Synthesis and magnetic properties of decamethylsamarocene composites of mesoporous niobium oxide
Xun
He
a,
Michel
Trudeau
b and
David
Antonelli
a
aDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Avenue, Windsor, Ontario, Canada N9B 3P4
bEmerging Technologies, Hydro-Québec Research Institute, 1800 Boulevard Lionel-Boulet, Varennes, Québec, Canada J3X 1S1
First published on 8th November 2002
Abstract
Mesoporous niobium oxide with a pore size of 22 Å was treated with excess decamethylsamarocene THF complex in THF to give a new mesoporous niobium oxide composite with a mixed oxidation state organosamarium phase in the pores. XRD and nitrogen adsorption studies confirmed that the mesostructure was retained on reduction of the framework, showing the decrease in surface area and pore volume expected as a result of occlusion of the pores by the organosamarium phase. Elemental analysis suggested that the structural integrity of the sandwich complex was largely retained upon intercalation, with a small amount of decomposition by loss of ligand. XPS studies confirmed the presence of the decamethylsamarocene THF complex and its corresponding cation. The Nb 3d region of the XPS spectrum exhibited 5/2, 3/2 emissions at a lower binding energy than the unreduced oxide, consistent with reduction to a state between Nb(V) and Nb(IV). The EPR spectrum showed a broad resonance at g = 2.00, indicative of free electrons in the walls of the mesoporous framework. Plots of magnetization versus temperature and magnetization versus field demonstrated that the material was paramagnetic, with a small contribution from a superparamagnetic phase, although no blocking temperature was observed. There was also evidence for spin glass behavior at low temperature, a phenomenon that is common in reduced samples of mesoporous transition metal oxides.
Introduction
The exploration of materials with two or more disparate phases interwoven into a periodic array on the nanometer scale is of current interest due to the potential of such materials to offer properties not observed in the corresponding pure bulk phases. Mesoporous silicas1–6 and transition metal oxides7–12 are attractive building blocks for materials with nanoscale periodicity because the regular and controllable voids can be filled with a wide variety of molecular or continuous phase substances to give composites with low dimensional nanowires encapsulated within the pore structure.13,14 Recent work in our group has demonstrated that mesoporous niobium, tantalum, and titanium oxides15–17 can act as electron acceptors because of the variable oxidation states available to the transition metal centers in the walls of the structure. While this property is of key importance to the design of Li battery materials,18 the oxidizing ability also enables host–guest intercalation reactions to occur in which quantum confinement and introduction of hole defects in the guest phase by electron transfer to the walls can be achieved in a single step, a process similar to the intercalation of molecules into layered host structures.19 This leads to materials with negatively charged walls and positively charged guest phases alternating in a periodic array. Exploitation of these properties and has already led to a new family of mesoporous composites with mixed oxidation state alkali metal fulleride,20 cobaltocene,21 nickelocene,22 vanadocene,23 chromocene,23 bis(benzene)vanadium,24 and bis(benzene)chromium25 nanowires in the pores. The Ni and Co materials are insulating superparamagnetic or spin glasses, depending on the loading level. Superparamagnetism is a property associated with fine grains of ferromagnetic materials in which the domain size is too small to overcome randomization introduced by temperature.26 Bulk samples of cobaltocene are not ferromagnetic, so the source of the superparamagnetism in these mesoporous niobium oxide–cobaltocene composites is not known. Since lanthanide elements have interesting optical and magnetic properties quite different from transition metals, due to the high density of localized f-states in the materials,27 we endeavored to synthesize reduced mesoporous composites with mixed oxidation state organolanthanide species in the pores, and to investigate what effect changing from a transition metal to a lanthanide has on the physical properties. In particular, we were interested in magnetic properties because lanthanide dopants can have a dramatic effect on the remnant magnetism of ferromagnetic materials. For example, SmCo5 is one of the best magnetic materials known. The physical properties of such mesoporous oxide–organolanthanide composites may also shed some light on the source of the superparamagnetism in the related cobaltocene composites.Experimental
Materials and equipment
All chemicals were obtained from Aldrich, unless otherwise stated. Samarium(II) iodide was obtained from Strem Chemicals. Trimethylsilyl chloride was obtained from Aldrich and distilled over calcium hydride. Mesoporous niobium oxide (Nb-TMS1) was obtained from Alfa-Aesar and used without further purification. Decamethylsamarocene, (C5Me5)2Sm(THF)2, was synthesized by the literature method28 and characterized by 1H-NMR. Anal. calcd for C28H46O2Sm: C, 59.52; H, 8.21; O, 5.66; Sm, 26.61; found: C, 59.31; H, 8.01; O, 5.76; Sm, 26.77%.Nitrogen adsorption and desorption data were collected on a Micromeritics ASAP 2010 instrument. X-Ray diffraction (XRD) patterns (Cu-Kα) were recorded from samples sealed in glass capillaries on a Siemens D-500 θ–2θ diffractometer. All X-ray photoelectron spectroscopy (XPS) peaks were referenced to the carbon C-(C, H) peak at 284.8 eV and the data were obtained using a Physical Electronics PHI-5500 instrument and charge neutralization. The DC conductivity measurements were recorded on a Jandel 4-point universal probe head combined with a Jandel resistivity unit. The equations used for calculating the resistivity were as follows. For pellets of <0.5 mm thickness: ρ = t{(π/log n2)(V/T)}, and for pellets of >0.5mm thickness: ρ = 2π(S)V/I, where ρ = resistivity, π/log n2 = sheet resistivity, V = voltage, I = current, t = thickness of the pellet, and S = the spacing of the probes (0.1 cm).
The electron paramagnetic resonance (EPR) powder samples were prepared under vacuum and the data were collected on a Bruker X-band ESP 300E EPR spectrometer. Magnetic measurements were conducted on a Quantum Design SQUID magnetometer MPMS system with a 5 T magnet. All elemental analysis data (conducted under an inert atmosphere) were obtained from Galbraith Laboratories (Knoxville, TN, USA).
Synthesis
Mesoporous niobium oxide–decamethylsamarocene composites were synthesized by addition of trimethylsilyl chloride-treated mesoporous niobium oxide (Nb-TMS1) to a THF solution of excess decamethylsamarocene, calculated on the basis of the molar ratio of Sm to Nb according to the inductively coupled plasma elemental analysis of the parent mesoporous oxide. The mesoporous solid immediately went from faun to black in color. After several days of additional stirring to ensure complete absorption of the organometallic complex, the reduced material was collected by suction filtration and washed several times with THF. The material was dried in vacuo at 10−3 Torr on a Schlenk line until all the condensed volatiles had been removed. The elemental analysis gave: Sm, 9.65; Nb, 41.52; O, 32.53; C, 11.78; H, 1.90; Si, 2.60%; I, less than 19 ppm; N, less than 0.05%. On the basis of these data, the formula of this material can be written as Nb1.0O4.5Sm0.14Si0.21C2.2H4.2.Results and discussion
The reduction of mesoporous niobium oxide by organometallic transition metal sandwich complexes has been studied extensively by our group. These topotactic host–guest inclusion reactions generally lead to materials with a reduced niobium oxide mesostructure and one or more organometallic species in the pores. Most commonly, mixtures of the neutral organometallic complex and its corresponding cation are observed, imparting a net positive charge to the encapsulated phase which balances out the negative charge of the framework. Of special interest are mesoporous niobium oxide–cobaltocene composites because these materials exhibit superparamagnetism. This is surprising given that this phenomenon is normally observed in fine grains of materials which are ferromagnetic in the bulk state, since pure cobaltocinium is paramagnetic. This suggests that different magnetic coupling processes are operative in the mesoporous composites, possibly involving interactions between the spins in the walls and those in the pores. Because lanthanides are often added to ferromagnetic metals in order to improve magnetic properties, the reduction of mesoporous niobium oxide with an organolanthanide complex could lead to mesoporous composites with novel magnetic properties and possibly help clarify the origin of superparamagnetism in the cobaltocene analogues. Decamethylsamarocene is a highly reactive, one-electron reducing reagent28 and, for this reason, is an ideal candidate for electron transfer-driven intercalation into mesoporous niobium oxide. The steric shielding afforded by the methyl groups should provide greater thermal stability with respect to decomposition of the corresponding cation in the pores. In some cases, most notably in the reaction of bis(toluene)niobium with mesoporous niobium oxide, the organometallic complex decomposes to leave a low valent metallic coat on the inner pore surface.29When a sample of trimethylsilated Nb-TMS1, giving an X-ray powder diffraction peak centered at d(100) = 40 Å, an Horwath–Kawazoe (HK) pore size of 23 Å, a Brunauer–Emmett–Teller (BET) surface area of 520 m2 g−1, and a pore volume of 0.287 cm3 g−1, was treated with two molar equivalents of decamethylsamarocene with respect to Nb in dry THF over several days under nitrogen, a new black material was formed. Fig. 1 shows the XRD spectra of the material before and after treatment with decamethylsamarocene. The broad reflection centered at d(100) = 35 Å in the product material demonstrates that this material retains its wormhole mesostructure on reaction with the organometallic complex, although the reduction in intensity with respect to the starting material suggests some loss of long range order on reduction. The nitrogen adsorption and desorption isotherms of the material before and after treatment with decamethylsamarocene are shown in Fig. 2(a). The BET surface area of the treated sample dropped to 283 m2 g−1, while the HK pore size and pore volume decreased to 20.8 Å and 0.151 cm3 g−1, respectively. Plots of incremental pore volume versus average pore diameter, as calculated from the adsorption strings of the isotherms, are shown in Fig. 2(b), demonstrating that the pore size in the decamethylsamarocene composite is smaller than in the parent material. These data are consistent with partial filling of the mesopores by the encapsulated organometallic. The elemental analysis of this material showed an increase from 5.73% C and 1.43% H in the starting material to 11.78% C and 1.90% H in the product, with 9.65% Sm, as determined by inductively coupled plasma spectrometry. The loading level of Sm in the pores (Sm∶Nb = 0.14∶1) is greater than in the analogous bis(benzene)chromium composites (0.07∶1) , but less than that in the cobaltocene composites (0.5∶1). The percentage of C is consistent with the intercalation of organometallic complex into the pores with some loss of ligand, since the expected value on the basis of the Sm content is over 20%. Loss of ligand in organolanthanide complexes is often more facile than in the analogous organotransition metal species because the larger coordination sphere around the metal center and lack of π bonding to the ligand enables low-energy dissociation processes to occur.
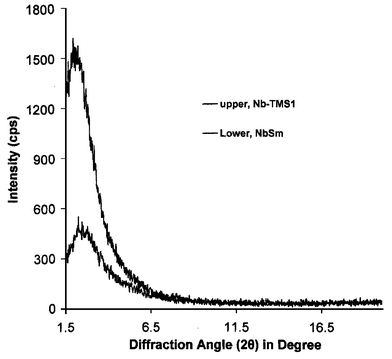 | ||
| Fig. 1 XRD patterns of trimethylsilated mesoporous niobium oxide before (upper trace) and after (lower trace) treatment with excess (C5Me5)2Sm(THF)2. | ||
 | ||
| Fig. 2 (a) Nitrogen adsorption (B, D) and desorption (A, C) isotherms of trimethylsilated mesoporous niobium oxide before (A, B) and after (C, D) treatment with excess (C5Me5)2Sm(THF)2. (b) Plots of incremental HK pore volume vs. average pore diameter before (upper trace) and after (lower trace) treatment with excess (C5Me5)2Sm(THF)2. | ||
The powder electron paramagnetic resonance spectrum of the composite material is shown in Fig. 3. This spectrum contains a single peak at 3340 G (g = 2.00) which originates from the electrons in the reduced mesoporous niobium oxide walls and is observed in many reduced mesoporous niobium oxide composites. The lack of Nb hyperfine splitting indicates that either the reduction electrons are not closely associated with the Nb centers or that the material has a broad array of surface sites because of the amorphous wall structure, however, the g value of 2.00, almost identical to that of a free electron, favors the former explanation. The unpaired 4f electrons from the Sm species were not detected from room temperature down to liquid nitrogen temperature because of the strong shielding of the samarium 5s and 5p electrons.30 The unsymmetrical nature of this resonance may suggest a contribution from a second paramagnetic species present in lower concentrations.
 | ||
| Fig. 3 Powder EPR spectrum of trimethylsilated mesoporous niobium oxide after treatment with excess (C5Me5)2Sm(THF)2. | ||
Fig. 4(a) shows the Nb 3d region for the reduced material and mesoporous niobium oxide (Nb-TMS1). The 5/2, 3/2 peaks move from 207.8 and 210.6 eV in the starting material to 207.3 and 209.8 eV in the reduced material, with small shoulder peaks appearing at 205.8 and 208.7 eV. This is consistent with reduction of the niobium oxide framework to a state between Nb(V) and Nb(IV), with a small amount of a more highly reduced phase.24Fig. 4(b) shows the Sm 3d 5/2, 3/2 region for (C5Me5)2Sm(THF)2 and its reduced mesoporous niobium oxide composite; the spectra exhibit emissions which can be assigned to divalent Sm(II) at 1080.2 and 1107.1 eV, and trivalent Sm(III) at 1083.5 and 1110.4 eV,31 respectively. A simulation revealed a 4∶1 ratio of Sm(III) to Sm(II). The emissions assigned to divalent Sm match the 3d 5/2, 3/2 emissions observed for pure decamethylsamarocene, which fall at 1080.3 and 1107.0 eV [Fig. 4(b), lower trace]. There is currently no spectroscopic or synthetic data available on the decamethylsamarocinium cation to verify the nature of the Sm(III) species, however, the low intensity of the Sm(II) emission in the composite relative to the Sm(III) emission suggests that much of the ligand was retained on oxidation, because there is not enough Sm(II) present to account for the high percentage of C in the elemental analysis. On the basis of the molecular weight of the Sm(II) complex, the Sm content in the sample, and the XPS simulation, only 3.08% of the carbon in the composite could be due to the neutral Sm(II) complex. The region near the Fermi level is shown in Fig. 4(c), revealing a distance to the Fermi level of 3.1 eV from the oxygen 2p valence emission, and small peak centered at 1 eV due to the Sm 4f emission.32
 | ||
| Fig. 4 XPS spectra of (a) the Nb 3d 3/2, 5/2 region for trimethylsilated mesoporous niobium oxide before (lower trace) and after (upper trace) treatment with (C5Me5)2Sm(THF)2, (b) the Sm 3d 3/2, 5/2 region for pure (C5Me5)2Sm(THF)2 (lower trace) and decamethylsamarocene-treated mesoporous niobium oxide (upper trace), and (c) the region near the Fermi level for mesoporous niobium oxide before (lower trace) and after (upper trace) treatment with decamethylsamarocene. | ||
Room temperature dc conductivity measurements on samples of the decamethylsamarocene-reduced mesoporous niobium composites, conducted using the four-point method under argon, showed that these materials are semiconducting with a conductivity as high as 3 × 10−6 Ω−1 cm−1. In previous work, we attributed conductivity in metallocene-doped mesoporous niobium oxide composites to an electron hopping mechanism through the mixed oxidation state dopant phase in the pores, with minimal involvement of the walls of the mesostructure.23,24 Variable temperature conductivity measurements on related bis(benzene)chromium composites show increasing resistivity with decreasing temperature. The low conductivities are expected on the basis of the values obtained for other reduced mesoporous oxides and the fact that the 4f electrons from the Sm dopant are core-like and tightly bound to the nuclei. Because of strong electron localization, few lanthanide-based materials exhibit high conductivities.
Complex magnetic behavior is expected in these materials because of the many different magnetic species present, including the reduced niobium oxide walls, Sm2+ with four unpaired electrons, and Sm3+ with five unpaired electrons. Fig. 5(a) shows the superconducting quantum interference device (SQUID) magnetometer plots of magnetic susceptibility versus temperature for the composite at 500 G. The shape of this plot suggests contributions to the magnetization from a temperature-dependent Langevin free-spin term and a temperature-independent term, most likely arising from Van Vleck paramagnetism, a phenomenon observed in many other reduced mesoporous transition metal oxides studied by our group.17 Pauli paramagnetism is ruled out because these materials are not metallic. The plots of magnetic susceptibility versus temperature recorded at 100 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 G show similar curves, ruling out any field-dependent magnetic transitions across this region. Fig. 5(b) shows the variation of the molar magnetic susceptibility (corrected by subtraction of the temperature-independent term obtained by extrapolating the Mvs.T plot to T
= 300 K, χg
= 9.15 × 10−4 emu g−1) with inverse temperature over the temperature range 6–200 K. The linearity over this region indicates that the temperature-dependent term in this material obeys Curie law (magnetic susceptibility, χm
=
C/T, where C
=
NμB2μeff2/3κ) above 20 K, with a tailing off of the magnetic susceptibility below this temperature. This is consistent with spin glass behavior, in which electron spins become frozen and begin to cancel out the contributions to the magnetization from other isolated spins. Spin glass behavior is common in amorphous alloys with a dilute magnetic component and has been observed in several other reduced mesoporous oxides, most notably in a mesoporous niobium oxide–nickelocene composite, which undergoes an Ni-dependent superparamagnet-to-spin glass transition.22 From the region between 100 and 200 K, a Curie constant (C) of 3.18 emu mol K−1 can be calculated. The effective magnetic moment (μeff ) can also be calculated as 1.59, close to the theoretical result of 1.41 based on the elemental analysis, assuming every divalent Sm(II) atom donates one electron to niobium and becomes trivalent Sm(III). Because each trivalent Sm(III) possesses five unpaired electrons, the average number of unpaired electrons per metal atom is 0.73 for a material with the formula Nb1.0O4.5Sm0.14Si0.21C2.2H4.2.
000 G show similar curves, ruling out any field-dependent magnetic transitions across this region. Fig. 5(b) shows the variation of the molar magnetic susceptibility (corrected by subtraction of the temperature-independent term obtained by extrapolating the Mvs.T plot to T
= 300 K, χg
= 9.15 × 10−4 emu g−1) with inverse temperature over the temperature range 6–200 K. The linearity over this region indicates that the temperature-dependent term in this material obeys Curie law (magnetic susceptibility, χm
=
C/T, where C
=
NμB2μeff2/3κ) above 20 K, with a tailing off of the magnetic susceptibility below this temperature. This is consistent with spin glass behavior, in which electron spins become frozen and begin to cancel out the contributions to the magnetization from other isolated spins. Spin glass behavior is common in amorphous alloys with a dilute magnetic component and has been observed in several other reduced mesoporous oxides, most notably in a mesoporous niobium oxide–nickelocene composite, which undergoes an Ni-dependent superparamagnet-to-spin glass transition.22 From the region between 100 and 200 K, a Curie constant (C) of 3.18 emu mol K−1 can be calculated. The effective magnetic moment (μeff ) can also be calculated as 1.59, close to the theoretical result of 1.41 based on the elemental analysis, assuming every divalent Sm(II) atom donates one electron to niobium and becomes trivalent Sm(III). Because each trivalent Sm(III) possesses five unpaired electrons, the average number of unpaired electrons per metal atom is 0.73 for a material with the formula Nb1.0O4.5Sm0.14Si0.21C2.2H4.2.
 | ||
| Fig. 5 SQUID magnetometer plots of (a) gram magnetic susceptibility vs. temperature (Mvs. T), (b) variation of molar magnetic susceptibility with inverse temperature, and (c) gram magnetic susceptibility vs. magnetic field (Bvs. H) for trimethylsilated mesoporous niobium oxide after treatment with excess (C5Me5)2Sm(THF)2. | ||
Fig. 5(c) shows the plots of magnetic susceptibility versus field (Bvs.H) for the samarocene-treated material. The slight S-shape in the plot provides evidence for superparamagnetism, since a straight line is expected for a classical paramagnet. There is also a small hysteresis, indicative of some degree of domain-dependant behavior in this material. The absence of a superparamagnetic blocking temperature (Tb) in the plot in Fig. 5(a) suggests that this may fall below 4 K, or that the superparamagnetic phase is not large enough to dominate over the paramagnetic domains. Analogous cobaltocene21 and nickelocene22 composites show Tb values of 8 and 22 K, respectively. The small μeff value is consistent with either very small superparamagnetic particles or superparamagnetism in only a small percentage of the material, since superparamagnetic particles have large μeff and this value represents an average across the entire sample. The observation of superparamagnetism in this system is surprising because lanthanides rarely show cooperative magnetism in the pure form, although they are often added to transition metals as a dopant to improve the remanent magnetism. Because of the broad bandwidths of early transition metal compounds, which tend to favor metallic or paramagnetic states with no domain-dependent magnetic ordering, and the lack of superparamagnetism in alkali metal reduced mesoporous niobium oxides, superparamagnetism in these samarocene composites is not expected to arise solely from the reduced niobium oxide walls. This suggests a cooperative mechanism in which the Sm phase in the walls is somehow influenced by the confinement in the reduced mesostructure to give magnetic coupling interactions leading to the spin alignment necessary in nanoscale domains for superparamagnetism to occur.
Conclusion
Mesoporous niobium oxide was reduced by decamethylsamarocene to give a new semiconducting composite with a mixture of decamethylsamarocene and a second Sm(III) species in the pores. Partial loss of the organic ligand was observed, suggesting that some of the Sm(II) complex did not survive oxidative intercalation. This is the first example of a reduced mesoporous transition metal complex with an organolanthanide in the pores. This is important because lanthanide elements possess many unusual magnetic and optical absorption properties due to the large number of unpaired electrons in the core of the f shell. The material was characterized by XPS, XRD, EPR, EA, and nitrogen adsorption. SQUID magnetometry studies revealed that this composite was largely paramagnetic, with some evidence for spin glass and superparamagnetic behavior at temperatures below 40 K. This is surprising given that very few Nb- or Sm-based materials display ferromagnetism, and superparamagnetism normally occurs in fine grains of ferromagnetic materials. Studies are ongoing to elucidate the source of cooperative magnetism in these materials.Acknowledgements
The Petroleum Research Fund administered by the American Chemical Society is thanked for funding. NSERC and the Ontario Premier's Research Excellence Program are also thanked for financial support.References
- (a) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartulli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS; (b) J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T-W. Chu, D. H. Olson, E. W. Shepard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- (a) Q. Huo, D. I. Margolese, U. Ciesla, D. G. Demuth, P. Feng, T. E. Gier, P. Sieger, A. Firouzi, B. F. Chmelka, F. Schuth and G. D. Stucky, Chem. Mater., 1994, 6, 1176 CrossRef CAS; (b) A. Firouzi, D. Kumar, L. M. Bull, T. Besier, P. Sieger, Q. Huo, S. A. Walker, J. A. Zasadzinski, C. Glinka, J. Nicol, D. Margolese, G. D. Stucky and B. F. Chmelka, Science, 1995, 267, 1138 CAS.
- C.-Y. Chen, S. L. Burkette, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 2, 27 CrossRef CAS.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321 CrossRef CAS.
- (a) D. M. Antonelli and J. Y. Ying, Curr. Opin. Colloid Interface Sci., 1996, 1, 523 CAS.
- P. Behrens, Angew. Chem., Int. Ed. Engl., 1996, 35, 515 CrossRef CAS.
- (a) D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed. Engl., 1996, 35, 426 CrossRef CAS; (b) D. M. Antonelli, A. Nakahira and J. Y. Ying, Inorg. Chem., 1996, 35, 3126 CrossRef CAS; (c) D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed. Engl., 1995, 34, 2014 CrossRef CAS; (d) D. M. Antonelli and J. Y. Ying, Chem. Mater., 1996, 8, 874 CrossRef CAS.
- Z. R. Tian, J. Y. Wang, N. G. Duan, V. V. Krishnan and S. L. Suib, Science, 1997, 276, 926 CrossRef CAS.
- M. Mamak, N. Coombs and G. Ozin, Adv. Mater., 2000, 12, 198 CrossRef CAS.
- P. Liu, J. Liu and A. Sayari, Chem. Commun., 1997, 577 RSC.
- U. Ciesla, D. Demuth, R. Leon, P. Petroff, G. Stucky, K. Unger and F. J. Schuth, J. Chem. Soc., Chem. Commun., 1994, 1387 RSC.
- D. M. Antonelli and M. Trudeau, Angew. Chem., Int. Ed., 1999, 38, 1471 CrossRef CAS.
- (a) C. G. Wu and T. Bein, Science, 1994, 264, 1757 CAS; (b) C.-G. Wu and T. Bein, Chem. Mater., 1994, 6, 1109 CrossRef CAS.
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950 CrossRef CAS.
- X. He and D. M. Antonelli, Angew. Chem., Int. Ed., 2002, 41, 214 CrossRef CAS.
- M. Vettraino, M. Trudeau and D. M. Antonelli, Adv. Mater., 2000, 12, 337 CrossRef CAS.
- M. Vettraino, M. Trudeau and D. M. Antonelli, Inorg. Chem., 2001, 40, 2088 CrossRef CAS.
- S. L. Brock, N. Duan, Z. R. Tian, O. Giraldo, H. Zhou and S. L. Suib, Chem. Mater., 1998, 10, 2619 CrossRef CAS.
- D. O'Hare, Chem. Soc. Rev., 1992, 121 RSC.
- (a) B. Ye, M. Trudeau and D. M. Antonelli, Adv. Mater., 2001, 13, 29 CrossRef CAS; (b) B. Ye, M. Trudeau and D. M. Antonelli, Adv. Mater., 2001, 13, 561 CrossRef CAS; (c) B. Ye, M. Trudeau and D. M. Antonelli, Chem. Mater., 2002, 14, 2774 CrossRef CAS.
- (a) S. Murray, M. Trudeau and D. M. Antonelli, Adv. Mater., 2000, 12, 1339 CrossRef CAS; (b) S. Murray, M. Trudeau and D. M. Antonelli, Inorg. Chem., 2000, 39, 5901 CrossRef CAS.
- M. Vettraino, X. He, M. Trudeau and D. M. Antonelli, J. Mater. Chem., 2001, 11, 1755 RSC.
- X. He, M. Trudeau and D. M. Antonelli, Chem. Mater., 2001, 13, 4808 CrossRef CAS.
- X. He, M. Trudeau and D. M. Antonelli, Inorg. Chem., 2001, 40, 6463 CrossRef CAS.
- X. He, M. Trudeau and D. M. Antonelli, Adv. Mater., 2000, 12, 1036 CrossRef CAS.
- C.-W. Chen, Magnetism and Metallurgy of Soft Magnetic Materials, Dover Publications, New York, 1986 Search PubMed.
- P. A. Cox, The Electronic Structure and Chemistry of Solids, Oxford Science Publications, London, 1987 Search PubMed.
- W. J. Evans, J. W. Grate, H. W. Choi, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 941 CrossRef CAS.
- M. Vettraino, X. He, J. E. Drake, M. Trudeau and D. M. Antonelli, Adv. Funct. Mater., 2002, 12, 174 CrossRef CAS.
- J. E. Wertz and J. R. Bolton, Electron Spin Resonance: Elementary Theory and Practical Application, McGraw-Hill Inc., New York, 1972 Search PubMed.
- K. Suzuki, T. Enoki and S. Bandow, Phys. Rev. B, 1993, 48, 11077 CrossRef CAS.
- A. Franciosi, P. Perfetti, A. D. Katnani, J. H. Weaver and G. Margaritonda, Phys. Rev. B, 1984, 29, 5611 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |