Separation, purification and identification of the major selenium metabolite from human urine by multi-dimensional HPLC-ICP-MS and APCI-MS
Bente Gammelgaard, Kim Grimstrup Madsen, Jesper Bjerrum, Lars Bendahl, Ole Jøns, Jørgen Olsen and Ulrik Sidenius
Department of Analytical Chemistry, The Royal Danish School of Pharmacy, Universitetsparken 2, DK-2100 Copenhagen, Denmark. E-mail: bg@dfh.dk
First published on 27th November 2002
Abstract
When humans are supplied with selenium-containing nutritional preparations, one of the selenium-containing metabolites in urine increases relatively more than the other selenium metabolites. The purpose of this study was to identify this major selenium metabolite. Urine samples from six male volunteers were collected and analysed by ion-pair chromatography with ICP-MS detection for this major selenium metabolite. Samples containing the metabolite were pooled and solid phase extracted to remove ionic substances. The extracted pool was purified and preconcentrated twice by preparative reversed-phase chromatography. The fractions containing the selenium metabolite were collected and further purified by size exclusion chromatography. It was not possible to ionize the selenium metabolite by electrospray ionization mass spectrometry, ESI-MS. Instead, atmospheric pressure chemical ionization, APCI, was applied. The m/z of the selenium metabolite was 300 for the 80Se isotope. MS/MS experiments indicated that the metabolite was a selenosugar, and it is proposed that the selenium metabolite is a Se-methyl-N-acetylselenohexosamine.
Introduction
Selenium is an essential element that plays an important role in the antioxidative processes in the human body. Selenium functions in the selenoproteins where it is incorporated as the amino acid selenocysteine.1 However, selenium has additional health effects that are probably not linked to the function of the selenoproteins, among which is a possible cancer protective effect.2 It has been suggested that the cancer protective effect is due to methylated selenium compounds3 as studies have shown that some methylated selenium compounds exert an anticarcinogenic effect.4The metabolism of selenium from the administration of a selenium compound until the incorporation of selenium in the essential selenoproteins is fairly well elucidated and understood. The metabolism is often described by a model developed by Ganther.5,6 According to this model, the ingested selenium is converted to the hydrogen selenide ion (HSe−) prior to incorporation as selenocysteine into the essential selenoproteins. Excess selenium is then excreted after consecutive methylation processes to monomethylselenol (CH3SeH), dimethyl selenide ((CH3)2Se) and dimethyl diselenide ((CH3)2Se2), which are exhaled during respiration. The only suggested urinary excretion product in this model is the further methylated trimethylselenonium ion, TMSe ((CH3)3Se+). This model, however, is primarily based on experiments with rats after supplementation of their diet with large doses of selenium.
After the introduction of hyphenated techniques with element selective detection, especially HPLC-ICP-MS, several reports on selenium speciation in biological material using different chromatographic systems appeared. Extensive reviews on selenium speciation in biological material have been given by Lobinski et al.7and Uden.8 From these studies it appears that TMSe is not the only selenium excretion product in human urine. Several selenium metabolites have been separated in basal human urine9–12 as well as human urine from individuals supplemented with different selenium preparations.13–16
The identities of some of these metabolites were suggested on the basis of co-elution with available standards in various chromatographic systems. Thus, the selenium species suggested to be present in human urine are TMSe11–13,14,17 in small amounts and not in all samples, SeMet14,15,16 after supplementation with SeMet, and selenite in low concentrations in some samples of basal human urine.18
The complex urine matrix and the low concentrations of selenium in basal human urine makes purification and preconcentration of the urine samples mandatory prior to identification by mass spectrometry. However, Cao et al. identified selenomethionine and selenocystamine in a human urine sample from an individual who was supplemented with 400 µg of SeMet. They used tandem MS in the multi-reaction monitoring mode, in which one or more transitions of precursor to product ions were monitored in tandem.16 In a recent study, Ogra et al.19 identified a novel metabolite in rat urine by ESI tandem MS. The authors suggested that the metabolite was a Se-methyl-N-acetylselenohexosamine. They also synthesized 2-acetamide-1,2-dideoxy-β-D-glucopyranosyl methylselenide and observed that the mass spectrum of this compound was identical to the spectrum of the metabolite.
In previous studies we observed that although several selenium metabolites are present in human urine, the amount of one of these metabolites increased distinctively more than the rest after supplementation with selenomethionine.15 This unidentified selenium metabolite did not co-elute with any available standards in chromatographic systems. Thus, the purpose of this work was to separate, purify and preconcentrate urine in order to identify this metabolite by mass spectrometry.
Experimental
Apparatus
The ICP-MS instrument was a PE SCIEX Elan 6000 (PerkinElmer, Norwalk, CT, USA).Sampler and skimmer cones were made of platinum. The plasma and auxilliary argon flow rates were 14 and 1.2 l min−1, respectively. The nebulization argon flow was optimized in each eluent. The RF power was 1400 W.
The data acquisition parameters were: dwell time, 500 ms; sweeps/reading, 1; readings/replicate, between 400 and 700. The 78Se and 82Se isotopes were monitored.
The Agilent 1100 series HPLC system consisted of a G1376A capillary pump, a G1313A autosampler, G1314A wavelength detector, G1316A column compartment and a G1379A de-gasser controlled by the ChemStation software, all from the Agilent 1100 series (Agilent Technologies, Waldbron, Germany). The UV detector was operated at 254 nm. The outlet of the analytical columns was coupled to the ICP-MS system by a laboratory made direct injection nebulizer, which has previously been described,20via a 65 µm id, 508 µm od length of capillary PEEK tubing (Upchurch Scientific, Oak Harbor, WA, USA).
The pump for preparative chromatography was a Jasco 880-PU (Jasco, Gross-Umstadt, Germany) equipped with a Rheodyne 7125 injection valve (Rheodyne, Cotati, CA, USA).
Electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) mass spectrometry was performed by direct infusion into a Finnigan MAT LCQ ion-trap mass spectrometer (Finnigan, San Jose, CA, USA) using the following settings.
ESI: sheath/auxiliary gas flow 60/0, spray voltage: +4.5 kV, capillary temperature: 200 °C, flow rate: 5 µl min−1.
APCI: sheath/auxiliary gas flow 60/0, capillary temperature: 150 °C, octapole RF amplitude: 400, flow rate: 50 µl min−1, discharge current: 5 µA, vaporizer temperature: 490 °C.
Evaporations were performed on a rotary evaporator (Büchi Rotavapor, Büchi Laboratoriums Technik AG, Flawil, Schwitzerland) by heating to 45 °C.
Reagents
All reagents were of analytical grade. Purified water obtained from a Milli-Q de-ionization unit (Millipore, Bedford, MA, USA) was used throughout.Stock standard solutions of 10 mg l−1 Se were prepared in water from selenomethionine, SeMet, selenocystine, SeCys, Se-(methyl)selenocysteine hydrochloride, MeSeCys, selenourea, SeU, selenocystamine, SeCA, selenoethionine, SeEt (all Sigma, St. Louis, MO, USA), Se-(methyl)selenomethionine iodide, MeSeMet, synthesized according to ref. 21 and trimethylselenonium iodide, TMSe, synthesized according to ref. 22. The stock standard solutions were standardized against a 1.001 g l−1 PE pure atomic spectroscopy standard (PerkinElmer). Working standard solutions were prepared daily in water.
Chromatographic systems
Ion-pair chromatography. The chromatographic column was a Luna C8(2), 100 mm × 1.0 mm id, 3 µm particle size (Phenomenex, Aschaltenburg, Germany). The eluent consisted of 10 mM heptafluorobutanoic acid (HFBA) (Sigma) and 20% methanol (LabScan Analytical Sciences, Dublin, Ireland). The flow rate was 50 µl min−1 and the injection volume was 3 µl.Preparative reversed-phase chromatography. The column was a Luna C18(2) pore size 100Å, 250 mm × 15 mm id (Phenomenex) protected by a SecurityGuard Cartridge C18, 10 mm × 10 mm, in a SecurityGuard Cartridge Holder (Phenomenex). The eluent consisted of 10 mM ammonium formate (ammonium hydroxide 25% (Merck), formic acid (Ferak, Berlin, Germany)), pH 7, and 5% methanol. The flow rate was 10 ml min−1 and the injection volume was 1000 µl.
Size exclusion chromatography. The column was a Shodex Asahipak GS-320 HQ, 300 mm × 7.6 mm id. The eluent was 0.1% (v/v) formic acid in 3% methanol. The column temperature was kept constant at 40 °C. The flow rate was 600 µl min−1 and the injection volume was 100 µl.
Urine samples
Urine samples were collected from six healthy, male volunteers, whose diets had been supplemented with 200 µg of selenium as Selenoprecice (PharmaNord, Vejle, Denmark) for 7 d and with 500 µg on the day of urine collection. Samples were collected within 24 h from supplementation. Urine was stored at −18 °C until analysis. The urine samples were analysed by ion-pair chromatography with ICP-MS detection to identify samples with high contents of the metabolite. Prior to analysis, a part of the urine samples was filtered through a 0.45 µm cellulose acetate membrane (Cromacol Ltd., Trumbull, CT, USA), adjusted to pH 2 with HFBA and diluted 1 + 1 with eluent. Samples with high contents of the selenium metabolite were pooled. The total selenium concentrations in urine samples were determined according to a previously described procedure.23Procedure
Solid phase extraction
The extractions were performed using Oasis HLB cartridges 6CC/500 mg (Waters, Milford, MA, USA). Flow rates were below 4 ml min−1, controlled by a VacElut vacuum manifold (Varian, Harbor City, CA, USA)The cartridges were conditioned with 3 ml of methanol, followed by 3 ml of 10 mM ammonium formate, pH 7. 10 ml of urine pool diluted 1 + 1 with 10 mM ammonium formate, pH 7, was applied stepwise followed by elution with 6 ml of 25% methanol. The combined methanol eluates were evaporated to dryness and the residue was reconstituted in 20 ml of 10 mM ammonium formate in 5% methanol.
Preparative chromatography
The reconstituted residue was applied to the preparative column and the elution of the purified metabolite was monitored by ICP-MS detection after flow splitting of the eluate. Fractions of 15 s intervals around the the expected retention time were collected (corresponding to 2.5 ml fractions) and analyzed for selenium with graphite furnace atomic absorption spectrometry. The eluates were combined and evaporated to dryness. The residue was reconstituted in 10 ml of eluent, the preparative chromatography repeated once and the eluate evaporated to dryness.Size exclusion chromatography
The residue from the preparative chromatography was dissolved in 2 ml of the formic acid–methanol eluent and 100 µl samples were applied to the size exclusion column. The fractions containing the selenium metabolite were identified by ICP-MS and collected. The fractions were combined, the solvent evaporated and the residue dissolved in 1 ml of 50% methanol (HPLC grade, Rathburn Chemicals Ltd, Walkerburn, Scotland) for identification by mass spectrometry.Results and discussion
Following the supplementation with the selenium enriched yeast tablets to the healthy volunteers, urine samples were collected within 24 h. All samples were analysed for total selenium concentration and by ion-pairing chromatography to identify samples with high concentrations of the main metabolite. Previous studies had shown that the concentration of this compound increased distinctively compared with other selenium metabolites after supplementation with selenomethionine.15The total selenium concentrations in the urine samples varied between <2.2 and 144 µg l−1, while the concentrations of the metabolite estimated on the basis of the peak height varied between 0 and 100 µg l−1. The time for maximum excretion varied between 4 and 10 h and after 15 h the selenium concentration had reached the basal level for most individuals. There was no difference in the profiles of the excretion curves of total selenium and the selenium metabolite, i.e. high total selenium concentrations resulted in high concentrations of the metabolite.
An example of analysis of one of the urine samples by ion-pair chromatography with UV and ICP-MS detection is shown in Fig. 1. The selenium compound eluting at 2 min is the metabolite to be identified. It appears from the UV-absorbance that the selenium compound in this system eluted together with several other compounds in large concentrations as the UV absorbance exceeded 2 absorbance units.
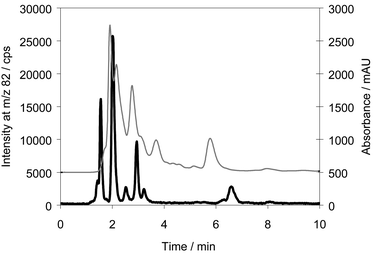 | ||
| Fig. 1 Chromatogram of a urine sample analysed by ion-pairing chromatography. Column: Luna C8, eluent: 0.1% HFBA in 20% methanol. Thin line: UV absorbance, bold line: 82Se. | ||
Solid phase extraction, SPE
The first step in the cleaning and preconcentration procedure was a solid phase extraction step as previous studies had shown that the metabolite was retained on C18 cartridges. The SPE cartridges used were chosen on basis of comparison of a C8, a C18, a copolymer based hydrophilic–lipophilic sorbent and a mixed mode cation-exchange/hydrophilic–lipophilic copolymer cartridge. The cartridges were compared for their ability to retain the unknown selenium metabolite. The retaining efficiency of the cartridges was controlled by analysing the effluent from the cartridge for the selenium metabolite by ion-pairing chromatography and ICP-MS detection. Furthermore, the pH of the samples and the conditioning solutions were adjusted to 3, 7 and 10, respectively. Neither the pH of the urine sample, nor the addition of the ion-pairing agent, HFBA, to the urine sample, influenced the retainment of the selenium metabolite. Hence, the metabolite apparently had no acidic or alkaline functional groups.Elution with 100% methanol resulted in very coloured effluents. Different concentrations of methanol were examined in the elution step in order to improve the selectivity of the purification step and thus reduce the elution of the coloured compounds. 25% methanol was chosen as the eluent as a change in the eluent from 100% methanol to 25% methanol did not decrease the yield of the SPE step, while the cartridge still retained a large part of the coloured compounds. Hence, the cartridges were eluted with 6 ml of 25% methanol in the following procedures. However, the total yield of the SPE step is only about 30% as part of the metabolite runs through the cartridge during application of the urine sample. A washing step between sample application and elution was not included in the procedure as this resulted in substantial loss of the selenium metabolite owing to its hydrophilic properties. The sodium and potassium concentrations of the urine pool were compared to the concentrations of the SPE eluate by flame emission spectrometry analysis. The concentrations of sodium and potassium in the eluate were 41 mM and 28 mM, respectively, which corresponded to 50% of the salt concentrations in the urine pool.
The eluates were combined and evaporated to dryness on a rotary evaporator. The evaporation process did not decompose the selenium metabolite as chromatography of the sample before and after evaporation did not change the retention time of the compound.
Preparative chromatography
A reversed-phase chromatographic system was developed to improve the separation of the selenium compound from potential interfering compounds. A C18 column showed better retention of the selenium species than various C8 columns. A neutral pH of the eluent was chosen as stability experiments had shown that the selenium metabolite disappeared within 24 h in the acidic environment of the HFBA eluent: instead two other unknown selenium containing compounds appeared in the chromatogram of the sample. The retention times of these compounds were increased as compared with the selenium metabolite in the ion-pairing chromatographic system, indicating that the compounds products were either more cationic and thereby forming a more retainable ion-pair or were more lipophilic than the original metabolite.A chromatogram of the urine pool of the collected samples analysed in the reversed phase system is shown in Fig. 2. It appears that separation of the main selenium metabolite from several other selenium species could be achieved. Thus, urine samples contain several selenium species which is in accordance with our earlier findings and results by others.9,11,12,13,16 The species that have previously been suggested to be present in human urine-selenite, TMSe and SeMet, are charged compounds which would elute before this unknown, probably neutral, compound. This was confirmed by analysing standards. In this system, selenite, TMSe and SeCys eluted in the front just after 1.7 min, selenourea eluted after 2.5 min and SeMet after 3.7 min (results not shown). Selenocystamine, which was identified by Cao et al. in a human urine sample by electrospray ionization mass spectrometry,16 eluted at 9.3 min. However, in the ion-pair chromatographic system, selenocystamine eluted after 16 min. We have never detected selenium species in urine with that long retention time in this chromatographic system.
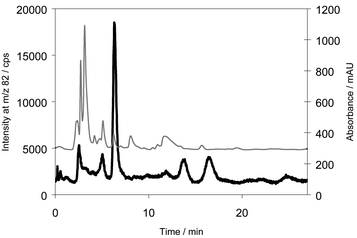 | ||
| Fig. 2 Chromatogram of the urine pool analysed by reversed phase chromatography. Column: Luna C18, Eluent: 10 mM ammonium formate, pH 7 in 5% methanol. Thin line: UV-absorbance, bold line: 82Se. | ||
The retention time of the main selenium metabolite was 6 min. This was confirmed by analysing the urine pool after solid-phase extraction in the HFBA system. Only the peak eluting after 2 min was now present. Analysing the same extract in the reversed phase system resulted in a peak eluting after 6 min. A chromatogram of the eluate from the solid phase extraction together with the trace from the UV absorbance is shown in Fig. 3. When comparing Figs. 2 and 3, it appears that the selenium concentration had increased about 20 fold while the UV absorbance only increased by 50%. However, the selenium metabolite eluted together with some UV-absorbing species. Hence, attempts to identify the selenium metabolite by ESI-MS at this stage were not successful because of extensive background.
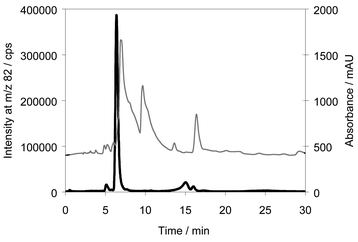 | ||
| Fig. 3 Chromatogram of eluate from the solid phase extraction analysed by reversed phase chromatography. Column: Luna C18, Eluent: 10 mM ammonium formate, pH 7 in 5% methanol. Thin line: UV absorbance, bold line: 82Se. | ||
When the eluate from the solid phase extraction was applied to the preparative system, the collected fractions were pale yellow, but after repeating this preparative chromatographic step once the collected fractions were colourless, indicating a further purification of the sample.
In preparative chromatography, long retention times are preferable as this often improves the probability of obtaining a selective separation. When analysing simultaneously for sodium during the preparative chromatography, it appeared that the sodium concentration was efficiently reduced during the first 6 min of elution, resulting in a concentration less than 0.3 µM of Na in the eluate when fractions were collected.
Size exclusion chromatography
To further improve the purity of the sample, size exclusion chromatography was applied to the reconstituted evaporation residue of the selenium-containing fraction from the preparative reversed phase chromatography. Chromatograms of the purified sample before and after the size exclusion chromatography step are shown in Fig. 4, together with the corresponding UV-absorbance traces. The retention time of the purified metabolite was 18 min and it appeared that several UV-absorbing compounds were still present in the sample after preparative chromatography; however, the majority of these compounds were separated from the selenium metabolite in the size exclusion step and the absorbance scale was diminished from 3.0 before purification (Fig. 1) to 60 mAU The elution order of standards in the size exclusion system was SeCys, MeSeCys, SeCA, SeMet, SeEt, unknown selenium metabolite. Hence, the separation was not entirely based on molecular size.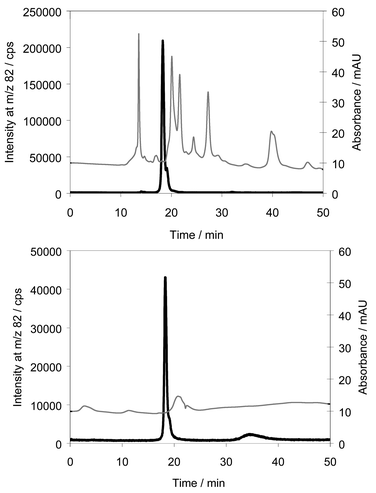 | ||
| Fig. 4 Chromatograms of selenium fraction collected from reversed phase chromatography before and after size exclusion chromatography. Column: Asahipak GS-320 HQ, Eluent: 0.1% formic acid in 3% methanol. Thin line: UV-absorbance, bold line: 82Se. | ||
Mass spectrometry
The instrumental settings of the mass spectrometer were optimized by direct infusion of the selenium standards MeSeCys, SeCys, SeMet and TMSe in increasing amounts of methanol.However, any attempt to detect the unidentified selenium metabolite by ESI-MS was in vain, probably because the metabolite is a neutral compound and difficult to ionize.
An attempt to identify the metabolite with electron impact MS was not succesful as no selenium isotope pattern was detectable. This fierce ionization mode probably totally decomposed the molecule. However, the compounds suggested by the instruments database showed ring structures containing oxygen indicating the compound could contain a sugar moiety.
The mass spectrum obtained with APCI ionization is shown in Fig. 5. The m/z of the 80Se metabolite was 300 and the intensities of the m/z 298, 297, 296 and 302 peaks corresponds to natural abundances of the selenium isotopes (76Se: 9%, 77Se: 8%, 78Se: 24%, 80Se: 50% and 82Se: 9%). MS-MS fragmentation on m/z 298, 300 and 302 all resulted in the same product ion, m/z 204, indicating that the MS-MS fragment did not contain selenium. This means that the selenium containing moiety was lost by the first fragmentation, probably as CH3SeH. Further fragmentation on m/z 204 resulted in the fragment ions m/z 186, 168, 144 and 126. This is shown in Fig. 6. The m/z of the parent ions together with the product ions and the possible leaving groups are summarized in Table 1.
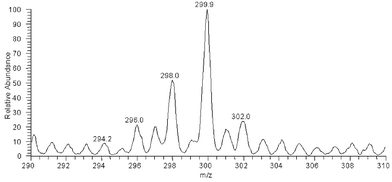 | ||
| Fig. 5 APCI mass spectrum of selenium containing fraction from size exclusion chromatography. | ||
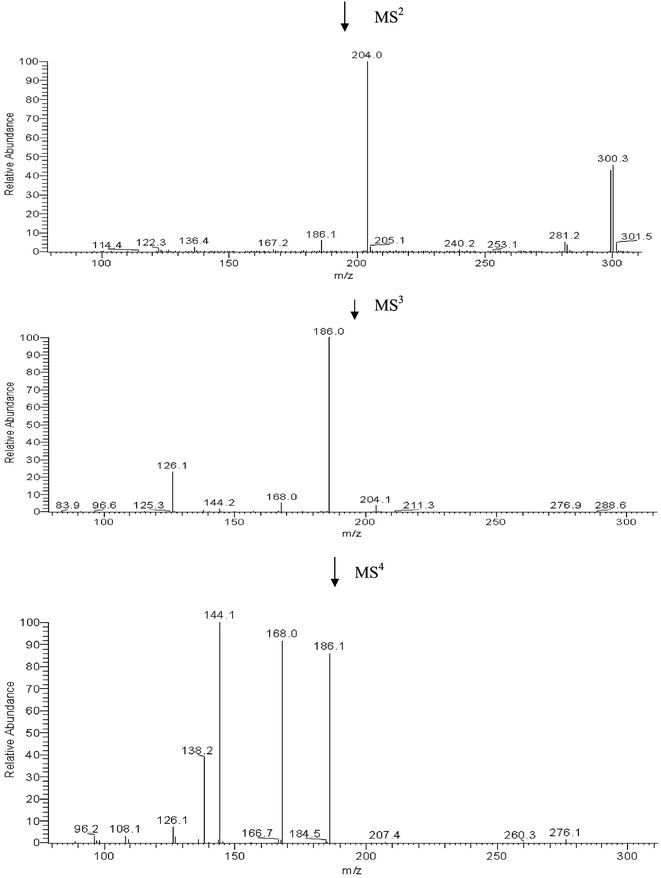 | ||
| Fig. 6 Fragmentation of m/z 298, 300 and 302 by (MS)2, (MS)3 and (MS)4. | ||
| Parent ion m/z | Product ion m/z | Product |
|---|---|---|
| M | 298, 300, 302 | [M + H]+ |
| 298, 300, 302 | 204 | [M − CH3Se]+ |
| 204 | 186 | [204 − H2O]+ |
| 186 | 168 | [186 − H2O]+ |
| 186 | 144 | [186 − CH3CO + H]+ |
| 186 | 126 | [144 − H2O]+ |
| 186 | 138 | ? |
A selenium metabolite with the same mass has very recently been identified in rat urine by Ogra et al.19 by ESI tandem MS. The authors suggested that the rat metabolite was a Se-methyl-N-acetyl-selenohexosamine. They also synthesized 2-acetamide-1,2-dideoxy-β-D-glucopyranosyl methylselenide and observed that the mass spectrum of this compound was identical to the spectrum of the metabolite. However, there was a slight difference in the chromatographic retention times of the metabolite and the synthetic product.19
The fragmentation of the compound identified by Ogra et al.19 is in accordance with our results as we also observed m/z 204 as the first fragmentation product, which is known as the marker ion of hexose-N-acetyl+ in the ESI-MS detection of glycopeptides.24 Hence, the metabolite in human urine is probably the same as the metabolite from rat urine.
The (MS)2 and (MS)3 of m/z 204 to 186 and 168, respectively, corresponding to loss of water from hydroxy groups, and the (MS)3 fragmentation of m/z 186 to 144, indicating the presence of an acetyl group, are in accordance with the results obtained by Ogra et al.,19 although their results were obtained by a general increase in collision energy. The m/z of the parent ion and all product ions are odd numbers, indicating an uneven number of nitrogen atoms in the parent ion as well as in the product ions. The presence of an amide group is supported by the experiments with retainment on solid phase extraction cartridges, which showed that the molecule had no acidic or alkaline functional groups.
The metabolite is not a conjugation product of glucuronic acid, which is a common metabolic product, as incubation of urine samples with glucuronidases did not change the chromatographic behaviour of the selenium metabolite. Hence, our experiments support that the structure of the human selenium metabolite is a Se-methyl-N-acetylselenohexosamine.
The pathways for formation of aminosugars and subsequent acetylation of the amino group via the UDP-N-acetylhexosamines is well known as being a part of the biosynthesis of glycoproteins and glycolipids. Also, the conversion of galactose to glucose via the activated UDP-glucose is described.25 Hence, a possible way of formation of the selenosugar is addition of selenide to the UDP-carbon resulting in the leaving of the UDP-group followed by a methylation of the selenium or direct addition of methylselenide to the UDP-carbon. Identification of the sugar, however, is not possible by MS techniques as the N-acetylglucosamine as well as the N-acetylgalactosamine would generate the m/z 204.
Hence, the metabolite now found in human urine is probably the same as the metabolite from rat urine. This suggests that rat selenium metabolism is an appropriate model for human selenium metabolism. However, it seems as if the rat metabolism is more simple as it has been reported that in same chromatographic system rat urine showed only one peak, while human urine showed three peaks.10
The rat in the work of Ogra et al.19 was supplied with selenium as 2 mg l−1 sodium selenite in the drinking water for 5 weeks. The volunteers in this work were supplied with 200 µg of selenium daily for a week and 500 µg on the day of delivery as Selenoprecise, which are selenium enriched yeast tablets. In spite of this, the rats and the humans produced the same metabolite. This supports the view that selenium metabolism is independent of the ingested chemical form of selenium as ingested selenium is reduced to selenide and traverses this selenide pool before incorporation in selenoproteins or excretion.5
In conclusion, a selenosugar, probably a Se-methyl-N-acetylselenohexosamine has, for the first time, been isolated and identified in human urine.
References
- M. P. Rayman, Lancet, 2000, 356, 233 CrossRef CAS.
- C. Clark, G. F. Combs, B. W. Turnbull, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glower, G. F. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, H. K. Park, B. B. Sanders, C. L. Smith and J. R. Taylor, JAMA, 1996, 276, 1957 Search PubMed.
- H. E. Ganther and J. R. Lawrence, Tetrahedron, 1997, 53, 12299 CrossRef CAS.
- C. Ip, J. Nutr., 1998, 128, 1845 Search PubMed.
- H. E. Ganther, Trace Elem. Anal. Chem. Med. Biol., 1984, 3, 3 Search PubMed.
- H. E. Ganther, J. Am. Coll. Toxicol., 1986, 5, 1 Search PubMed.
- R. Lobinski, J. S. Edmonds, K. T. Suzuki and P. C. Uden, Pure Appl. Chem., 2000, 72, 447 Search PubMed.
- P. C. Uden, Anal. Bioanal. Chem., 2002, 373, 422 Search PubMed.
- J. M. G. Lafuente, M. Dlaska, M. L. F. Sanchez and A. Sanz-Medel, J. Anal. At. Spectrom., 1998, 13, 423 RSC.
- J. M. G. Lafuente, J. M. Marchante-Gayon, M. L. F. Sanchez and A. Sanz-Medel, Talanta, 1999, 50, 207 CrossRef CAS.
- M. A. Quijano, A. M. Gutierrez, M. C. Pérez-Conde and C. Camara, Talanta, 1999, 50, 165 CrossRef CAS.
- J. Zheng, M. Ohata and N. Furuta, J. Anal. At. Spectrom., 2002, 17, 730 RSC.
- J. M. Marchante-Gayon, I. Feldmann, C. Thomas and N. Jakubowski, J. Anal. At. Spectrom., 2001, 16, 457 RSC.
- B. Gammelgaard, O. Jøns and L. Bendahl, J. Anal. At. Spectrom., 2000, 16, 339 Search PubMed.
- B. Gammelgaard, L. Bendahl, U. Sidenius and O. Jøns, J. Anal. At. Spectrom., 2002, 17, 570 RSC.
- T. H. Cao, R. A. Cooney, M. M. Woznichak, S. W. May and R. F. Browner, Anal. Chem., 2001, 73, 2898 CrossRef CAS.
- B. Gammelgaard, K. Jessen, F. Kristensen and O. Jøns, Anal. Chim. Acta, 2000, 404, 47 CrossRef CAS.
- B. Gammelgaard and O. Jøns, J. Anal. At. Spectrom., 2000, 15, 945 RSC.
- Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, J. Chromatogr. B, 2002, 767, 301 CrossRef CAS.
- L. Bendahl, B. Gammelgaard, O. Jøns, O. Farver and S. H. Hansen, J. Anal. At. Spectrom., 2001, 16, 38 RSC.
- T. W. M. Fan, A. N. Lane, D. Martens and R. M. Higashi, Analyst, 1998, 123, 875 RSC.
- S. J. Foster and H. E. Ganther, Anal. Biochem., 1984, 137, 205 CrossRef CAS.
- B. Gammelgaard and O. Jøns, J. Anal. At. Spectrom., 1999, 14, 867 RSC.
- C. Dass, Principles and Practice of Biological Mass Spectrometry, Wiley Interscience, John Wiley, New York, 2001 Search PubMed.
- C. K. Mathews and K. E. Van Holde, Biochemistry, The Benjamin/Cummings Publishing Company, Inc., Menlo Park, California, 1996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |