A thermodynamic investigation of solvent-free reactions
Waldo H. Correaa, J. Kate Edwardsab, Adam McCluskeyb, Ian McKinnonc and Janet L. Scott*a
aCentre for Green Chemistry, Monash University, P.O. Box 23, Victoria, 3800, Australia. E-mail: janet.scott@sci.monash.edu.au; Fax: 61 3 9905 4597; Tel: 61 3 9905 4600
bChemistry, School of Environmental and Life Sciences, The University of Newcastle, Callaghan, NSW 2308, Australia
cSchool of Chemistry, P.O. Box 23, Monash University, Victoria, 3800, Australia. E-mail: janet.scott@sci.monash.edu.au; Fax: 61 3 9905 4597; Tel: 61 3 9905 4600
First published on 18th November 2002
Abstract
A series of solvent-free condensation reactions have been studied calorimetrically, with the goal of determining feasibility of scale-up in the realisation of potentially greener processes, in which the use of auxiliary compounds, such as solvents, are obviated. Knoevenagel reactions leading to intermediates used in the production of 1,4-dihydropyridine drugs, and in drug libraries currently under testing, exhibit a reaction exotherm ascribed almost entirely to the formation of the imminium ‘Knoevenagel’ intermediate, which may be controlled by regulation of the quantity of catalyst. Reactions to form stable imines and cyanoamide products are also demonstrated to be exothermic.
Green ContextThe safe elimination of solvents from reactions implies an alternative heat sink is required to keep the reaction under control. How major a factor this is is obviously dependent on the thermodynamics and kinetics of the reaction. This paper provides details of heat flows and thermodynamic information for a range of Knoevenagel reactions, allowing a better understanding of the likely heat evolution, and thus providing a basis for proper design and scale-up of these processes.DJM |
Introduction
A number of authors have described protocols for the solvent-free synthesis of a wide range of organic compounds and the number of reports of rapid, selective and efficient transformations, occurring with a high degree of conversion of reactants to products, grows daily.1,2 Such protocols bear further investigation as they may offer distinct advantages such as: improved atom utilisation3 or atom economy4 by avoidance of common derivatisation procedures;5 decreased by-product formation and hence decreased waste (improved E-factors6) resulting from purification procedures required to separate the desired product from the impurities; and, in many cases, reduced energy utilization both in the reaction and purification stages as well as opportunities for process intensification. Some specific reactions targeted include solventless aldol reactions,5 sequential aldol and Michael addition as a route to symmetrical and unsymmetrical 1,5-diketones leading to Krohnke type pyridines,7 synthesis of 3-carboxycoumarins8 and tetrahydroquinazolines,9 oligomerisation reactions,10 calix[4]resorcinarene11 synthesis and, the focus of this paper, the 4-substituted-1,4-dihydropyridine compounds,12 such as the commonly used cardiovascular drug Felodipine, and some highly functionalized cyanoamido benzaldehyde derivatives of interest as dynamin inhibitors.13These (and numerous other) examples indicate that solvent-free reaction conditions may provide one tool for addressing waste-reduction and energy efficiency, but there are obvious and important questions to be asked about whether or not such procedures are hazard-free. One of the reasons that solvent use has become ubiquitous in organic synthetic chemistry relates to the very effective role of solvents (usually present in huge excess compared to reagents) as heat transfer media and, more specifically, as heat sinks in exothermic reactions. The consequences of loss of control of exothermic reactions are well known: thermal runaways, resulting from the mismatch between rate of heat evolution (which increases exponentially) and rate of heat removal (which increases linearly) may result in a rapidly rising temperature, leaving little time for correction. Reaction vessels may be at risk from over-pressurisation due to violent boiling or rapid gas generation and elevated temperatures may initiate secondary, more hazardous runaway reactions or decompositions. Clearly a greater understanding of the thermal characteristics of solvent-free reactions is required. Thus, suitable import must be given to the phrase ‘fundamental knowledge of chemical processes’ in the definition of green chemistry.
To this end we have undertaken studies of phase changes and the role of eutectic melts in solvent-free reactions between solid reactants,14,15 used combined differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) to detect reaction endotherms or exotherms and thus optimize the temperature of reaction12 and we now report a microcalorimetric investigation designed to allow rational, informed decision-making in determining the feasibility of scale-up of some useful solvent-free transformations.
Results and discussion
The Knoevenagel reactions of benzaldehydes 1 with ethyl acetoacetate 2 or cyanoamide derivatives 4, leading to intermediates of known12 or potential13 drug substances (Scheme 1), were chosen as model reactions for this study as there are a large number of 1,4-dihydropyridine drugs currently produced in tonne quantities16 and, should any of our library compounds prove to have promising activity, larger quantities would be needed and effective scale-up of these reactions required.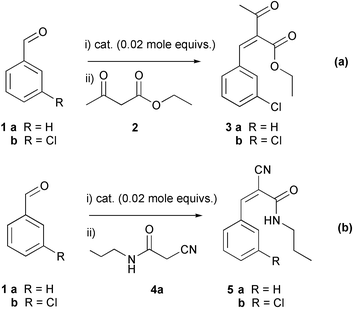 | ||
| Scheme 1 | ||
Reaction (b) is the second of two solvent-free reactions in the synthesis of these potential dynamin inhibitors 5 and thus reaction (c) (Scheme 2) was included in the study.
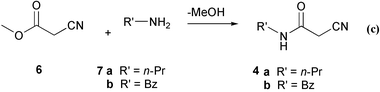 | ||
| Scheme 2 | ||
The first exploratory calorimetric experiment revealed the occurrence of a rapid exothermic reaction on addition of premixed ethylacetoacetate 2 and catalyst to benzaldehyde 1a (mol ratio 1∶0.02∶1). Quantities of reagents were scaled down significantly to achieve a heat flow deflection within the measurement range of the microcalorimeter and benchtop experiments confirmed the exothermic nature of the reaction. The shape of the heat flow curve, depicted in Fig. 1, implies a rapid, highly exothermic event followed by (or overlaid on) a slower process. The time taken for the initial process is comparable to the instantaneous reaction between HCl and NaOH used to calibrate the instrument.
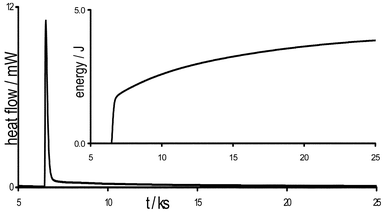 | ||
| Fig. 1 Heat flow trace for benzaldehyde added to premixed catalyst and ethyl acetoacetate (mol ratio 1∶0.02∶1; tmixing = 6.5 ks). Inset, integrated curve clearly showing a rapid first process overlaid on a slower second process.17 | ||
The Knoevenagel reaction is a two-step process18 and, since two distinct processes were detected in the heat flow curve, reaction (d) (Scheme 3), the formation of the active imminium intermediate, was investigated and compared to the formation of stable imines, reaction (e). (The facile and almost completely waste-free solventless synthesis of such stable imines has been reported by Kaupp et al.19 and we have found this a suitable method for preparation of, otherwise difficult to isolate, imines.20 Thus, this too, represents a useful chemical transformation worthy of investigation of scale-up feasibility.)
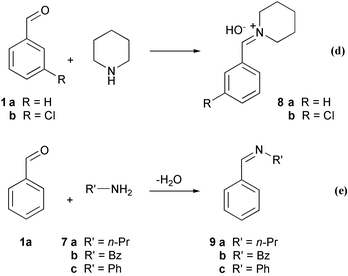 | ||
| Scheme 3 | ||
Values of the measured enthalpies of reaction obtained in each case are listed in Table 1. Clearly these are net reaction enthalpies, incorporating contributions from all processes including mixing, formation of active intermediates and reaction. In all cases quantities of excess reactant are chosen such that no precipitation or crystallisation of product occurs, although, in the stoichiometric reactions described previously,12,13,19 this may provide an added contribution.
| Reactants | |||||
|---|---|---|---|---|---|
| Reaction | Limiting | Excess | Product | Catalyst | ΔH/kJ mol−1 |
| a piperidine;b single determination;c calculated value (difference) | |||||
| (a) | 2 | 1a | 3a | pipa | −26.3 ± 0.9 |
| 2 | 1b | 3b | pip | −31.0 ± 1.7 | |
| 2 | 1a | 3a | pip/CH3COOH | −26.7 ± 0.6 | |
| 2 | 1b | 3b | pip/CH3COOH | −30.1 ± 1.1 | |
| (b) | 4a | 1a | — | 15.5b | |
| 4a | 1a | 5a | pip | −31.5c | |
| 4a | 1b | — | 22.3b | ||
| 4a | 1b | 5b | pip | −36.0c | |
| (c) | 6 | 7a | 4a | — | −11.7 ± 0.5 |
| 6 | 7b | 4b | — | −68.8 ± 1.4 | |
| (d) | pip | 1a | 8a | — | −46.9 ± 1.6 |
| pip | 1b | 8b | — | −47.5 ± 2.5 | |
| (e) | 7a | 1a | 9a | — | −5.1 ± 0.2 |
| 7b | 1a | 9b | — | −30.4 ± 1.5 | |
| 7c | 1a | 9c | — | −24.8 ± 2.3 | |
The major contribution to the exothermic nature of the Knoevenagel reactions studied ((a) and (b)) is from the preliminary reaction between benzaldehyde or 3-chlorobenzaldehyde and the piperidine catalyst to form the ‘imminium’ intermediate 8. This imminium species subsequently reacts with the ethyl ester to form the benzylidene product, as described by Knoevenagel in one of the original publications on the topic.18 The piperidine catalytic cycle, depicted in Scheme 4, is traversed a number of times in the experiments described here. Reexpressing the measured enthalpy in terms of the number of moles of piperidine21 undergoing reaction (d) during the process of reaction (a), R = H and R = Cl yields values of −38 kJ mol−1 and −39 kJ mol−1, respectively, indicating that the second process i.e. addition of ethylacetoacetate and elimination of piperidine is, if anything, endothermic. The choice of catalyst (piperidine or piperidine acetate) has no apparent affect on the enthalpy of reaction and benchtop experiments confirm that the product produced is identical in each case.
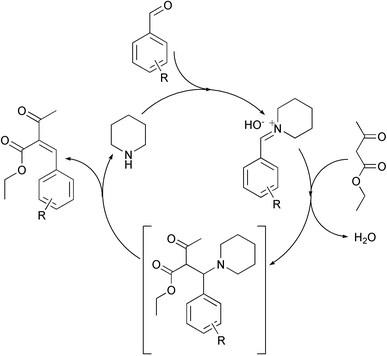 | ||
| Scheme 4 Catalytic cycle with respect to piperidine. The reaction proceeds via the Knoevenagel mechanism and the formation of the imminium intermediate from piperidine and the benzaldehyde derivative accounts for significant quantities of heat evolved during the reaction. | ||
The second Knoevenagel reaction studied involves reaction of a solid cyanoamide derivative 4a with liquid benzaldehydes 1a and 1b and this, not surprisingly, yields a thermal trace indicative of an exothermic process overlaid on an endothermic one as illustrated in Fig. 2.
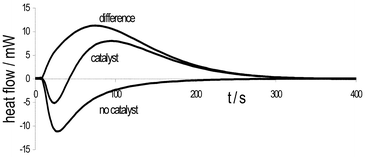 | ||
| Fig. 2 Heat flow traces for the reaction of 1b and 4a. Bottom: without catalyst i.e. heat of mixing of reactants only; middle: with piperidine catalyst, showing exothermic process overlaid on endothermic process and top: the result of subtracting the heat of mixing from the heat flow measured during the reaction, allowing an estimate of the reaction exotherm. (Note: tmixing ≠ 0 s.) | ||
The endothermic process is ascribed to the dissolution of solid 4a in 1b and measurement of the heat flow upon mixing of 4a in 1a or 1b indeed yields a distinct endotherm (and no product on the timescale of the measurement). This allows estimation of the ΔH of dissolution and subtraction of this (after correction for specific experimental quantities) from the measured heat flow during reaction yields a calculated heat flow curve representing the reaction exotherm as illustrated in Fig. 2. While these values are determined for one experiment only, it is gratifying to note that the ΔH of reaction for reaction (b) is similar to that determined for reaction (a).
If the measured exotherm is due largely to the heat generated upon formation of the imminium intermediate then control of the reaction exotherm may be effected by regulation of the quantity of piperidine added. Thermal control of reaction (c) would be effected by slow addition of one reactant and, in this case the use of reactors engineered to have small mixing chambers, such as extrusion reactors, which would be preferable to bulk batch processes. The same is true of the formation of stable imines, reaction (e), as these processes are also exothermic. In all cases the formation of the more stable product, leads, not surprisingly to a higher measured reaction exotherm.
This microcalorimetric study of solvent-free reactions demonstrates that it is possible to determine, by quite simple experiments, using readily available equipment, the overall ΔH of reaction to allow sensible decisions relating to scale-up and exotherm control. In addition, while this is not intended to comprise a comprehensive study of the mechanism of the Knoevenagel reaction or the thermochemistry of each discrete process, information gained about the source of the reaction exotherm allows one to effect control by controlling the order and rate of addition of reactants. Such studies are required if solvent-free reactions are to become useful tools in the green chemistry toolbox.
Experimental
Instrumentation
All calorimetry measurements for this work were carried out using a SETARAM micro DSC III calorimeter. The instrument incorporates the use of two mixing batch vessel one of which acts as a reference during a trace. The vessels comprise a cylinder with two separate chambers which are separated by parts mounted on a rod which pass through the cylinder. The upper chamber has a capacity of 0.2 cm3 and the lower chamber has a capacity of 0.55 cm3. A composite o-ring on the obturator ensures the tightness of the upper chamber. These were removed after each experiment as in almost each case the o-rings would swell. The upper viton stopper ensures the tightness of the vessel and these did not need to be replaced as frequently. This method allows for non-invasive and non-destructive analysis of the samples and their interactions.All volumes were delivered using calibrated micro-syringes. All reagents were of 98% purity or greater and used as purchased from the supplier unless noted otherwise.
The multi-task software package provided by SETARAM recorded heat flow (thermal power) as a function of time from which ΔH values were determined by integration.
General procedure for determination of ΔHreaction for reactions (a)–(e)
For each set of experiments, the limiting reagent was injected into the lower chamber of the mixing vessel to ensure good mixing and completeness of reaction. Care was taken to avoid premature mixing of reactants (manufacturer supplied o-rings were replaced with o-rings of larger diameter) and, once a steady baseline was achieved, reaction was initiated by mixing of reactants on plunger depression. Baselines were modelled as either constant or sloping straight lines as appropriate. All experiments were conducted at 25 °C. On average all traces were completed within 3 h and each repeated at least three times.References
- For a recent review, see: F. Toda and K. Tanaka, Chem. Rev., 2000, 100, 1025–1074 Search PubMed.
- Some recent notable examples include: (a) H. Hagiwara, H. Nagamoto, S. Kazayama, H. Sakai, T. Hoshi, T. Suzuki and M. Ando, J. Chem. Soc., Perkin Trans. 1, 1999, 457–459 RSC; (b) G. Kaupp, J. Schmeyers, A. Kuse and A. Atfeh, Angew. Chem., Int. Ed., 1999, 38, 2896–2898 CrossRef CAS.
- B. M . Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- C. L. Raston and J. L. Scott, Green Chem., 2000, 2, 49–52 RSC.
- R. G. Sheldron, Chem. Industry, 1992, 903 Search PubMed.
- (a) G. W. V. Cave and C. L. Raston, Chem. Commun., 2000, 2199–2200 RSC; (b) G. W. V. Cave and C. L. Raston, J. Chem. Soc., Perkin Trans. 1, 2001, 3258–3264 RSC.
- J. L. Scott and C. L. Raston, Green Chem., 2000, 2, 245–247 RSC.
- W. H. Correa, S. Papadopoulos, P. Radnidge B. A. Roberts and J. L. Scott, Green Chem., 2002, 4, 245–251 RSC.
- J. L. Scott, D. R. MacFarlane, C. L. Raston and C. M. Teoh, Green Chem., 2000, 2, 123–126 RSC.
- B. A. Roberts, G. W. V. Cave, C. L. Raston and J. L. Scott, Green Chem., 2001, 3, 280–284 RSC.
- W. H. Correa and J. L. Scott, Green Chem., 2001, 3, 296–301 RSC.
- A. McCluskey, P. J. Robinson, T. Hill, J. L. Scott and J. K. Edwards, Tetrahedron Lett., 2002, 43, 3117–3120 CrossRef CAS.
- G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am Chem. Soc., 2001, 123, 8701–8708 CrossRef CAS.
- G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159–2169 RSC.
- Common cardiovascular drugs such as Felodipine, Nifedipine, Amlodipine and numerous others fall within this category.
- The first rapid peak may not be ascribed wholely to the reaction of benzaldehyde and piperidine to form the imminium intermediate but is, instead, the sum of this sharp exotherm, due to rapid formation of the imminium intermediate, superimposed on the slower second process, which we suggest should be ascribed to cycling of piperidine upon reaction with the active methylene compound (a process which will begin almost instantly once any imminium intermediate is formed, in this experiment). The energy released during the processes occurring before the inflection point (apparently 40% of the total energy) is thus not representative of imminium intermediate formation alone as the processes are not sequential but will occur concomitantly in this experiment. It is for this reason that the experiment was redesigned so that piperidine (or piperidine acetate) was added to the benzaldehyde derivative, which was, in turn, added to the active methylene compound.
- (a) L. F. Tietze and U. Beifuss, in Comprehensive Organic Synthesis, Pergamon Press, Oxford, New York, ed. B. M. Trost, 1991, vol. 2, 341–395 Search PubMed; (b) E. Knoevenagel, Chem. Ber., 1896, 29, 172–174 Search PubMed.
- J. Schmeyers, F. Toda, J. Boy and G. Kaupp, J. Chem. Soc., Perkin Trans. 2, 1998, 989–993 RSC.
- D. Saylik, C. R. Strauss, J. L. Scott, unpublished results.
- Note that the ratios of reactants are 1∶0.02∶0.1 benzaldehyde∶catalyst∶ethyl acetoacetate. Thus, the ratio of ethyl acetoacetate to piperidine is ca. 5∶1, but 1 mol of piperidine is already reacted with benzaldehyde thus the piperidine added will traverse the catalytic cycle ca. 4 times, hence one may reexpress the reaction enthalpy in terms of piperidine traversing the catalytic cycles as follows: (mol 2/(mol 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −mol piperidine) ×
ΔH and specifically for the two substrates tested in reaction (a): R = H: 1.48 × 10−4/(1.48 × 10−4−2.79 × 10−5) = −38 kJ mol−1; R = Cl: 1.31 × 10−4/(1.31 × 10−4−2.24 × 10−5) = −39 kJ mol−1.
−mol piperidine) ×
ΔH and specifically for the two substrates tested in reaction (a): R = H: 1.48 × 10−4/(1.48 × 10−4−2.79 × 10−5) = −38 kJ mol−1; R = Cl: 1.31 × 10−4/(1.31 × 10−4−2.24 × 10−5) = −39 kJ mol−1.
| This journal is © The Royal Society of Chemistry 2003 |