Determination of selenium in biological materials by isotope dilution analysis with an octapole reaction system ICP-MS
L.
Hinojosa Reyes
,
J. M.
Marchante Gayón
,
J. I.
García Alonso
* and
A.
Sanz-Medel
Department of Physical and Analytical Chemistry, University of Oviedo, C/Julián Clavería 8, 33006 Oviedo, Spain. E-mail: jiga@sauron.quimica.uniovi.es; Fax: +34 985103125
First published on 22nd November 2002
Abstract
An inductively coupled plasma quadrupole mass spectrometer (ICP-QMS) with an octapole reaction system (ORS) was used for the determination of selenium by isotope dilution analysis (IDA) in biological reference materials and serum samples from healthy subjects. The potentially interfering argon dimers (40Ar38Ar+ and 40Ar2+) at the selenium masses 78 and 80 were almost eliminated by the use of hydrogen as reaction gas. Thus, the detection limits were improved five times for 78Se and the measurement of 80Se (the main selenium isotope with an abundance of 49.61%) is now possible. An enriched 77Se solution was prepared and characterized by reverse IDA and isotope ratios measured were 78Se/77Se and 80Se/77Se. Instrumental parameters were optimised in order to obtain optimal precision and accuracy in the isotope ratio measurement of Se by ORS-ICP-QMS. The precision achieved for the isotope ratio measurements reached 0.2% (RSD for n = 5) for both ratios. Systematic errors, including detector dead time (47 ± 2 ns), mass bias effects (about 3%) and spectroscopic interferences due to the presence of bromine in the samples, were corrected. The accuracy of the measured selenium isotope ratios was improved by correcting the intensity signals of the selenium isotopes for SeH+ formation (about 3%). The proposed IDA method has been applied to the determination of Se in biological reference materials (serum, urine and tissues) and the results showed good agreement with the certified values.
Introduction
The interest in selenium determination in biological materials has continuously increased because it is considered to be an essential element occurring in inorganic and organic chemical forms.1 The concentration range in which selenium is physiologically essential (biological window) is narrower than for any other trace element.2,3 Therefore, the accurate determination of selenium levels in biological materials is required to understand the role of this element in health and disease.Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful technique for elemental analysis due to its multielemental capabilities, low detection limits and the possibility of measuring isotope ratios.4,5 In combination with isotope dilution analysis (IDA) highly accurate and precise determinations can be carried out for elements that have at least two isotopes free from spectral interferences.6 Since an isotope of the element is used as an “internal standard”, IDA will compensate for many sources of error, including sample loss during the preparation or pre-treatment processes. A major advantage of IDA over conventional calibration procedures is that it can provide compensation for a variety of physical and chemical interferences, such as plasma fluctuations, instrument instabilities and analyte suppressions by sample matrix7,8,9 This feature is of great importance for the determination of selenium in biological samples where serious matrix effects are commonly observed.
However, the accurate determination of selenium by ICP-MS has been hampered by the presence of spectroscopic interferences. Major isotopes of selenium (80Se, 78Se and 76Se) are all subject to severe Ar2+ interferences. Other Se isotopes of lower abundance (82Se or 77Se) can also be interfered by molecular ions from halogens (BrH+, ClO+) present in the sample. The main polyatomic interferences, which could occur in the determinations of selenium in biological samples, are listed in Table 1.10–13
| Se isotope | Abundance (%) | Interference |
|---|---|---|
| 74 | 0.89 | 38Ar36Ar+, 37Cl2+, 40Ar34S+ |
| 76 | 9.37 | 40Ar36Ar+, 40Ar36S+, 31P214N+ |
| 77 | 7.63 | 40Ar36ArH+, 38Ar2H+, 40Ar37Cl+ |
| 78 | 23.77 | 40Ar38Ar+, 31P216O+ |
| 80 | 49.61 | 40Ar2+, 79BrH+ |
| 82 | 8.73 | 40Ar2H2+, 12C35Cl2+, 34S16O3+, 81BrH+ |
The determination of selenium in biological matrices by IDA-ICP-MS has been reported using a range of sample introduction systems such as pneumatic nebulisation,14,15 hydride generation15,16 and, more recently, electrothermal vaporization.17,18 Nevertheless, these works have reported a series of disadvantages. Thus, Janghorbani et al.15 reported isotope ratio precisions around 1% using pneumatic nebulisation or hydride generation and measured the less abundant Se isotopes (74Se, 77Se and 82Se isotopes) in order to avoid Ar2 interferences. Hydride generation also required intensive sample pretreatment and suffered from reagent impurities and memory effects, as well as from interferences in the hydride generation step. On the other hand, electrothermal vaporization has proved to be effective for the reduction of polyatomic interferences caused from matrix components but not from those caused by polyatomic argon ions. In addition, the precision of such analysis was not as good as those obtained by simple nebulisation.
One way to reduce or eliminate spectral interferences is the use of a sector field mass spectrometer, operated at high resolution.19 However, drawbacks of the high resolution mass analyser are the inherent decrease in instrument sensitivity as the resolution is increased and its high cost.
The development of ICP-QMS equipped with collision/reaction cells20–23 is offering an interesting alternative to high-resolution systems for the removal of spectral interferences. The cells (quadrupoles, hexapoles or octapoles) are pressurized with a gas or a mixture of gases to reduce or eliminate the interfering polyatomic species by collisional dissociation and, mainly, by ion–molecule reactions.24 Helium25 is usually used as collision gas while H2,20–22 NH3,23 Xe25 or CH426 are usually employed as possible reaction gases. The collision/reaction cell can also increase the ion transmission efficiencies by collisional focusing.20,21,27
An analytical methodology for the accurate determination of traces of selenium by IDA-ICP-MS using a new octapole reaction system is described here using hydrogen as the reaction gas. The proposed method has been validated by the determination of selenium in a range of biological reference materials with satisfactory results.
Experimental
Instrumentation
The ICP-MS used was an Agilent, Model 7500c (Agilent Technologies, Tokyo, Japan), consisting of an ICP source with a plasma-shielded torch (grounded metal plate), an enclosed octapole ion guide operated in RF only mode and a quadrupole mass analyser with a secondary electron multiplier operating in a dual mode (i.e., either a pulse counting mode or analogue mode, depending on the ion intensity).Helium and hydrogen were introduced into the octapole cell as collision or reaction gases, respectively. The gases were introduced into the cell under mass flow control through stainless steel lines. The sample introduction system consisted of a Babington nebuliser with a Scott double-pass quartz spray chamber cooled down to 2 °C. The operating conditions (plasma and reaction/collision cell parameters) and acquisition parameters were optimised for the determination of selenium by IDA and they are summarized on Table 2.
| Plasma parameters— | |
| RF power | 1500 W |
| Plasma gas flow rate | 15 L min−1 |
| Auxiliary gas flow rate | 1.2 L min−1 |
| Sample uptake rate | 0.4 mL min−1 |
| Sampling depth | 7 mm |
| Sampler and skimmer | Ni, 1 and 0.4 mm id |
| Ions lens setting | Optimised for best sensitivity of 10 µg l−1 Li, Co, Y and Tl, 1% (w/w) HNO3 solution |
| Reaction/collision parameters— | |
| H2 gas flow | 4 mL min−1 |
| Octapole bias | −13 V |
| QP bias | −12 V |
| Data acquisition parameters | |
| Monitored isotopes | 76, 77, 78, 79, 80, 81, 82 and 83 |
| Points per peak | 3 |
| Acquisition time per point | 4 s |
| Replicates | 5 |
A Milestone (Socisole, Italy) Model 1200 microwave digester with an EM-457(A) extractor module and an AC-100 open/close module with medium pressure PTFE vessels were employed for the digestion of the solid samples. The following two steps program was used: (1) 250 W, 2 min and (2) 450 W, 5 min.
Reagents and materials
All reagents used were of analytical grade. Ultra-pure water was obtained from a Milli-Q System (Millipore Co., Bedford, MA, USA). A standard solution of 1000 mg L−1 of Se as SeO2 stabilized in 2–3% (v/v) nitric acid Suprapur® was purchased from Merck (Darmstadt, Germany). Hydrogen peroxide (Merck) and nitric acid from Merck (additionally purified by sub-boiling distillation) were used for sample digestions.Enriched 77Se was obtained from Cambridge Isotope Laboratories (Andover, MA, USA) as elemental powder and it was dissolved in a minimum volume of sub-boiled nitric acid and diluted to volume with ultra-pure water. The concentration of this solution was established by reverse isotope dilution analysis. Table 3 gives the isotopic composition of natural Se and the measured isotopic composition of the enriched 77Se spike, which was not certified for isotopic composition. The isotopic abundance of the Se standard solution was considered to be of natural isotopic abundance, as reported by Rosman and Taylor.28
| Mass | Natural | 77Se enricheda |
|---|---|---|
| a Determined after dead time, SeH+ formation and mass bias discrimination corrections using natural selenium (n = 5). | ||
| 74 | 0.89 ± 0.04 | 0.063 ± 0.009 |
| 76 | 9.37 ± 0.29 | 1.03 ± 0.07 |
| 77 | 7.63 ± 0.16 | 91.1 ± 0.7 |
| 78 | 23.77 ± 0.28 | 3.9 ± 0.3 |
| 80 | 49.61 ± 0.41 | 3.4 ± 0.2 |
| 82 | 8.73 ± 0.22 | 0.54 ± 0.04 |
The biological reference materials analysed in this work were: Seronorm™ Trace Elements Serum from ″Sero As″ (Billingstad, Norway), Bovine Liver NIST SRM 1577a, Freeze-Dried Urine NIST SRM 2670 from National Institute of Standards and Technology (Gaithersburg, MD, USA) and Horse Kidney IAEA H-8 from the International Atomic Energy Agency (Vienna, Austria). Solid and digested samples were kept refrigerated at 4 °C.
Healthy volunteers were selected from our research group for the human serum samples analysed. Blood samples were extracted from the arterial line using a standard syringe. The blood was allowed to clot and centrifuged at 2000 g for 3 h at 4 °C. All samples were kept refrigerated at 4 °C until measurement.
Procedures
The Seronorm™ Trace Elements Serum and Freeze Dried Urine reference materials were reconstituted as described in the certificate and directly analysed after appropriate dilution with ultra-pure water. Human serum samples were diluted 1 + 9 with ultra-pure water after the addition of a known amount of the 77Se spike solution.
Plastic polypropylene containers were used to prepare all solutions on a weight basis. Diluted standard solutions were prepared daily before the analysis with 1% (w/w) HNO3.
The correction for SeH+ and BrH+ formation was carried out using mathematical equations by monitoring also the signals at masses 76, 82 and 83 (for SeH+) and 79 and 81 (for BrH+). All isotope ratio measurements were corrected using a dead time of 47 ns. A natural standard of selenium was also measured between the samples to calculate the mass bias correction factor.
The optimum spike to sample ratio was calculated as described previously,29 using the random error propagation theory. Taking into account the natural abundances published by Rosman and Taylor28 and the isotope composition of the spike, the optimal isotope ratios were 0.49 for 80Se/77Se and 0.36 for 78Se/77Se.
Finally, the concentration of the analyte was calculated using the isotope dilution equation described previously.29
Results and discussion
Elimination of argon polyatomic interferences
As quadrupole ICP-MS instruments equipped with reaction cells can remove argon-containing polyatomic interferences,22 reaction cell parameters were tuned for optimum signal-to-noise (S/N) ratio by measuring 78Se and 80Se isotopes. The main instrumental parameter affecting the S/N ratio was the flow of hydrogen used as reaction gas while adequate settings of the octapole bias and QP bias were also important to reduce the background levels by energy discrimination. The optimal cell parameters selected are given in Table 2. When the reaction/collision cell was pressurized, the octapole bias was set at −13 V and the quadrupole bias was at −12 V in order to achieve the lowest background signal. As the electric potential of the quadrupole analyser is slightly more positive than the electric potential of the octapole, the residual gas and product ions generated in the cell have not enough energy to overcome this energy barrier. On the other hand, as polyatomic ions have larger collisional cross sections than those from monoatomic ions, more frequent interactions with the cell gases are produced and a significant reduction in kinetic energy relative to the analyte results.30 Therefore, energy filtering can also be used to help separate the analyte from the polyatomic interference.Next, the nature and optimum flow of the cell gas were evaluated: firstly, the effects of He, H2 and He–H2 mixtures, as reaction and/or collision gases, were tested to remove polyatomic interferences of argon at masses 78 and 80. It is important to recognize that the optimum cell gas flow (minimum limit of detection) does not necessarily correspond to the flow providing maximum net sensitivity (i.e., the detection limit is determined both by the sensitivity and by the noise on the background signal). For ion signals below ca. 104 cps the standard deviation of the background can be approximated by counting statistics. Therefore, the detection limit can be estimated according to:

 | ||
Fig. 1 Signal intensity of 80Se as a function of the hydrogen flow rate in the octapole reaction cell. (- - -) Signal intensity for the blank solution (1%
(w/w) HNO3). (—) Signal intensity for a 10 ng g−1 selenium standard. (![[circle, cut, short horiz bar]](https://www.rsc.org/images/entities/char_e0eb.gif) ) Estimated detection limit. ) Estimated detection limit. | ||
Fig. 1 shows the effect of H2 gas flow rate on the signals of both a standard solution of selenium and a blank solution at m/z 80. As can be seen, an important improvement in the EDL was obtained at mass 80. Fig. 1 shows that using a hydrogen flow rate of around 4 mL min−1, the minimum EDL for 80Se was obtained (14 pg g−1). The sensitivity for the 10 ng g−1 selenium standard was approximately 50000 counts s−1 at mass 80, while the count rate for the blank (1% (w/w) HNO3) was 300 counts s−1. This latter value represents a reduction of approximately six orders of magnitude in the intensity of the 40Ar2+ dimers compared with conventional ICP-QMS operation. Similar results were observed at m/z 78, where an improvement of five times in the EDL, compared with conventional ICP-QMS, was observed.
Precision of isotope ratio measurements
Acquisition parameters were optimised for the best precision of 77Se/80Se and 77Se/78Se isotope ratios using a standard solution of 100 ng g−1 natural Se. Based on our previous experience,31 three points per peak and five replicates were selected and only the influence of integration time was evaluated. The influence of the total integration time per mass unit on the relative standard deviation (RSD) (n = 5) of the 77Se/80Se ratio measurement is showed in Fig. 2. As can been seen, the measured RSD decreased from ca. 1% at 0.1 s integration time per unit mass to ca. 0.2% at 3–6 s total integration time per mass. An integration time of 4 s per mass was finally selected. A similar effect was also observed in the RSD of the 77Se/78Se ratio measurement.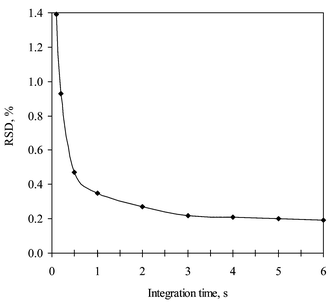 | ||
| Fig. 2 Influence of integration time on the isotope ratio precision. | ||
Accuracy of the measured isotope ratios
We observed that, when hydrogen was used to pressurize the octapole cell with a flow rate of 4 ml min−1, the measured 82SeH+/82Se+ ratio was around 3%, a lower figure than the value around 9% reported by Sloth26 or Boulyga.33
However, this effect must be taken into consideration because it affects the isotope ratio measurement accuracy by influencing ions of mass m + 1. The SeH+/Se+ ratio was evaluated with 82Se by measuring the m/z 83/82 ratio in the same run in which the mass discrimination per unit mass was determined. No correction for krypton was necessary as Kr ions were neutralized in the cell. A 100 ng g−1 Se standard of natural isotopic composition was nebulised for this purpose and the measured intensities for 77Se and 78Se were corrected by taking into account the formation of the hydrides 76SeH+ and 77SeH+, respectively. The intensity correction equations used were:
 | (1) |
 | (2) |
Finally, the mass bias correction was determined using the exponential model of mass bias.31Fig. 3 shows the plot of relative error ln(Rexp/Rtheo) in the experimental isotope ratios with respect to 80Se versus the mass difference between the measured isotopes, ΔM, using weighted linear regression calculations. The result was a linear relationship. The mass bias factor (K), derived from the slope of the regression line was of ca. −3% per mass unit. Similar mass bias factors have been reported for selenium26,33,34 by ICP-MS.
 | ||
| Fig. 3 Linear relationship between corrected measured isotopic ratios and the mass difference between the measured isotopes from the 80Se reference isotope. K = −0.0338. | ||
Once K has been determined, the corrected isotope ratios, Rcorr, can be calculated using:
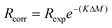
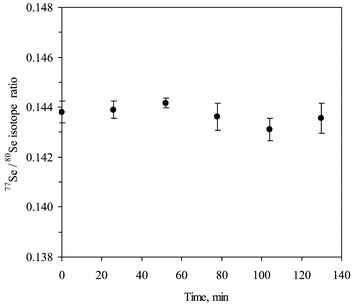 | ||
| Fig. 4 Stability of 77Se/80Se isotope ratio with time. | ||
 | ||
| Fig. 5 77Se/80Se isotope ratio as a function of bromine concentration. (—) Original data. (····) Corrected 77Se/80Se isotope ratio after proposed mathematical correction. | ||
This interference was corrected by monitoring also 79Br and 81Br (% abundance 50.69 and 49.31, respectively28) by considering equations (1) and (2) and the following equations for masses 79, 80, 81 and 82:
 | (3) |
 | (4) |
 | (5) |
 | (6) |
The corrected 77Se/80Se isotope ratios are plotted in Fig. 5 as a function of bromine concentration (broken line). As can be observed, the correction method used is adequate within the experimental uncertainty. It is worth mentioning here that, when the concentration of bromine increases, so does the uncertainty in the correction at mass 80. This fact was taken into account in the calculation of the uncertainties shown in Fig. 5, where the law of propagation of errors was applied to equations (1) to (6) and to the corrected 77/80 ratio.
Validation of the method and determination of selenium in real samples
Selenium was determined in four biological reference materials and in three human serum samples by the proposed IDA procedure. In all cases, the samples were spiked with the enriched isotope, digested (or simply diluted) and the ratios 80Se/77Se and 78Se/77Se evaluated after adequate interference correction. As an example, the obtained mass spectra in the range 76–83 for a serum and a blank are shown in Fig. 6. The presence of high levels of bromine is clearly seen from the spectrum. As similar levels of bromine were also detected in the reference materials, the correction of BrH+ had to be performed in all samples for the 80Se/77Se ratio. This was carried out by measuring a 1 ppm bromine standard and calculating the bromine factor from the measured 82/81 and 80/79 ratios. A bromine factor of 0.0415 was applied for the correction of all intensities in the real samples and reference materials. This factor was found to change slightly from day to day. The selenium factor was calculated using the mass bias correction solution (100 ppb natural selenium standard) from the measured 83/82 ratio. Then, all intensities measured were corrected using equations [1] to [6]. The final quantitative results using both ratios are given in Table 4. As can be observed, the results obtained for all reference materials for the 78/77 ratio lie within the ranges given for the certificate at the 95% confidence level. However, for Horse Kidney and Bovine Liver data for the 80/77 ratio show certain differences from the certified values, which could be a result of the low concentration of selenium in the digested samples and the high value of the bromine correction.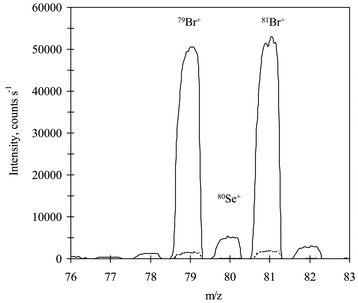 | ||
| Fig. 6 Mass spectrum of a serum sample diluted 1 + 9 (solid line) and an ultra pure water blank (broken line). | ||
| Material | Reference value | Concentration foundc | |
|---|---|---|---|
| 80Se/77Se | 78Se/77Se | ||
| a Concentrations in ng g−1. b Concentrations in µg g−1. c Average ± uncertainty at 95% confidence level. | |||
| Seronorm™ Human Seruma | 83 ± 3 | 83.6 ± 1.7 | 83.2 ± 2.0 |
| SRM-2670 Freeze Dried Urinea | 30 ± 8 | 31.1 ± 1.2 | 30.3 ± 0.5 |
| IAEA H-8 Horse Kidneyb | 4.67 ± 0.30 | 5.3 ± 0.5 | 4.59 ± 0.07 |
| NIST SRM 1577a Bovine Liverb | 0.71 ± 0.07 | 0.87 ± 0.05 | 0.70 ± 0.07 |
Real human serum samples were also analysed to compare our results with literature values and as a starting point to carry out an analytical survey of the levels of selenium in the population of Asturias (Spain). Results from 3 individuals are given in Table 5. As can be observed, the levels found are within the ranges given in the literature.35 The values obtained for the 80/77 ratio are somehow higher than those obtained for the 78/77 ratio, indicating the need for a better correction procedure for the interference of bromine on selenium. In the future this method will be applied to study selenium level changes in health and disease.
| Sample | Concentration found/ng g−1 | |
|---|---|---|
| 80Se/77Se | 78Se/77Se | |
| 1 | 77.8 ± 1.4 | 73.4 ± 2.2 |
| 2 | 97.6 ± 2.4 | 89.6 ± 2.0 |
| 3 | 87.3 ± 0.6 | 83.0 ± 0.6 |
Acknowledgements
The authors are grateful to Agilent Technologies for the loan of the Agilent 7500c to the Ministry of Science and Technology (Madrid, Spain), for financial support through project number BQU2000-0221 and to CONACYT (Mexico) for awarding a Doctoral Grant to L. Hinojosa Reyes.Healthy volunteers (V. Diaz, A. Rodríguez, L. Bravo, C. García and G. Centineo) are greatly acknowledged for blood donation and M. Serrano of the “Hospital Central de Asturias” for the blood extraction.
References
- L. H. Foster and S. Sumar, Crit. Rev. Food Sci. Nutr., 1997, 37(3), 211 Search PubMed.
- L. A. Daniels, Biol. Trace Elem. Res., 1996, 54, 185 Search PubMed.
- M. Navarro-Alarcón and M. C. López-Martínez, Sci. Total Environ., 2000, 249, 347 CrossRef CAS.
- M. Stastná, I. Nemcova and J. Zýka, Anal. Lett., 1999, 32(13), 2531 CAS.
- C. Sariego Muñiz, J. M. Marchante Gayón, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 1999, 14, 193 RSC.
- K. G. Heumann, Int. J. Mass. Spectrom. Ion Processes, 1992, 118(119), 575 CrossRef.
- F. Vanhaecke, L. Moens and R. Dams, J. Anal. At. Spectrom., 1998, 13, 1189 RSC.
- J. P. Valles Mota, M. R. Fernández de la Campa, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 1999, 14, 113 RSC.
- C. Sariego-Muñiz, J. M. Marchante Gayón, J. I. García Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 1999, 14, 1505 RSC.
- J. Goossens, F. Vanhaecke, L. Moens and R. Dams, Anal. Chim. Acta, 1993, 283, 137 CrossRef.
- S. H. Tan and G. Horlick, Appl. Spectrosc., 1986, 4, 445 Search PubMed.
- L. S. Zhang and S. M. Combs, J. Anal. At. Spectrom., 1996, 11, 1049 RSC.
- F. H. Konad and M. H. Yang, J. Anal. At. Spectrom., 1996, 11, 413 RSC.
- B. T. G. Ting, C. S. Mooers and M. Janghorbani, Analyst, 1989, 114, 667 RSC.
- M. Janghorbani and B. T. G. Ting, Anal. Chem., 1989, 61, 701 CrossRef CAS.
- W. T. Buckley, J. J. Budac and D. V. Godfrey, Anal. Chem., 1992, 64, 724 CrossRef CAS.
- J. W. H. Lam, R. E. Sturgeon and J. W. McLaren, Spectrochim. Acta, Part B, 1999, 54, 443 CrossRef.
- J. Turner, S. J. Hill, E. H. Evans, B. Fairman and C. S. J. Wolff Briche, J. Anal. At. Spectrom., 2000, 15, 743 RSC.
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53, 1739 CrossRef.
- I. Feldmann, N. Jakubowski and D. Stuewer, Fresenius’ J. Anal. Chem., 1999, 365, 415 CrossRef CAS.
- I. Feldmann, N. Jakubowski and D. Stuewer, Fresenius’ J. Anal. Chem., 1999, 365, 422 CrossRef CAS.
- P. Leonhard, R. Pepelnik, A. Prange, N. Yamada and T. Yamada, J. Anal. At. Spectrom., 2002, 17, 189 RSC.
- V. I. Baranov and S. D. Tanner, J. Anal. At. Spectrom., 1999, 14, 1133 RSC.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Spectrochim. Acta, Part B, 2002, 57, 1361 CrossRef.
- J. T. Rowan and R. S. Houk, Appl. Spectrosc., 1989, 43, 976 Search PubMed.
- J. J. Sloth and E. H. Larsen, J. Anal. At. Spectrom., 2000, 15, 669 RSC.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Fresenius’ J. Anal. Chem., 2001, 370, 454 Search PubMed.
- K. R. Rosman and P. D. P. Taylor, J. Anal. At. Spectrom., 1999, 14, 5N Search PubMed.
- J. I. García Alonso, Anal. Chim. Acta, 1995, 312, 57 CrossRef.
- N. Yamada, J. Takahashi and K. Sakata, J. Anal. At. Spectrom., 2002, 17, 1213 RSC.
- J. Ruiz Encinar, J. I. García Alonso, A. Sanz-Medel, S. Main and P. J. Turner, J. Anal. At. Spectrom., 2001, 16, 315 RSC.
- F. Vanhaecke, G. Wannemacker, L. Moens, R. Dams, C. Latkoczy, T. Prohaska and G. Stingeder, J. Anal. At. Spectrom., 1998, 13, 567 RSC.
- S. F. Boulyga and J. S. Becker, Fresenius’ J. Anal. Chem., 2001, 370, 618 Search PubMed.
- S. M. Gallus and K. G. Heumann, J. Anal. At. Spectrom., 1996, 11, 887 RSC.
- S. Caroli, A. Alimonti, E. Coni, F. Petrucci, O. Senofonte and N. Violante, CRC Crit. Rev. Anal. Chem., 1994, 24, 363 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |