here described have been prepared before, albeit for different applications. Compound 1 is an intermediate for, e.g., fungicides.17 Compound 2 has been disclosed in a patent on UV absorbers for the short wavelength UV regime18 and its thermal rearrangement was studied.19 The preparation of compounds 2 and 3 has been described in a different patent.20Thermal stability
Regarding the envisioned applications in polymer blend films, which are preferentially produced by common melt-processing schemes, investigations concerning the thermal stability of the latent UV absorbers were conducted using differential scanning calorimetry (DSC). All benzoyl-caged latent UV absorbers were found to be thermally stable up to a temperature of 250 °C, at least on the timescale of the DSC experiment. The t-Boc-caged latent UV absorber 4, however, was found to be thermolytically cleaved at temperatures of ca. 170 °C and higher. The DSC data suggest that heating 4 to 160 °C leaves the substance virtually unchanged, while heating to temperatures ranging from 170–250 °C leads to complete conversion to 7. This finding is in accordance with the commonly observed thermal stability of t-Boc-protected compounds.16Incorporation of latent UV absorbers in LLDPE films
The conversion of the latent UV absorbers to their parent UV absorbers was studied in linear low density polyethylene (LLDPE) blend films containing 0.2% w/w of the compound. In the case of the acid-labile latent UV absorber 4, the blend film additionally contained 3% w/w of benzenesulfonic acid phenyl ester as the PAG.21 The films were produced in a standard hot press and had a thickness of approximately 100 µm. The film thickness and chromophore contents employed allowed for experimentally convenient optical extinctions and reaction kinetics.Photophysical characteristics
UV-Vis spectrometry of the compounds in chloroform solutions revealed that the spectra of all the latent UV absorbers are characterized by a primary absorption maximum that is shifted to considerably shorter wavelengths when compared with the absorption of the parent hydroxy analog (Fig. 2). This bathochromic shift may be as large as 42 nm (in the case of 3). While the spectrum of hydroxybenzophenone (5) exhibits absorption maxima at 340 and 265 nm, that of its benzoyl ester (1) only exhibits a single absorption band centered at 244 nm. In the case of compound 6, two absorption maxima are also observed, at 341 and 301 nm. The spectrum of compound 2, on the other hand, exhibits a single absorption band centered at 303 nm. The absorber/latent absorber pair 7/3 exhibit similar spectral properties to the pair 6/2, with a slight bathochromic shift as a consequence of the chlorine substituent on the benzotriazole moiety. The two absorption maxima of 7 are located at 347 and 313 nm, while the spectrum of 3 exhibits a single absorption band centered at 305 nm. The sole absorption band of the acid-labile t-Boc-protected analog 4 is located at 309 nm. These key optical parameters and the corresponding extinction coefficients are collected in Table 1. When dispersed in LLDPE, the latent UV absorbers exhibit virtually the same absorption characteristics as in solution (see Table 1).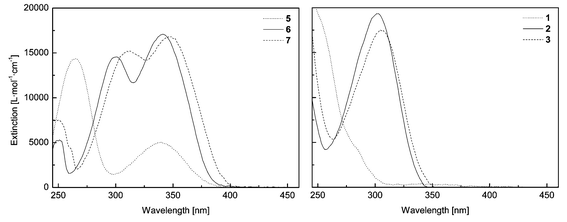 | ||
| Fig. 2 Absorption spectra of the UV absorbers 5–7 and of the latent UV absorbers 1–3 (all solutions 0.5 × 10−5 M in CHCl3). See Table 1 for key optical parameters. | ||
| Core | Compound | Caging group | λ max (CHCl3)/nm | ε (CHCl3)/L mol−1 cm−1 | λ max (LLDPE)/nm |
|---|---|---|---|---|---|
| Benzophenone | 5 | — | 340, 265 | 5100, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
340 |
| 1 | Benzoyl | 244 | 20900 |
243 | |
| Benzotriazole | 6 | — | 341, 301 | 17100, 14600 |
346 |
| 2 | Benzoyl | 303 | 19300 |
306 | |
| Substituted benzotriazole | 7 | — | 347, 313 | 16800, 15200 |
350 |
| 3 | Benzoyl | 305 | 17500 |
314 | |
| 4 | t-Boc | 309 | 19400 |
310 |
All benzoyl-caged latent UV absorbers were found to be virtually non-photoluminescent in solution, as well as when incorporated in LLDPE films. At first, this finding seems somewhat surprising considering the fact that these compounds cannot undergo radiationless energy dissipation by the ESIPT pathway, and especially in the view of the fact that both 6 in a non-polar configuration13 and the methyl ester22 of 6, which both lack an intramolecular hydrogen bond, were found to be photoluminescent in earlier studies. The observation that the benzoyl esters of the present UV absorbers are, in contrast, non-photoluminescent may be attributed to a relaxation pathway that is referred to as the ‘loose bolt’ effect:14 the benzoyl moiety provides multiple vibrational and rotational degrees of freedom, which allow for a radiationless transition from the excited to the ground state.14 We recently reported on the same phenomenon observed for aromatic esters of 2-(2-hydroxyphenyl)benzoxazole, which, consequently, form a novel class of caged photoluminescent dyes.9 Very much in accordance with the results presented in that study, the t-Boc-caged latent absorber 4 exhibits a fairly strong, blue photoluminescence, both in chloroform solution and when in a LLDPE matrix.
Activation of photo-labile latent UV absorbers
Upon exposure to 254 nm UV light from a laboratory UV lamp, chloroform solutions of the benzoyl-caged latent UV absorbers were found to reproduce the absorption characteristics of their parent hydroxy analogs. Importantly, restoration of the absorption characteristics of the parent was also observed when the latent absorbers were conatined in a polymer matrix. Thus, we observed that the spectra of LLDPE films containing 0.2% w/w of the 2-hydroxybenzophenone-based, benzoic acid-caged latent UV absorber 1 developed an absorption band centered around 340 nm upon UV irradiation. This is indicative of the regeneration of 2-hydroxybenzophenone, which exhibits the same absorption characteristics. In spectra of LLDPE films containing 0.2% w/w of the benzotriazole-based latent UV absorber 2, an absorption band centered at 346 nm was observed on UV irradiation, in accordance with the absorption band of its parent active UV absorber 6 (cf.Fig. 3). The increase of the developing absorption band appears to follow first order kinetics, as can be concluded from Fig. 4. Importantly, apart from the appearance of the respective parent UV absorbers' absorption bands, no change in the absorption characteristics (e.g. yellowing) was observed.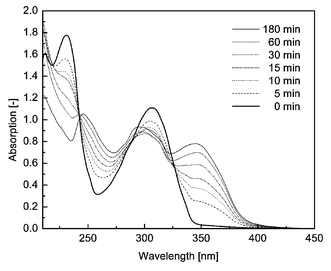 | ||
| Fig. 3 Activation of 2 in a LLDPE film (0.2% w/w, thickness 100 µm) upon irradiation at 254 nm. Absorption spectra prior to irradiation (bold line) and after different photoactivation intervals. | ||
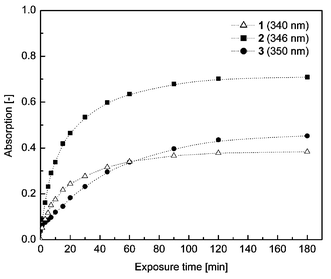 | ||
| Fig. 4 Development of the indicated UV absorption bands upon conversion of the three benzoyl-caged latent UV absorbers 1 (△, 340 nm), 2 (■, 346 nm) and 3 (●, 350 nm) in 0.2% w/w LLDPE blend films upon irradiation at 254 nm. Dotted lines are first order kinetic fits calculated from the experimental data. For comparison, the absorption data were scaled such that the absorbance of the peak in question in the spectrum of the pristine film was 1 in all cases. | ||
It should be noted that the concentration of latent UV absorber (0.2% w/w), matrix material (commercial grade LLDPE with some self-absorption at the activation wavelength) film thickness (100 µm), and optical power (340 µW cm−2) were chosen in order to conduct the photoactivation experiments on an experimentally convenient timescale. With the optical intensities usually employed for photolithograpy (e.g. laser systems),15 much shorter processing times can be expected.
As was reported by others, the acetate ester of 6 regenerates 6 upon irradiation, and not, as might be expected, a photo-Fries rearrangement product.22 However, in accordance with our previous findings concerning the cleavage of aliphatic esters,9 this compound only regenerates traces of its parent alcohol, even under the extremely harsh irradiation condition employed in the study.22 The aromatic ester 2, in contrast, quite efficiently releases 6. However, we found that 3 regenerated the absorption characteristics of the parent active UV absorber 7 only to some extent when exposed to short wavelength UV light. As can be seen from Fig. 4, the conversion of 2 to 6 proceeds faster and more completely than the conversion of 3 to 7. Note, that the absorption data plotted in Fig. 4 have been scaled such that the magnitudes of the primary absorptions of the pristine samples are 1 in all cases; since the extinction coefficients of the bands due to 6 and 7 are very similar, it is obvious from Fig. 4 that the conversion of 3 to 7 is much less efficient than the conversion of 2 to 6. The conversion of 1 to 5 (Fig. 4) is also faster and more complete than the conversion of 3 to 7, taking into account the fact that the extinction of the absorbance due to 5 is only about one third of that for 6 and 7. We attribute the fact that the benzoic acid-caged latent UV absorbers 1 and 2 in LLDPE films are readily converted to their active parent hydroxyl analogs, hydroxybenzophenone and 6, respectively, while conversion of the benzoic acid-caged latent UV absorber 3 to 7 is less easy, to critical differences in the molecular structures of these compounds. In 1 and 2, the only substituent having rotational and vibratory degrees of freedom is the benzoyl moiety, while in 3, the two tert-butyl groups offer additional degrees of freedom. When excited with UV light, 1 and 2 can dissipate the excess energy and return to the ground state by vibrational relaxation due to the benzoyl moiety, the ‘loose bolt’ effect referred to above.14 Since the benzoyl ester bond is the weakest bond in these molecules, ester cleavage is likely to occur as a consequence of the vibrational relaxation, yielding a biradical which quickly converts to the hydroxyl-substituted UV absorber and benzoic acid by hydrogen abstraction. Evidence supporting this hypothesis can be found elsewhere.9 In the case of 3, the benzoyl moiety as well as the two tert-butyl substituents allow for vibrational energy dissipation, which consequently leads to reduced sensitivity for 3 because energy dissipation does not exclusively occur through vibration of the benzoyl moiety and consequent ester cleavage.
This finding evidently has implications for the design of other latent UV absorbers. Most commercial UV absorbers comprise substituents that allow for vibrational energy dissipation and, thus, enhance the photostability of the molecule. As shown for 3, and for the reasons given above, derivatization of such UV absorbers with benzoic acid yields latent UV absorbers with reduced photosensitivity. In order to also make the latter UV absorbers accessible from a latent precursor, we employed the concept of protection of the hydroxy group with the acid-labile t-Boc protective group10 (see below).
GC-MS studies
The change in the absorption characteristics of the photo-labile, benzoyl-caged latent UV absorbers upon UV exposure clearly suggests the release of the parent UV absorbers. In order to obtain further evidence for the formation of the UV absorbers and to identify possible additional by-products, GC-MS studies were conducted. For that purpose, LLDPE blend films containing the latent UV absorbers were exposed to 254 nm UV light for 3 h and the reaction products were extracted with chloroform (films containing 1 and 3) or toluene (film containing 2). In all solutions containing extracts from the exposed latent UV absorber blend films, the corresponding parent UV absorber could indeed be detected by GC-MS. Moreover, in the case of 1, benzoic acid was identified as a byproduct, which is in accordance with previously published results.9 We thus conclude that the regeneration of the UV absorbers occurs by homolytic α-cleavage14 of the respective benzoyl esters, followed by hydrogen abstraction from the environment.Activation of acid-labile latent UV absorbers
The activation behavior of 3 in LLDPE blend films conatining 0.2% w/w of the latent UV absorber and 3% w/w of benzenesulfonic acid phenyl ester as the PAG was studied. While the sensitivity of this particular PAG (which is used in microlithography)21 is usually too limited for most applications,11 it allowes the acid-catalyzed conversion experiments described here to be conducted on a suitable timescale, especially in the rather low concentration15,16 employed. Conveniently, the absorption band of the PAG is located at a lower wavelength than the absorption band of 4, so that the presence of 4 does not considerably affect the photocleavage of the PAG.The acid-labile latent UV absorber 4 is converted to the UV absorber 7 in the polyethylene film via a two-step process: first, the strong sulfonic acid is released by irradiation of the blend film; second, the acid catalytically cleaves the t-Boc protective group of 4 in a post-exposure heat treatment sometimes referred to as ‘post-exposure bake’,23 quantitatively restoring the UV absorber 7. LLDPE film samples containing 0.2% w/w of 4 and 3% w/w of the PAG were irradiated at 25 °C with 254 nm UV light for a pre-determined time interval. After the irradiation step, the samples were heated to 100 °C for 1 h. UV-Vis absorption spectra were collected prior to and after this treatment. The progress of the conversion as a function of the exposure time is represented in Fig. 5 in terms of the absorbance of the developing absorption maximum at 350 nm. As can be seen from the data in this figure, upon activation of the PAG at 25 °C, some conversion of 3 to 7 can already be observed. In contrast, blend films not containing the PAG did not exhibit similar absorption characteristics (spectra not shown). The same applies to reference blend films containing 3% w/w of the PAG only, and no 3. Direct photolysis of 4 or absorption of photo-generated acid can therefore be excluded as causes of the observed appearing absorption band. Upon heat treatment, the characteristic absorption of 7 was restored. With the relatively thick samples (100 µm) and the relatively low PAG concentration (3% w/w) employed in this series, an irradiation time of 30 min was required to obtain essentially complete conversion to 7 after treatment at 100 °C for 1 h. It is expected that this processing time can dramatically be reduced by increasing the PAG content, employing a more potent or more sensitive PAG, and by using more powerful light sources.
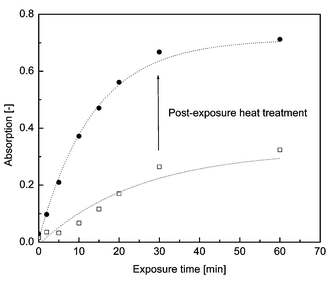 | ||
| Fig. 5 Conversion of a LLDPE film containing 0.2% w/w 4 and 3% w/w PAG upon irradiation at 254 nm and subsequent post-exposure heat treatment. The data points represent the scaled absorption at 350 nm after irradiation (□) and after subsequent post-exposure heat treatment (●). Dotted lines are first order kinetic fits calculated from the experimental data. | ||
Although the photo-labile systems presented here may at first glance seem more convenient, the acid-labile systems may have a number of advantages. As already mentioned above, it appears that with the latter approach, many UV absorbers that rely on internal conversion involving ESIPT may be derivatized to yield a latent UV absorber. Since only the photo-acid generator is UV sensitive, not the latent absorber itself, these systems can be expected to be less subject to unwanted conversion, especially in applications where long wavelength UV light is selectively blocked. Although mainly sensitive to short wavelength UV light, the photo-labile systems could suffer some unwanted conversion in these applications. In acid-labile systems, only the PAG is light sensitive and, in addition, a subsequent heat-treatment step is required to cleave the latent UV absorber. Thus, even if some PAG should involuntarily be generated during operation, no significant quantity of additional UV absorber is released. As mentioned above, the PAG may be removed after the desired patterning is obtained, thus rendering the device ultimately insensitive to further unwanted conversion.
Practical application
The latent UV absorbers presented above have the property of being able to transmit UV light with wavelengths longer than 350 nm, while the UV absorber they release very efficiently absorbs UV light in the range 275–400 nm (Fig. 2). Since UV radiation of that latter wavelength regime is commonly employed for excitation of photoluminescent (PL) materials, the latent UV absorbers can be used to selectively screen out the excitation radiation and, therefore, block photoluminescence of the underlying PL material. Accordingly, PL patterns and images can be produced. Such images are of interest in a number of practical applications, including personalized, ready-made items. Similar PL images currently are produced by lithography of a pigmented photoresist,24 deprotection of caged PL dyes through photogenerated acid catalysts,15,16 selective photoactivation,9 or selective photobleaching.25 The latent UV absorbers presented here provide an alternative to these methodologies. In order to illustrate this, a demonstration device was produced by selectively activating two pieces of LLDPE film containing 0.2% w/w of 4 and 3% w/w PAG by exposure to 254 nm UV light through different photomasks and subsequent post-exposure heat treatment. Lamination of the two films imprinted with different latent images onto each side of a PL polyethylene sheet [cf. exploded view in Fig. 6(A)] yielded an essentially colorless, transparent object that revealed either one or other of the latent patterns as a photoluminescent image under UV illumination, depending on the side of the film that was exposed. A series of photographs of this object is presented in Fig. 6.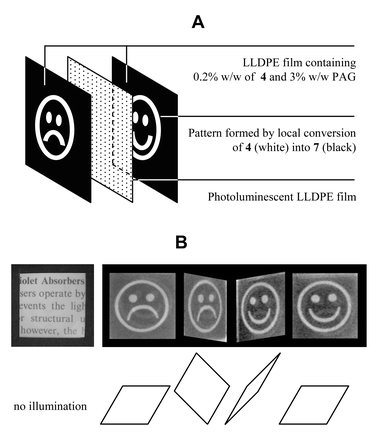 | ||
| Fig. 6 Demonstration object produced from two patterned LLDPE films containing 0.2% w/w 4 and 3% w/w PAG. The patterned films are laminated on either side of a photoluminescent LLDPE film (A). Under normal illumination, the object appears essentially colorless and transparent. Under UV irradiation, the latent image imprinted in the layer facing the UV light source appears on the photoluminescent layer (B). Photographs were taken under UV irradiation at 365 nm. | ||
Clearly, with this approach, many different PL systems can be patterned, including oriented systems displaying polarized PL, which cannot be obtained in a patterned fashion by common printing techniques.25,26 Thus, this technology offers the possibility of readily creating high information-content structures which may readily be personalized27 by very simple means.
Conclusions
We have synthesized and characterized a number of compounds capable of releasing classical UV absorbers. Two classes of such latent UV absorbers were produced, namely, photo-labile and acid-labile systems. LLDPE blend films containing photo-labile latent UV absorbers or the acid-labile variety in combination with a photoacid generator allowed for spacially resolved conversion and, thus, for the generation of absorbing–non-absorbing patterns. The present systems convert upon UV irradiaton at short wavelengths (250–350 nm), which may make these compounds particularly interesting for optical data storage systems with high storage densities. Moreover, the liberated UV absorber exhibits a dramatically shifted absorption with respect to the latent UV absorber, which allows for a highly wavelength-selective activation and easy information read-out by simple and cost-effective means. We believe that the present systems represent a potentially valuable extension to the available chromogenic dyes working in the visible range of the electromagnetic spectrum.Experimental
Materials
All UV absorbers were purchased from Aldrich and used as received. All solvents were of analytical grade. For spectroscopic experiments, chloroform (stabilized with 0.8% v/v ethanol) was used. Pyridine was dried by refluxing over KOH and subsequently distillied onto molecular sieves. For the preparation of polymer blend films, a commercial-grade LLDPE obtained from Dow Chemicals was used (Dowlex BG 2340).Synthesis
Methods
Acknowledgements
The authors are indebted to Sharon Ohba and Prof. Hatsuo Ishida (both of Case Western Reserve University) for the extensive GC-MS studies and for providing access to the GC-MS equipment, respectively.References
- Y. Atassi, J. Chauvin, J. A. Delaire, J. F. Delouis, I. Fanton-Maltey and K. Nakatani, Pure Appl. Chem., 1998, 70(11), 2157 CAS.
- S. Kuranda, G. W. Rankin and K. Sridhar, Opt. Lasers Eng., 1994, 20, 177 CrossRef.
- R. Ao, L. Kümmerl and D. Haarer, Adv. Mater., 1995, 7(5), 495 CrossRef CAS.
- N. S. Allen, Polymer Photochemistry, Applied Science Publishers, London, 1983, p. 167 Search PubMed.
- N. S. Allen, Developments in Polymer Photochemistry, Applied Science Publishers, London, 1982, p. 304 Search PubMed.
- J. F. McKellar and N. S. Allen, Photochemistry of Man-Made Polymers, Applied Science Publishers, London, 1979, p. 216 Search PubMed.
- R. M. Tarkka and S. A. Jenekhe, Mater. Res. Soc. Symp. Proc., 1996, 413, 97 Search PubMed.
- M. Wiechmann, H. Port, F. Laemer, W. Frey and T. Elsaesser, Chem. Phys. Lett., 1990, 165(1), 28 CrossRef CAS.
- C. Kocher, P. Smith and C. Weder, J. Mater. Chem., 2002, 12, 2620 RSC.
- T. W. Greene and G. M. Wuts, in Protective Groups in Organic Synthesis, John Wiley and Sons, New York, 1999 Search PubMed.
- E. Reichmanis, F. M. Houlihan, O. Nalamasu and T. X. Neenan, Chem. Mater., 1991, 3, 394 CrossRef CAS.
- J. E. Hanson, K. H. Jensen, N. Gargiulo, D. Motta, D. A. Pingor, A. E. Novembre, D. A. Mixon, J. M. Kometani and C. Knurek, ACS Symp. Ser., 1995, 614, 137 Search PubMed.
- J. Catalán, P. P. Pérez, F. Fabero, J. F. K. Wilshire, R. M. Claramunt and J. Elguero, J. Am. Chem. Soc., 1992, 114, 964 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, CA, 1978 Search PubMed.
- J. M. Kim, T. E. Chang, J. H. Kang, D. K. Han and K. D. Ahn, Adv. Mater., 1999, 11, 1449.
- J. M. Kim, J. H. Kang, D. K. Han, C. W. Lee and K. D. Ahn, Chem. Mater., 1998, 10, 2332 CrossRef CAS.
-
E. J. Stronjy and J. R. Dewald
(Dow Chemical Co.), US Pat.,3598863, 1971 Search PubMed.
-
H. Heller, E. Keller, H. Gysling and F. Mindermann
(J. R. Geigy A.G.), US Pat., 3004896, 1961 Search PubMed.
- I. N. Smolenskii, S. S. Gontar and V. I. Markov, Vopr. Khim. Khim. Tekhnol., 1985, 78, 112 Search PubMed.
- K. Iwakura (Fuji Photo Film Co.), Jpn. Pat., 06184121, 1994 Search PubMed.
-
H. Binder, R. Schwalm and D. Funhoff
(BASF Aktiengesellschaft), Eur. Pat., 0540965, 1993 Search PubMed.
- L. Xiao-Bai, M. A. Winnik and J. E. Guillet, Polym. Sci. (Polym. Chem.), 1983, 21, 1263 Search PubMed.
- A. S. Gozdz and J. A. Shelburne, Polymer, 1992, 33(21), 4653 CrossRef CAS.
- C. Zhang, A. M. Vekselman and G. D. Darling, Chem. Mater., 1995, 7, 850 CrossRef CAS.
- C. Kocher, A. Montali, P. Smith and C. Weder, Adv. Funct. Mater., 2001, 11, 31 CrossRef CAS.
- S. Holdcroft, Adv. Mater., 2001, 23, 1753 CrossRef CAS.
-
C. Kocher
(Landqart), Eur. Pat., 02007000.9, 2002 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |