Conversion of the hydroxyl group in 1-hexyl alcohol to an amide group in supercritical water without catalyst
Mitsuru Sasaki*a, Junko Nishiyamab, Munehiro Uchidab, Kohtaro Gotoc, Kiyohiko Tajimac, Tadafumi Adschirib and Kunio Arai*b
aGenesis Research Institute, Inc, 4-1-35 Noritake-shinmachi, Nishi-ku, Nagoya, 451-0051, Japan. E-mail: sasaki@arai.che.tohoku.ac.jp; Fax: +81 22 217 7246; Tel: +81 22 217 7248
bDepartment of Chemical Engineering, Tohoku University, 7 Aza-Aoba, Aramaki, Aoba-ku, Sendai, 980-8579, Japan. E-mail: karai@arai.che.tohoku.ac.jp; Fax: +81 22 217 7246; Tel: +81 22 217 7245
cNoguchi Institute, 1-8-1 Kaga, Itabashi-ku, Tokyo, 173-0003, Japan
First published on 27th January 2003
Abstract
The main reaction pathway in the reaction between 1-hexyl alcohol and acetamide in subcritical and supercritical water without catalyst is proposed and the optimum conditions where amidation of 1-hexyl alcohol can selectively occur are explored.
Green ContextSupercritical water is a fascinating material, which has been used for several organic transformations recently (see e.g. Hamley et al., Green Chem., 2002, 4, 235). Here, it is shown that it is possible to alkylate amides at the N atom using alcohols in supercritical water without catalyst. Selectivity was very good, especially considering the potential number of side-reactions which could occur.DJM |
Water can be considered as a promising solvent that is benign to the environment.1 If water can be utilized under its supercritical condition (Tc = 374.2 °C, Pc = 22.1 MPa, ρc = 0.32 g cm−3), it can act like most organic solvents by manipulating the reaction temperature and pressure. Recently, a large number of researches regarding not only hydrolysis of polymers (e.g. polyethylene terephthalate2 and cellulose3) and hazardous organic chemicals,4 but also synthesis of organic materials5 in subcritical and supercritical water have been reported. Katritzky et al.6 and Chandler et al.7 found that Friedel–Crafts alkylation of phenol and p-cresol occurred at 275 °C under a catalyst free condition by adding tert-butyl alcohol and isopropyl alcohol. Sato et al.8 found that reaction of phenol and isopropyl alcohol could successfully form isopropyl phenol under non-catalytic conditions in supercritical water. Akiya et al.9 reported that dehydration of cyclohexanol to cyclohexene took place in near-critical water in a selective manner. Experimental results for Diels–Alder reactions,10 aldol condensation,11 Claisen rearrangement,11 and the Rupe rearrangement11 in supercritical water have also been reported.
Notably, Ikushima et al.12 reported that ε-caprolactam could be quantitatively synthesized by the Beckmann rearrangement of cyclohexanone oxime in supercritical water in the absence of catalyst. This finding is of great importance to solve the problem of emission of ammonium sulfate, which is a by-product of the conventional synthetic processes, and to develop an efficient method of ε-caprolactam production. Another production method for ε-caprolactam is a UCC (Union Carbide Co.) method,13 in which cyclohexanone is oxidized by Baeyer–Villiger oxidation to form ε-caprolactone, followed by the formation of ε-caprolactam via the reaction between ε-caprolactone and ammonia under hydrothermal conditions. This method seemed to be promising from the viewpoint of green chemistry,14 but the yield and purity of ε-caprolactam obtained were low. Ito et al.15 carried out this reaction in supercritical water and proposed that in the main reaction pathway ε-caprolactone hydrolyzed to 6-hydroxyhexanoic acid and subsequently reacted with ammonia to form 6-hydroxyhexanamide. They also found that a high yield of ε-caprolactam (87%) could be achieved at temperatures of 400–450 °C, over 10–60 min using water densities of 0.3–0.5 g cm−3.
In the above study, an interesting point is that an amide bond can form by intramolecular dehydration between a hydroxyl group and an amide group in 6-hexylhexanamide. In general, this reaction does not occur in water under catalyst free conditions. This finding suggests that hydroxyl groups in primary alcohols can be converted to amide groups in supercritical water without catalyst. In this communication, we report results of the reactions between 1-hexyl alcohol and acetamide in subcritical and supercritical water without catalyst for determining the optimum conditions where amidation can occur in a selective manner in the absence of catalyst.
Reactions were conducted using a 6.0 cm3 stainless steel 316 tube bomb reactor. 1-Hexyl alcohol (99.0% purity, Wako Pure Chem. Ind., Ltd.) and acetamide (99.0% purity, Wako Pure Chem. Ind., Ltd.) were loaded into the reactor and a given amount of distilled water, which corresponds to a density of water of 0–0.5 g cm−3, was introduced. The air in the reactor was substituted to argon gas by repeated purging and then the reactor was sealed. The molar ratio of acetamide to 1-hexyl alcohol was 5–50, corresponding to concentrations of 0.33 mol L−1 for 1-hexyl alcohol (1) and 1.66–16.6 mol L−1 for acetamide (7). The reactor was submerged into a sand-bath heated to 300–450 °C. After 5–60 min, the reactor was quickly quenched. Products were identified by GC-MS and by comparing their GC and HPLC retention times with those of standards. Quantitative analysis of the products was performed using HPLC. The conversion of 1 (X) and the yield of product i (Yi) are defined as X = ([1]0 − [1])/[1]0) and Yi = [i]/[1]0. The selectivity of the product i was also calculated using Si = Yi/X.
Fig. 1 shows the yield of the main products derived from 1 at 400 °C, 0.5 g cm−3 and [7]0 = 1.66 mol L−1. Products identified in this study were N-hexylacetamide (2), hexanal (3), hexene (4), hexylamine (5), hexylacetate (6), acetic acid (8) and hexanoic acid (10). Conversion X increased with time and reached about 60% at 60 min. The yield Y2 was 21.9% at 60 min while the yields Y3 and Y4 reached about 20% and 8% at 30 min and then decreased with time. Yield Y6 was 4.4% at 10 min and then decreased to 0.8% at 60 min. The yield of 5, a hydrolysis product of 2, was low (1.5%), although it was expected that 2 should easily hydrolyze to 5 in supercritical water. The reason for the low level of 5 may be that 2 is stablized over 5 due to solvation in supercritical water, or that 2 is converted to 1 and 7 by the reverse reaction of amidation. Under the used reaction conditions, about 70% of 7 hydrolyzed to form 8 at 10 min and the yield Y8 scarcely changed even for extended reaction times. In summary, amidation between 1 and 7 took place in supercritical water. Moreover, we evaluated any possibility of the formation of 2 and/or 5 from the reaction between 4 and 7 in subcritical and supercritical water and found that they did not form.
![Yields of the main products at 400 °C, 0.5 g cm−3, 1-hexyl alcohol [1]0 = 0.33 mol L−1 and acetamide [7]0 = 1.66 mol L−1: (○) 1-hexyl alcohol (1); (□) N-hexylacetamide (2); (△) hexanal (3); (◇) hexene (4); (×) hexylamine (5); (▽) hexanoic acid (10); (+) hexyl acetate (6).](/image/article/2003/GC/b211451h/b211451h-f1.gif) | ||
| Fig. 1 Yields of the main products at 400 °C, 0.5 g cm−3, 1-hexyl alcohol [1]0 = 0.33 mol L−1 and acetamide [7]0 = 1.66 mol L−1: (○) 1-hexyl alcohol (1); (□) N-hexylacetamide (2); (△) hexanal (3); (◇) hexene (4); (×) hexylamine (5); (▽) hexanoic acid (10); (+) hexyl acetate (6). | ||
Based on these results, we propose the main reaction pathway in the 1-hexyl alcohol–acetamide–water system in subcritical and supercritical water as shown in Scheme 1. The main reaction paths are (a) intermolecular dehydration between 1 and 7, (b) a reverse Cannizzaro reaction between 1 and 8 formed by the dissociation of 7, (c) intramolecular dehydration to 4, (d) intermolecular dehydration (esterification) between 1 and 8, and (e) hydrolysis of 7 to 8 and ammonia (11). It has already been reported that the Cannizzaro reaction takes place in supercritical water under catalyst free conditions.16 From a preliminary experiment, we also confirmed that the reverse Cannizzaro reaction can take place in supercritical water.13
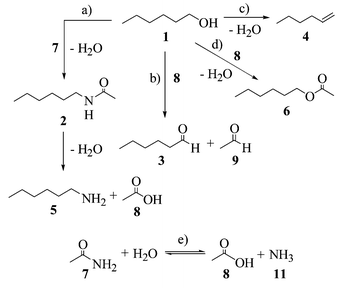 | ||
| Scheme 1 Main reaction pathway in the 1-hexyl alcohol (1)–acetamide (7)–water system in subcritical and supercritical water. | ||
The effect of the density of water on amidation was examined under the conditions: 400 °C, 60 min and [1]0 = 0.33 mol L−1. Fig. 2(a) and (b) shows the results at [7]0 = 1.66 and 16.6 mol L−1, respectively. At low [7]0, as shown in Fig. 2(a), gaseous products were mainly formed by pyrolysis of 1 in argon atmosphere and low water densities of 0.1 and 0.3 g cm−3. By contrast, at a higher water density (0.5 g cm−3), gaseous products were scarcely detected and liquid products, especially 2, mainly formed. With increasing [7]0 (see Fig. 2(b)), the yield Y2 became higher at 0.3 and 0.5 g cm−3. This experimental finding demonstrates that amidation of 1 can be promoted at high water densities in supercritical water.
![Water density dependence of the product distributions at 400 °C, 60 min, [1]0 = 0.33 mol L−1 and water densities ranging 0 to 0.5 g cm−3: (a) [7]0 = 1.66 mol L−1; (b) [7]0 = 16.6 mol L−1: (○) 1-hexyl alcohol (1); (□) N-hexylacetamide (2); (△) hexanal (3); (◇) hexene (4); (×) hexylamine (5); (▽) hexanoic acid (10); (+) hexyl acetate (6).](/image/article/2003/GC/b211451h/b211451h-f2.gif) | ||
| Fig. 2 Water density dependence of the product distributions at 400 °C, 60 min, [1]0 = 0.33 mol L−1 and water densities ranging 0 to 0.5 g cm−3: (a) [7]0 = 1.66 mol L−1; (b) [7]0 = 16.6 mol L−1: (○) 1-hexyl alcohol (1); (□) N-hexylacetamide (2); (△) hexanal (3); (◇) hexene (4); (×) hexylamine (5); (▽) hexanoic acid (10); (+) hexyl acetate (6). | ||
Next, the effect of the reaction temperature on amidation was examined under the condition of [1]0 = 0.33 mol L−1, [7]0 = 1.66 mol L−1 and 0.3 g cm−3. Table 1 shows the experimental results conducted at temperatures of 300, 400 and 450 °C. At 300 °C, X was 31% and S2 was low (29%). 3 was formed at a similar level to 2 by a reverse Cannizzaro reaction between 1 and 8. In this case, the yield Y5 was 4.0%. At 400 °C, X was over 96% and Y2 reached 74.5% (selectivity: 77%) at 60 min with 5 not forming at all. At 450 °C and 10 min, X became even higher while Y2 was comparatively high (57.2%). In this case, however, the carbon recovery of liquid products was low. This is probably because gaseous products form by the thermal degradation of 1 and its primary products. From these experimental results, it was confirmed that the amidation of 1-hexyl alcohol in supercritical water was more suitable than that in subcritical water from the view point of a rapid and selective process. Also, it was suggested that subcritical water might be applicable as a reaction medium for hydrolyzing amides to amines.
| Y (mol%) | ||||||
|---|---|---|---|---|---|---|
| T/°C | 100 − X (mol%) | 2 | 3 | 4 | 5 | 6 |
| 300 | 60.8 | 11.3 | 14.2 | n.d. | 4.0 | 7.1 |
| 400 | 3.6 | 74.5 | 9.6 | n.d. | n.d. | 2.0 |
| 450 | 3.8 | 57.2 | 10.1 | n.d. | n.d. | n.d. |
Further, reactions of 1 with other amidation reagents (benzamide and hexanamide) were conducted and results are shown in Table 2. With both reagents, the amide compounds (N-hexylbenzamide and N-hexylhexanamide, respectively) could be synthesized, but the selectivity of these products were low compared with the case of acetamide. This is probably because the thermal stabilities of the amide compounds are not high under the used conditions.
| R | [R]0/[1]0 | 100 − X (mol%) | YAmide (mol%) | SAmide (mol%) |
|---|---|---|---|---|
| Benzamide | 10 | 25.3 | 18.6 | 24.9 |
| Hexanamide | 20 | 4.1 | 16.7 | 17.4 |
In summary, we have demonstrated the conversion of the hydroxyl group in 1-hexyl alcohol to an amide group in supercritical water without catalyst. This experimental finding leads to the development of a new method for efficient production of amide compounds from primary alcohols. Future efforts will be directed to elucidate the reaction mechanism further and to explore optimum conditions for rapid and selective amidation in supercritical water.
Acknowledgements
The authors gratefully acknowledge support for a Grand-in-Aid for Scientific Research on Priority Area (Grant 11450295) from the Ministry of Education, Culture, Sports, Science and Technology.References
- (a) J. M. DeSimone, Science, 2002, 297, 799 CrossRef CAS; (b) M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS.
- (a) T. Adschiri, O. Sato, K. Machida, I. Saito and K. Arai, Kagaku Kogaku Ronbunshu, 1997, 23(4), 505 Search PubMed; (b) Z. Fang, R. L. Smith, H. Inomata and K. Arai, J. Supercrit. Fluids, 1999, 15(3), 229 CrossRef CAS; (c) S. G. Kazarian and G. G. Martirosyan, Phys. Chem. Chem. Phys., 2002, 4(15), 3759 RSC; (d) A. T. Quitain, M. Faisal, K. Kang, H. Daimon and K. Fujie, Hazard. Mater., 2002, 93(2), 209 Search PubMed.
- (a) T. Adschiri, S. Hirose, R. M. Malaluan and K. Arai, J. Chem. Eng. Jpn., 1993, 26(6), 676 Search PubMed; (b) O. Bobleter, Prog. Polym. Sci., 1994, 19, 797 CrossRef CAS; (c) T. Sakaki, M. Shibata, T. Miki, H. Hirosue and N. Hayashi, Energy Fuels, 1996, 10, 684 CrossRef CAS; (d) T. Sakaki, M. Shibata, T. Miki, H. Hirosue and N. Hayashi, Bioresour. Technol., 1996, 58, 197 CrossRef CAS; (e) M. Sasaki, B. M. Kabyemela, R. M. Malaluan, S. Hirose, N. Takeda, T. Adschiri and K. Arai, J. Supercrit. Fluids, 1998, 13, 261 CrossRef CAS; (f) S. Saka and T. Ueno, Cellulose, 1999, 6(3), 177 Search PubMed; (g) K. Sakanishi, N. Ikeyama, T. Sakaki, M. Shibata and T. Miki, Ind. Eng. Chem. Res., 1999, 38(6), 2177 CrossRef CAS; (h) M. Sasaki, Z. Fang, Y. Fukushima, T. Adschiri and K. Arai, Ind. Eng. Chem. Res., 2000, 39(8), 2883 CrossRef CAS; (i) X. Y. Lu, A. Sakoda and M. Suzuki, Chinese J. Chem. Eng., 2000, 8(4), 321 Search PubMed; (j) M. Sasaki, K. Iwasaki, T. Hamaya, T. Adschiri and K. Arai, Kobunshi Ronbunshu, 2001, 58(10), 527 Search PubMed; (k) T. Sakaki, M. Shibata, T. Sumi and S. Yasuda, Ind. Eng. Chem. Res., 2002, 41(4), 661 CrossRef CAS.
- (a) J. C. Meyer, P. A. Marrone and J. W. Tester, AIChE J., 1995, 41(9), 2108 Search PubMed; (b) J. D. Taylor, J. I. Steinfeld and J. W. Tester, Ind. Eng. Chem. Res., 2001, 40(1), 67 CrossRef CAS; (c) T. Moriyoshi, K. Sakai and Y. Uosaki, High Pressure Res., 2001, 20(1–6), 491 Search PubMed; (d) R. Lachance, J. Paschkewitz, J. DiNaro and J. W. Tester, J. Supercrit. Fluids, 1999, 16(2), 133 CrossRef CAS; (e) J. D. Taylor, F. A. Pacheco, J. I. Steinfeld and J. W. Tester, Ind. Eng. Chem. Res., 2002, 41(1), 1 CrossRef CAS.
- (a) J. B. Dunn and P. E. Savage, Ind. Eng. Chem. Res., 2002, 41(18), 4460 CrossRef CAS; (b) P. A. Hamley, T. Ilkenhans, J. M. Webster, E. Garcia-Verdugo, E. Venardou, M. J. Clarke, R. Auerbach, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2002, 4(3), 235 RSC; (c) M. Sasaki, K. Goto, K. Tajima, T. Adschiri and K. Arai, Green Chem., 2002, 4(3), 285 RSC.
- A. R. Katritzky, S. M. Allin and M. Siskin, Acc. Chem. Res., 1996, 29(8), 399 CrossRef CAS.
- K. Chandler, F. Deng, A. Dillow, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 1997, 36(12), 5175 CrossRef CAS.
- T. Sato, G. Sekiguchi, T. Adschiri and K. Arai, Chem. Commun., 2001, 1566 RSC.
- N. Akiya and P. E. Savage, Ind. Eng. Chem. Res., 2001, 40(8), 1822 CrossRef CAS.
- J. An, L. Bangnell, T. Cablewski, C. R. Strauss and R. W. Trainor, J. Org. Chem., 1997, 62(8), 2505 CrossRef CAS.
- M. B. Korzenski and J. W. Kolis, Tetrahedron Lett., 1997, 38(32), 5611 CrossRef CAS.
- Y. Ikushima, K. Hatakeda, O. Sato, T. Yokoyama and M. Arai, J. Am. Chem. Soc., 2000, 122, 1908 CrossRef CAS.
- J. Nishiyama, M.S. Thesis, Tohoku University, Sendai, Japan, 2002.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998; ch 1 & 2 Search PubMed; (b) D. T. Allen and D. R. Shonnard, Green Engineering: Environmentally Conscious Design of Chemical Processes, Prentice Hall PTR, Upper Saddle River, NJ, 2001 Search PubMed.
- H. Ito, J. Nishiyama, T. Adschiri and K. Arai, Kobunshi Ronbunshu, 2001, 58(12), 679 Search PubMed.
- Y. Tsujino, C. Wakai, N. Matsubayashi and M. Nakahara, Chem. Lett., 1999, 83.
| This journal is © The Royal Society of Chemistry 2003 |