Tuning the emission of cyclometalated iridium complexes by simple ligand modification
Andrew
Beeby
*a,
Sylvia
Bettington
a,
Ifor D. W.
Samuel
b and
Zhenjia
Wang
b
aDepartment of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE. E-mail: andrew.beeby@durham.ac.uk
bSchool of Physics and Astronomy, University of St. Andrews, North Haugh, St. Andrews, Fife, UK KY16 9SS. E-mail: physics@st-and.ac.uk
First published on 15th November 2002
Abstract
The photo-excitation and relaxation of a new cyclometalated complex, Ir(ppy)2fppy, is investigated through the measurement of absorption and photoluminescence spectra. Comparison of Ir(ppy)2fppy with Ir(ppy)3 reveals that the lowest energy excited state of Ir(ppy)2fppy is localised on the aldehyde-substituted ligand, thus giving the complex its characteristic orange emission (λmax ≈ 600 nm). OLEDs containing Ir(ppy)2fppy doped into poly(vinylcarbazole) (PVK) exhibit the same characteristic emission as found in solution, implying efficient energy transfer between the PVK and the iridium complex. Our results provide the potential for the synthesis of iridium complexes with different emission wavelengths through the modification of only one ligand.
Introduction
Organic light-emitting diodes (OLEDs) have attracted a great deal of interest due to their potential applications in flat panel displays.1,2 Much effort has been focussed on increasing the efficiency of these OLEDs by improving the materials and device structures. The operation of OLEDs is based on the injection of opposite charges, which combine to form either singlet or triplet excitons. Singlet excitons can decay radiatively, whilst in most materials triplet excitons decay non-radiatively. The formation of triplet excitons therefore places a serious restriction on the efficiency of OLEDs. A simple argument based on spin statistics suggests that only 25% of the excitons formed are in the singlet state, and this has been confirmed experimentally in the case of OLEDs made from molecular materials.3,4 Therefore the study of materials which are capable of generating emission from predominantly triplet excitons is potentially very useful. Such electrophosphorescence has been demonstrated by the inclusion of dyes based on an organometallic complex.5–9 One of the most promising results in terms of efficiency and brightness has been obtained in OLEDs doped with fac-tris(2-phenylpyridine)iridium(III), Ir(ppy)31. These devices exhibited a peak external quantum yield of 13.7%, which led to an estimated internal quantum yield of 40%,10 a value which is similar to the photoluminescence (PL) quantum yield observed in degassed toluene.11 Recent devices containing tertiary butyl substituted 2-phenylpyridines doped into poly[2-(6-cyano-6-methyl)heptyloxy-1,4-phenylene], CNPPP, were found to show higher quantum efficiencies than that of the Ir(ppy)3 doped devices.12 More importantly, the addition of the butyl group on the 2-phenylpyridine ligand was found to significantly suppress the decay of the device efficiency at high current density.In this paper we explore how the emission wavelength of iridium complexes can be tuned, and report the photophysics of a new cyclometalated iridium complex. Ir(ppy)3 is an efficient green emitter, but for display applications red and blue are also required. It is important to understand how the ligands affect the properties of these materials, and one approach is to replace all the phenylpyridine ligands with entirely different ligands.13,14 In order to tune the emission towards the red we studied a material which differs from Ir(ppy)3 by the addition of a formyl group on just one of the ligands. This gave the new iridium complex fac-bis(2-phenylpyridyl)-2-(4′-formylphenyl)-pyridyliridium(III), Ir(ppy)2fppy 2, illustrated in Fig. 1. Our results show that modification of just one ligand provides a powerful way of tuning the properties of these materials. We also show that the emission wavelength of this new iridium complex is sensitive to the polarity of its surroundings.
 | ||
| Fig. 1 Structures of 1, Ir(ppy)3, and 2, Ir(ppy)2fppy. | ||
Dilute solution state work
The absorption spectra of dilute solutions of Ir(ppy)3 and Ir(ppy)2fppy in dichloromethane are shown in Fig. 2. There are two main features visible in both spectra. The strong absorption bands at ca. 245 nm and 285 nm are assigned to ligand centred π*←π transitions on the 2-phenylpyridine ligands, whilst the broad absorption bands at lower energy are typical of spin-allowed metal to ligand charge-transfer (MLCT) transitions.13,14 | ||
| Fig. 2 Absorption spectra of 1 (offset for clarity) and 2 in aerated dichloromethane at 298 K. | ||
In general the absorption spectrum of 2 is similar to that of 1 except that the MLCT transition is lower in intensity, extends to longer wavelengths and exhibits less fine structure. Ir(ppy)2fppy also exhibits less fine structure in the MLCT region than Ir(ppy)3. The similarity between their absorption spectra indicates that they have similar electronic excitation configurations. Fig. 3 shows the PL excitation and emission spectra of 1 and 2 in aerated dichloromethane solution.
![(a) PL excitation [λem(1a) = 517 nm, λem(2a) = 612 nm] and (b) emission spectra of 1 and 2 in aerated dichloromethane, λex
= 350 nm. Spectrum 1a is offset for clarity.](/image/article/2003/JM/b207502d/b207502d-f3.gif) | ||
| Fig. 3 (a) PL excitation [λem(1a) = 517 nm, λem(2a) = 612 nm] and (b) emission spectra of 1 and 2 in aerated dichloromethane, λex = 350 nm. Spectrum 1a is offset for clarity. | ||
For both solutions the emission intensity was found to increase upon degassing but with unchanged spectral profile. This is indicative of oxygen quenching of the relatively long lived triplet MLCT states. The PL excitation spectra are similar to the corresponding absorption spectra.
In order to learn more about the nature of the excited state, the effect of solvent on the emission was studied and the results are shown in Table 1 and Fig. 4. The peak of the emission spectrum of 2 shifts from 580 nm in toluene to ∼615 nm in acetonitrile and other polar solvents. In contrast, the emission spectrum of 1 shows no discernible shift upon changing the solvent polarity.
 | ||
| Fig. 4 Emission spectra of Ir(ppy)3 and Ir(ppy)2fppy in aerated solvents at 298 K, λex = 350 nm. Spectra are numbered as follows: (1) Ir(ppy)3 in dichloromethane, (2) Ir(ppy)2fppy in toluene, (3) Ir(ppy)2fppy in dichloromethane, (4) Ir(ppy)2fppy in acetonitrile and (5) Ir(ppy)2fppy in ethanol. | ||
| Complex | Solvent | λ max/nm |
|---|---|---|
| Ir(ppy)3 | Dichloromethane | 517 |
| Ir(ppy)2fppy | Toluene | 580 |
| Ir(ppy)2fppy | Dichloromethane | 612 |
| Ir(ppy)2fppy | Acetonitrile | 616 |
| Ir(ppy)2fppy | Ethanol | 619 |
| Ir(fppy)3 | Toluene | 565 |
| Ir(fppy)3 | Dichloromethane | 586 |
| Ir(fppy)3 | Acetonitrile | 596 |
| Ir(fppy)3 | Ethanol | 605 |
We attribute the solvatochromism of 2 and Ir(fppy)33 to the greater delocalisation of charge in the excited 3MLCT state. The formyl group acts as an electron acceptor and hence stabilises the negative charge that can be considered to reside on the ligand. The degree of stabilisation, and hence the extent of the red shift, depends upon the polarity of the environment. This observation of solvatochromism is also important for understanding the differences observed between the solution and film spectra (see below).
Previous investigations of Ru(bpy)32+ (bpy = 2,2′-bipyridine) using various absorption,15,16 electroabsorption17 and Raman techniques18 indicated that the lowest MLCT state was localised on a single ligand. Time-resolved studies of the formation of the triplet MLCT excited state with transient absorption pump-probe spectroscopy showed the initial delocalisation of the excited state over all three ligands, followed by the charge localising onto a single ligand.19 In the cyclometalated iridium complexes the excitation processes can be both ligand-centred and MLCT in nature. Following excitation the emissive 3MLCT state is formed, and by analogy with the Ru(bpy)32+ complexes, it is proposed that this is localised on a single ligand. In the Ir(ppy)3 complex this can be any one of the three identical 2-phenylpyridine ligands. However, in the case of 2, the presence of the carbonyl moiety on the 2-(4′-formylphenyl)pyridine ligand lowers its energy in comparison with the 2-phenylpyridine ligand. Hence, it is upon this ligand that the final excitation resides. Subsequent emission from the complex is characterised by the substituted ligand. Luminescence measurements of fac-tris[2-(4′-formylphenyl)-pyridyl]iridium(III), Ir(fppy)33, show similar solvatochromism to the singly substituted complex 2, although in all solvents the emission from 3 is blue shifted by ca. 15–25 nm compared with 2, as is shown in Table 1.
Thin-film studies
The photoluminescence spectra of spin-coated poly(vinylcarbazole) (PVK) and poly(methyl methacrylate) (PMMA) films containing 0.5–5% wt./wt. of the complex 2 show similar emission spectra to those observed from toluene solutions following excitation at 350 nm, Fig. 5. | ||
| Fig. 5 (a) PL spectra of a 5% w/w Ir(ppy)2fppy doped PVK film, offset for clarity, and (b) an aerated solution of Ir(ppy)2fppy in toluene at 298 K, λex = 350 nm. | ||
The emission spectra observed from the PVK films show none of the characteristic PVK fluorescence, indicating there to be efficient energy transfer from the host to guest, and suggesting that iridium complexes could be used as phosphorescent dopants in polymer films. There is a small difference in peak position between the toluene solution and PVK and PMMA films, which we attribute to differences in the polarity of the medium surrounding the iridium complex. Following low-energy pulsed excitation (0.1 mJ cm−2) PMMA films containing low concentrations of the complex showed single exponential decays with a lifetime of τ = 1.35 ± 0.1 µs (λem = 580 nm). As the pulse energies and/or concentrations were increased the observed decays became progressively non-single exponential, showing evidence of short-lived components, possibly arising from increased excited state–excited state quenching processes. These could not be fitted adequately to a sum of two exponentials, nor to a second order decay. PVK films doped with Ir(ppy)2fppy also showed more complex behaviour, indicating an energy transfer process which occurs by several pathways.
OLED studies
Single layer OLEDs consisting of Ir(ppy)2fppy doped into PVK were made with indium tin oxide (ITO) and aluminium contacts. Light emission was observed and it can be seen in Fig. 6 that the spectra obtained by PL (in aerated toluene) and electroluminescence (EL) spectra are identical. | ||
| Fig. 6 (a) EL spectra of Ir(ppy)2fppy doped into PVK with 6 wt.%, offset for clarity, and (b) PL spectra of Ir(ppy)2fppy in aerated toluene. | ||
The current–voltage–light output characteristics are shown in Fig. 7. Luminance as high as 300 cd m−2 at 34 V was obtained and the peak quantum efficiency was found to be 0.02% using aluminium as the cathode. The low efficiency and high operating voltage are due to the very large barrier to electron injection from aluminium into PVK.
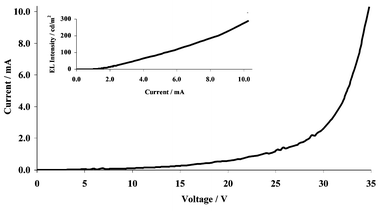 | ||
| Fig. 7 Current versus voltage for ITO/PEDOT/PVK: Ir(ppy)2fppy 6% wt/wt/Al. Inset: EL brightness versus current. | ||
The imbalance of charge injection can be seen in Fig. 7, where charge injection begins at 8 V, but 21 V is required to obtain light emission. High efficiency would not be expected for the simple device structure we have studied; the important point is that we have demonstrated tuning of the emission, and also the possibility of using a new iridium complex as a phosphorescent dopant in polymer OLEDs.
The photoluminescence quantum yield (PLQY) can be a useful guide to the maximum conceivable efficiency for materials for OLEDs. We have measured this for Ir(ppy)3 and Ir(ppy)2fppy doped into PVK (6% wt.) using an integrating sphere with CCD detection.20,21 For Ir(ppy)3 we obtain a value of 35 ± 3%, which is similar to that in toluene solution.12 For Ir(ppy)2fppy we obtain 60 ± 3% which is very encouraging and suggests that it may perform even better than Ir(ppy)3 in optimised devices. An attractive method for fabricating full colour displays would be to spin-cast a high-quality blue-emitting polymer film and then use screen printing,22 microcontact printing23 or hybrid inkjet printing24 to deposit green Ir(ppy)3 and orange Ir(ppy)2fppy. In addition, it is worth noting that we can develop different emission colours by modifying these phenylpyridine–iridium complexes.
Conclusions
We have synthesised and characterised a new phosphorescent iridium(III) complex, Ir(ppy)2fppy 2, which shows a strong orange emission in the region of 600 nm. Our results show that a small modification of one ligand is sufficient to cause a substantial change in the emission spectrum, and a large increase in the photoluminescence quantum yield. Our approach provides a powerful way of tuning the properties of iridium complexes, which are a very important class of materials for OLEDs. The lifetime, effect of oxygen and solvatochromism show that a triplet MLCT is responsible for the light emission. We have shown that this complex can be used successfully as a phosphorescent dopant in polymer OLEDs, and we expect that it would be suitable for use in OLEDs made via evaporation.Experimental
Complexes 1, 2 and 3 were synthesised using a procedure adapted from the literature.25 The synthesis of bis-(2-phenylpyridyl)-2-(4′-formylphenyl)pyridineiridium(III), Ir(ppy)2(fppy) 2 was carried out as follows: [Ir(ppy)2Cl]2 (214 mg, 0.2 mmol), 4-(2′-pyridyl)benzaldehyde (0.366 g, 2 mmol, Aldrich, 97%), AgCF3SO3 (0.109 g, 0.4 mmol, Lancaster, 98%) and diglyme (2 ml) were placed in a reaction vessel and degassed. The reaction mixture was heated at 110 °C whilst under continuous stirring for 24 h. The resulting dark orange–red solution was cooled to room temperature and water was added. The suspension was filtered and the residue dissolved in dichloromethane. This solution was then chromatographed using SiO2–dichloromethane, where the product was found in the middle fractions. The pure product was isolated as a red solid, in 27% yield (73 mg).1H-NMR (300 MHz). δH (CD2Cl2): 9.64 (1H, s), 8.01(1H, d), 7.92 (2H, d), 7.78 (1H, d), 7.67 (6H, m), 7.53 (2H, q), 7.36 (1H, dd), 7.22 (1H, d), 7.02 (1H, m), 6.84 (7H, m), 6.65 (1H, dd).
MS. (EI+) 683 (M)+, 655 (M–CO)+, 605 (M–C5H4N)+, 529 (M–C11H8N)+, 499 (M–C12H9NO)+ and (HR-EI+) 683.1538 (M)+ (C34H24N3IrO = 683.1548).
Absorption spectra were recorded on a ATI–Unicam UV-2 spectrophotometer. The photoluminescence spectra were recorded on a PerkinElmer LS-50B and were corrected for the spectral response of the machine. OLEDs were fabricated on ITO substrates, which were cleaned by sonication in acetone and isopropanol, air dried and coated with a PEDOT hole-injecting layer. Ir(ppy)2fppy doped into PVK was spin-coated on top of the ITO layer, with a typical layer thickness of 100 nm and capped with aluminium cathodes. Device testing was performed in a vacuum using a source measure unit for dc operation. The EL spectra were measured using a CCD spectrophotometer.
Acknowledgements
I. D. W. S. is a Royal Society University Research Fellow. The authors thank EPSRC and Opsys Ltd. for financial support.References
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature (London), 1999, 397, 121 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature (London), 2000, 403, 750 CrossRef CAS.
- M. A. Baldo, D. F. O’Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B, 1999, 60, 14422 CrossRef CAS.
- A. R. Brown, K. Pichler, N. C. Greenham, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Chem. Phys. Lett., 1993, 210, 61 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature (London), 1998, 395, 151 CrossRef CAS.
- V. Cleave, G. Yahioglu, P. LeBarny, R. H. Friend and N. Tessler, Adv. Mater., 1999, 11, 285 CrossRef CAS.
- Y. G. Ma, C. M. Che, H. Y. Chao, X. M. Zhao, W. H. Chan and J. C. Shen, Adv. Mater., 1999, 11, 852 CrossRef CAS.
- D. F. O'Brien, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 74, 442 CrossRef.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS.
- T. Tsutsui, M. Yang, M. Yahiro, K. Nakamura, T. Watanabe, T. Tsuji, Y. Fukuda, T. Wakimoto and S. Miyaguchi, Jpn. J. Appl. Phys., 1999, 38, L1502 CrossRef CAS.
- (a) K. A. King, P. J. Spillane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431 CrossRef CAS; (b) E. V. Donckt, B. Camerman, F. Hendrick, R. Herne and R. Vandeloise, Bull. Soc. Chim Belg., 1994, 103, 207 Search PubMed.
- W. Zhu, Y. Mo, M. Yuan, W. Yang and Y. Cao, Appl. Phys. Lett., 2002, 80(12), 2045 CrossRef CAS.
- M. Maestri, D. Sandrini, V. Blazani, U. Maeder and A. von Zelewsky, Inorg. Chem., 1987, 26, 1323 CrossRef CAS.
- M. G. Colombo, A. Hauser and H. U. Güdel, Inorg. Chem., 1993, 32, 3088 CrossRef CAS.
- H. Riesen, L. Wallace and E. Krausz, Inorg. Chem., 1996, 35, 6908 CrossRef CAS.
- J. S. Gold, S. J. Milder, J. W. Lewis and D. S. Kliger, J. Am.Chem.Soc., 1985, 107, 8285 CrossRef CAS.
- D. H. Oh, M. Sano and S. G. Boxer, J. Am. Chem. Soc., 1991, 113, 6880 CrossRef CAS.
- P. J. Carroll and L. E. Brus, J. Am. Chem. Soc., 1987, 109, 7613 CrossRef CAS.
- A. T. Yeh, C. V. Shank and J. K. McCusker, Science (Washington, D. C.), 2000, 289, 935 Search PubMed.
- N. C. Greenham, I. D. W. Samuel, G. R. Hayes, R. T. Phillips, Y. A. R. R. Kessener, S. C. Moratti, A. B. Holmes and R. H. Friend, Chem. Phys. Lett., 1995, 241, 89 CrossRef CAS.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230 CrossRef CAS.
- F. Pschenitzka and J. C. Sturm, Appl. Phys. Lett., 1999, 74, 1913 CrossRef CAS.
- J. A. Rogers, Z. Bao and V. R. Raju, Appl. Phys. Lett., 1998, 72, 2716 CrossRef CAS.
- S. Chang, J. Bharathan, Y. Yang, R. Helgeson, F. Wudl, M. B. Ramey and J. R. Reyolds, Appl. Phys. Lett., 1998, 73, 2561 CrossRef CAS.
- M. G. Colombo, T. C. Brunhold, T. Riedener and H. U. Güdel, Inorg. Chem., 1994, 33, 545 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |