A rapid and accurate method for the determination of plutonium in food using magnetic sector ICP-MS with an ultra-sonic nebuliser and ion chromatography†
P.
Evans
*,
S.
Elahi
,
K.
Lee
and
B.
Fairman
LGC Ltd, Queens Road, Teddington, UK TW11 0LY. E-mail: Peter.Evans@lgc.co.uk
First published on 7th January 2003
Abstract
In the event of a nuclear incident it is essential that analytical information on the distribution and level of contamination is available. An ICP-MS method is described which can provide data on plutonium contamination in food within 3 h of sample receipt without compromising detection limits or accuracy relative to traditional counting methods. The method can also provide simultaneous determinations of americium and neptunium. Samples were prepared by HNO3 closed-vessel microwave digestion, evaporated to dryness and diluted into a mobile phase comprising 1.5 M HNO3 and 0.1 mM 2,6-pyridinedicarboxylic acid. A commercially available polystyrene–divinylbenzene ion chromatography column provides on-line separation of 239Pu and 238U reducing the impact of the 238U1H interference. Oxidation of the sample using H2O2 ensures all Pu is in the Pu+4 state. The oxidation also displaces Np away from the solvent front by changing the oxidation state from Np+3 to Np+4 and produces the insoluble Am+4 ion. Simultaneous Pu, Am and Np analyses therefore require omission of the oxidation stage and some loss of Pu data quality. Analyses were performed using a magnetic sector ICP-MS (Finnigan MAT Element). The sample is introduced to the plasma via an ultrasonic nebuliser–desolvation unit (Cetac USN 6000AT+). This combination achieves an instrumental sensitivity of 238U > 2 × 107 cps/ppb and removes hydrogen from the sample gas, which also inhibits the formation of 238U1H. The net effect of the improved sample introduction conditions is to achieve detection levels for Pu of 0.020 pg g−1 (4.6 × 10−2 Bq kg−1) which is significantly below 1/10th of the most stringent EU (European Union) legislation, currently 0.436 pg g−1 (1 Bq kg−1) set for baby food. The new method was evaluated with a range of biological samples ranging from cabbage to milk and meat. Recovery of Pu agrees with published values (100% ± 20%).
Introduction
Plutonium (Pu) is present in the environment due to the use and manufacture of nuclear weapons and from industrial incidents during the past 50 years. Pu comprises five isotopes of which 239Pu is the most significant. It has a half life of 24110 years and has multiple industrial and military applications.1 Pu represents a significant danger to health both in the short and long-term and it has become the focus of extensive legislation. Further artificial radionuclides of the actinide series such as americium (Am) and neptunium (Np) pose similar problems.In the event of a nuclear incident it is essential that rapid data is available concerning the distribution of radioactive particles of Pu, Am and Np to ensure that they do not enter the food chain. Current legislation imposes strict control over the maximum permitted levels (MPL) of Pu and Am in food.2 The most stringent MPL of Pu for the UK is in baby food where the level is set at 1 Bq kg−1 equivalent to only 0.436 pg g−1. At these concentrations the radioactivity is undetectable from background using field-portable spectrometers whilst laboratory based analyses are traditionally slow and complex. For example, α-spectrometry required sample digestion, ion separation and ion counting. It is capable of excellent precision but the lead-time from sample receipt to data analysis is around 3 days.3
In recent years ICP-MS has replaced many of the actinide analyses because of good sensitivity and improved sample throughput. However, conventional quadrupole ICP-MS has several disadvantages when measuring sub-pg abundance of Pu in the environment. Firstly, the instruments are not sufficiently sensitive without extensive sample pre-concentration (requiring ion separation) and secondly, the small 239Pu ion signal is interfered with by the ubiquitous presence of the polyatomic adduct 238U1H which cannot be resolved at the current levels of mass resolution technology.
Recent developments in ion chromatography permit U–Pu separation on-line for ICP-MS analysis.4 However, in order to achieve the required detection limits for food MPL a complex 2-column system has been used to pre-concentrate the artificial radionuclides prior to separation.5 Consequently, due to the complexity of the sample introduction, the technique has remained primarily academic and unsuitable for emergency situations.
A method is described, based upon the ion separation of Truscott et al.4 that overcomes the need for on-line sample concentrations by using the improved sample transmission efficiency from an ultra-sonic nebuliser attached on-line with an ion chromatography column and magnetic sector ICP-MS. The combined performance of the technique permits analysis of Pu and other actinides in food samples at levels that are significantly lower than 1/10th of the MPL.
From sample receipt to first sample analysis the new method requires approximately 3 h and then continues to yield 4 analyses per hour thereafter. This permits fast, high volume, low-level actinide analyses to underpin response to a nuclear incident.
Experimental
Instrumentation
All analyses were carried out using a Finnigan MAT Element magnetic sector ICP-MS. The Element has an argon plasma source coupled to a magnetic sector mass spectrometer with reverse Niers Johnson geometry. A differential entrance slit with 3 factory-set resolutions (10% valley definition) of 300 (low), 3000 (medium) and 7500 (high) permits greater separation of mass peaks.6 At low-resolution mode the Element has higher ion transmission than conventional ICP-MS and a flat topped peak geometry. The net effect is more accurate scanning of low intensity ion signals and improved detection levels.Samples were introduced to the plasma via an ultrasonic nebuliser–desolvation system (Cetac USN 6000AT+). Aqueous samples are nebulised by a transducer prior to the removal of water vapour and some volatile elements using an Ar desolvation membrane and transmission to the plasma.7 The effect is to improve sample introduction efficiency to the plasma by up to an order of magnitude whilst reducing the formation of hydride and oxide interferences that derive from the aqueous media.
Standard operating conditions for the plasma and nebuliser are shown in Table 1. The combined effect of the magnetic sector ICP-MS and the ultrasonic nebuliser is to yield instrument sensitivity for 238U of 2 × 107 cps ppb−1 whilst reducing hydride formation to <0.02%. This is particularly relevant to the accurate analysis of 239Pu where the interfering ion 238U1H cannot be resolved from 239Pu even when the instrument is operating in high resolution mode (requiring R > 37,000).
| Finnigan MAT Element magnetic sector ICP-MS | |
|---|---|
| Forward power/W | 1205 |
| Ar coolant gas flow rate/1 min−1 | 14 |
| Ar auxiliary gas flow rate/1 min−1 | 1.0 |
| Ar nebulizer gas flow rate/1 min−1 | 1.5 |
| Mass resolution (10% valley definition) | 300 |
| Samples per peak | 20 |
| Setting time/ms | 5 |
| Total analysis time per sample/min | 16 |
| Mass window | 20% |
| Intergration window | 80% |
| Scan mode | E-Scan |
| Detection mode | Dual (ion counting and analogue) |
| Cetac USN 6000AT+ ultrasonic nebuliser | |
|---|---|
| Heater 1/°C | 120 |
| Cooler/°C | 5 |
| Heater 2/°C | 140 |
| Ar sweep gas/1 min−1 | 2.75 |
| Chromatography | |
|---|---|
| HPLC pump rate/ml min−1 | 1.0 |
| Mobile phase | 1.5 mM dipicolinic acid (2,6-pyridinecarboxylic acid) |
| Loop volume/µl | 350 |
Chromatography
The chromatographic separation of the actinides is an essential stage in accurate Pu analysis. It is required to separate the small 239Pu signal from the 238U ion signal from which the interference 238U1H is derived. Even with the benefits of hydride reduction achieved using the desolvating nebuliser it is still necessary to minimise the contribution to the 239Pu signal by separating the peaks. In addition, the process of ion separation concentrates the minor actinide signals into a narrow window of integration which reduces the effects of baseline noise on detection limits.The chromatographic separation is modified from that of Truscott et al. (2001).4 A column was prepared from PLRP-S (PS–DVB) polystyrene–divinylylbenzene substrate (5 µm, Polymer Laboratories, UK) packed into a 100 × 4.6 mm id PEEK column. The mobile phase comprises 1.5 M HNO3 and 0.01 mM dipicolinic acid (2,6 pyridinedicarboxylic acid) (Aldridge, Dorset, UK). Interaction of the aromatic group of the dipicolinic acid and benzene in the substrate are thought to result in a dynamic equilibrium state. The +3 and +2 ions are not retained by the column.4 The effect is the separation of actinide elements and in particular the elution of uranium prior to the elution of Pu.
The ion column and ICP-MS were interfaced using a high-performance liquid chromatography pump, a Rheodyne model 9010 injection valve (Rheodyne, Cotati, CA, USA) and a 350 µl PEEK sample loop. Standard operating conditions are shown in Table 1.
The selection of a column procedure that only uses commercially available equipment and reagents was an essential step in the provision of a method that can be used in the event of a nuclear incident. Commercial columns are fully interchangeable therefore the measurement protocol does not have to differ between laboratories or in the event of contamination or column clogging. The method can be fully standardised and comply with normal accreditation procedures (e.g. BS EN ISO/IEC/17025).
Reagents
High purity acids and hydrogen peroxide were used throughout the experimentation to reduce the effects of blank contribution (Romil, UK). Radionuclide standards (239Pu, 243Am, 237Np and natural U) were obtained from Amersham Laboratories (Amersham, UK) and diluted to working concentrations by sequential gravimetric dilution in 1% HNO3. A mixed calibration standard was prepared by combining the radionuclides, heating in concentrated HNO3 and H2O2 to fix Pu, Am and Np into a single oxidation state (+4, +3 and +4 respectively) followed by sequential dilution to the required calibration concentrations in the mobile phase. U was included in the calibration blend to ensure realistic separation conditions and baseline noise was maintained throughout.Samples
There is a general paucity of available reference materials for validating actinide analysis in biological materials. Samples of dried, homogenised lamb, milk and cabbage were obtained that comprise part of the 1999 MAFF (UK Ministry of Agriculture Fisheries and Food) sponsored proficiency testing scheme of UK radiochemistry departments.8 The Pu content of the lamb, milk and cabbage are 0.525, 0.512 and 1.072 Bq kg−1 (equivalent to 0.229, 0.223 and 0.467 pg g−1 respectively). These materials are not CRMs but the extensive comparative information that is available makes them an ideal test of Pu analysis capability. The 239Pu content is close to the maximum permitted levels (MPL) for baby food (1 Bq kg−1) and significantly lower than the MPL for dairy produce and liquid foodstuffs (20 Bq kg−1).Bovine liver (SRM1566, NIST, Gaitherburg, USA) and rye grass (BCR 281, Geel, Belgium) were also used as part of this study. These material are not certified for actinide content. However, the homogenised form of these materials make them ideal candidiates on which to perform spike recovery tests and to extend the range of biological matrices in this study. Aliquots of the bovine liver, rye grass and the MAFF cabbage, lamb and milk were ‘spiked’ with known amounts of actinides (equivalent to 1.5 pg g−1) prior to sample digestion. Spike recoveries were calculated relative to results for un-spiked aliquots.
In selecting a series of reference materials the study has used a ‘worse case scenario’ for each matrix type. Where target detection levels are set they correspond to natural samples, which contain large amounts of water. The reference materials have been dried which means that the amount content of matrix components is higher than for the majority of ‘real’ samples, which will be dominated by water. Dried samples are harder to digest in a microwave. A technique that works for dried reference material will equally handle ‘real-world’ sample where the interference from the matrix is less.
The MAFF samples were calculated on a dry weight basis. This was achieved by separately drying 1 g aliquots of each of the materials at 85 °C for 24 h. The experimental results were then converted to a dry mass value prior to assessment. The moisture contents of the cabbage, milk, and lamb at the time of the experiments were determined to be 6.8%, 4.8% and 3.8% respectively.
The wider application of the technique was assessed by measuring the Pu recovery for SRM4353 (NIST, Rocky flats soil). This is a Pu enriched environmental matrix at Pu levels substantially higher than in the food samples (7.65 Bq kg−1, 3.3 pg g−1).
Sample digestion
Samples were mineralised by closed vessel microwave digestion using concentrated HNO3 as the reagent. Two different microwave systems were used to test the sample preparation method under varying laboratory conditions and to increase the sample throughput.Two microwave systems were used during this investigation both of which use sealed vessels to accelerate the digestion time by raising the pressure of reaction. This system also reduces the amount of reagent required and therefore constrained acid blank contribution. The CEM MDS 2000 (CEM instruments, USA) uses Teflon vessels whereas the Paar Multiwave (Perkin Elmer Instruments, Beaconsfield, UK) uses quartz vessels.
The first stage of the digest is to accurately weigh ∼0.5 g of sample into the digestion vessels. 5 g of concentrated HNO3 was added and the vessels sealed. Each microwave uses a stepped heating program from 0–600 W for the CEM microwave and 0–1000 W for the Multiwave. The total digestion time is 1.3 and 1 h respectively. Both digestion protocols leave a clear solution and no residue for all matrix types except for rye grass, which leaves a small residue of silicates. Following digestion the samples were transferred into pre-weighed Pyrex beakers and pre-concentrated by heating to dryness before re-dilution into the mobile phase. In the results section the role of the differing oxidation states of the actinides is discussed and the implications for designing an effective simultaneous actinide analysis. Where determination of Pu is the primary objective, the samples are oxidized using a mixture of concentrated HNO3 and H2O2 to leave a single Pu oxidation state (Pu4+), however, this leads to poor Am recovery. Good Am recovery is only obtained by omitting the oxidation stage.
From each sample digest a solution of ∼3 g is generated. It is therefore possible to split a digested sample and only oxidize one portion (for Pu analysis) retaining the second aliquot for a combined analysis of Np, Am and Pu. In addition, where rapid data is the imperative a digest is diluted into the mobile phase to provide the ultimate in rapid screening for all of the actinides where higher levels of contamination have been anticipated.
The Rocky flats soil SRM4353 was prepared by extraction of Pu into HNO3 by repeated heating of an aliquot with HNO3 on a hot plate. The extracted fluid was separated by centrifugation before evaporation to dryness after which the material was processed in the same way as the microwave preparations. A total digest of SRM4353 could not be achieved without using an HF acid blend followed by silicate removal, a significantly different and more time consuming method than for the biological samples.
Results
Actinide elution and the effect of oxidation states
The effect of oxidation with H2O2 is illustrated in Fig. 1. The elution profile of a 1 pg g−1 standard is shown in Fig. 1a after heating with HNO3. The effect of adding H2O2 to the heating step is shown in Fig. 1b. In Fig. 1a the Pu ion signal is split to both sides of the U elution. The first peak is the +3 ion which is poorly retained by the column and the second peak is the Pu4+ ion. Heating with peroxide oxidises all of the Pu to the +4 state providing a more satisfactory chromatographic conditions. In addition, the Np peak is displaced away from the solvent front. This is a potentially beneficial effect as the solvent front can be associated with complex polyatomic interferences and instrument detector noise. The figures shown represent standard solutions which are largely free of matrix interferences, greater noise is experienced for samples, as seen in Fig. 1c. The slight broadening of the Np peak permits accurate calculation of total signal intensity. The Am peak remains unchanged with regards to elution profile indicating that the oxidation state is unaltered. Fig. 1c shows the same elution profile for a complex matrix sample, cabbage after heating with peroxide during the sample preparation stage. The elution times are unaffected by the complex cabbage matrix and the Pu elutes after the main U signal. Based on a hydride formation of <0.02% the 238U1H ion signal at the point of maximum 239Pu elution is less than 10 cps, a negligible contribution to the peak area. The hydride formation was measured on a pure uranium standard monitoring the m/z 238/239 ratio. However, in matrix samples the Am appears to oxidise to the insoluble +4 ion and it is lost from the chromatographic separation.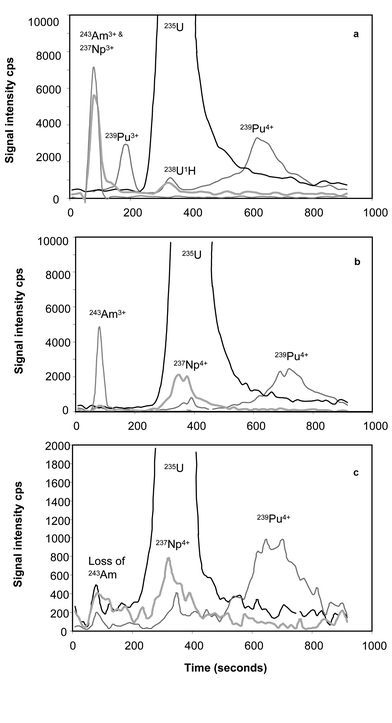 | ||
| Fig. 1 Chromatographic separation of Pu. 1a, 1 pg g−1 mixed actinide standard without oxidation of Pu and Np using H2O2. 1b, 1 pg g−1 mixed standard that has been oxidised with H2O2. The Pu forms a single peak and the Np shifts to a different elution point away from the solvent front. 1c, Elution profile of a MAFF cabbage sample that has been spiked with Am, Np and Pu at 1.5 pg g−1 prior to digestion. | ||
Optimum analysis of Pu and Np are therefore achieved by the application of H2O2 oxidation during the sample preparation stage. However, the trade-off is the potential loss of Am from solution. Each digest produces 2–3 ml of sample digest. Where the analyst is required for screening of all of the man-made actinides the sample does not require peroxide oxidation and information can be obtained for Am, Np and Pu simultaneously. A further sample, or sub-sample of the digest can be subsequently oxidised where more precise measurment of Pu and Np are required.
Detection limits and calibration
A calibration range was prepared by diluting a combined standard of 239Pu, 243Am, 237Np and natural uranium in a matrix of 1.5 M HNO3 and 0.01 mM dipicolinic acid (to give equivalent matrix conditions to the samples). Each analyte ion exhibits a linear calibration with an R2 linear regression of >0.99 and with zero intercept.Multiple measurements of a blank solution are used to calculate the detection limit in a typical sample as detailed in Table 2. The results are obtained from a blank standard that has undergone all of the sample preparation steps and as such will reflect the true detection limits of the method as opposed to basic instrument performance. The results for Pu are significantly better than the target detection limit of 0.1Bq kg−1 (10% of the MPL for baby food).2 A detection limit for 241Am can be interpolated from the measured results for 243Am (2.45 × 10−1 Bq kg−1). Assuming an equivalent mass detection limit (0.033 pg g−1) derives the activity detection limit is 4.2 Bq kg−1 which is a reflection of the short half life of 241Am (432 years). Unlike α-spectrometry the instrumental detection limits of ICP-MS do not differ significantly between isotopes as they are not a function of decay properties. Therefore, fast decaying isotopes are no easier to measure than the longer-lived ones and this will be reflected in relatively higher detection limits when quoted in terms of becquerels. The detection limit for 241Am is above the MPL for baby food but would be sufficient for normal foodstuffs. There is no Np specific legislation, however the detection limits are very low and could account for even a trace contribution (<20 fg g−1) where total α-emitting radiation data is required.
| Isotope | Detection limit in samples | |
|---|---|---|
| pg g−1 | Bq kg−1 | |
| 243Am | 0.033 | 2.45 × 10−1 |
| 237Np | 0.011 | 3 × 10−4 |
| 239Pu | 0.020 | 4.6 × 10−2 |
Sample analyses
The results for the analysis of MAFF lamb, milk and cabbage are shown in Table 3 along with data for SRM4353 rocky flats soil. The results are in agreement with the MAFF trial with an average Pu recovery for the three sample types that is in agreement with the trial values. The standard deviation of the measurements based upon 7–9 replicate analyses is 12–32% increasing for the lower level samples but comparable to data quality typical of significantly more abundant analytes. The recovery for SRM4353 agrees well with certified values and previous analyses.5 The standard deviation of the mean is smaller than for the food materials (despite the small number of replicates) which indicates how data precision improves with greater ion signal intensity.Table 4 details the recovery of biological samples spiked with Np and Pu equivalent to 1.5 pg g−1 in the sample. All samples underwent H2O2 oxidation to yield a single oxidation state of Pu and to displace the Np away from the solvent front. All samples, with the exception of rye grass, have a recovery that is indistinguishable from 100% at a precision of ±20%. Low recovery for rye grass may be a function of adsorption of actinides onto the residual silicate phase left after digest. Samples were also spiked with Am. As discussed previously, the Am is lost in the oxidation method and therefore this data is not shown. However, samples of milk that were not oxidised yield a 243Am recovery of 120%. The resultant trade-off is a loss of Np and Pu accuracy, reducing recovery in this small sub-sample to 37 and 56% respectively.
| Matrix type | Source | No. replicates | Pu spike recovery (1.5 pg g−1) (%) | Np spike recovery (1.5 pg g−1) (%) |
|---|---|---|---|---|
| Cabbage | MAFF | 4 | 107 | 106 |
| Milk | MAFF | 2 | 111 | 116 |
| Lamb | MAFF | 3 | 110 | 106 |
| Bovine liver | SRM1566 | 4 | 97 | 113 |
| Rye grass | BCR218 | 6 | 72 | 73 |
No difference was observed between the samples prepared in the Teflon and quartz microwave vessels. This indicates a robust digestion protocol that is transferable between laboratories.
A time-line for providing rapid analytical data for Pu in food
Using the sample preparation, separation and analysis protocol outlined in this paper the analysis of actinides in food samples can be achieved within 3 h of sample receipt. Fig. 2 is a hypothetical time-line of the operations required to provide actinide analyses. The model assumes that the instrument needs to be turned on, tuned and configured for the column separation conditions, as would be realistic requirements in the event of an incident. The model is based upon 2 staff members working as a team, one operating the ICP-MS and the other processing samples. The process shown is for a fully quantitative analysis. The time-scale of analysis can be further reduced by opting for a rapid screening variant in which the digested sample is diluted in mobile phase and injected directly into the ion-chromatography-ICP-MS system. Under these conditions the Pu signal may be split between the +3 and +4 oxidation states thus reducing overall precision. However, a simultaneous measurement of Am, Pu and Np is possible from which the most critical samples can selected for fully quantitative analysis.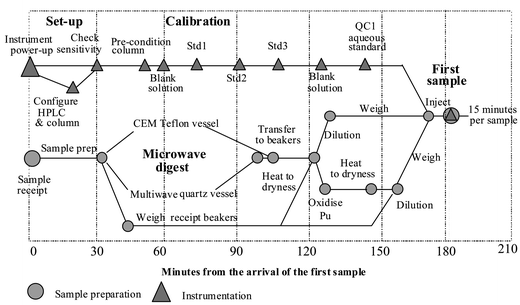 | ||
| Fig. 2 A time-line diagram analysis of actinide analysis under ‘emergency’ conditions. | ||
Upon receipt of the samples the SF-ICP-MS instrument is connected to the ultrasonic nebuliser and HPLC column. After standard instrument checks for sensitivity (during which time the column is being conditioned with the mobile phase) the instrument begins to analyse standards. The instrument is analysing the first blank standard within the first hour. A full calibration takes a further 90 min. Once calibrated the instrument can provide 4 analyses per hour (samples or standards).
The role of the second staff member is to prepare the samples. Samples are prepared by closed vessel microwave digestion in batches of 6–8. Depending upon the requirements of the incident samples can be screened for actinides or dried and oxidised to give full quantitative analysis of Pu and Np. The first sample is fully quantified for Pu within 3 h of receipt.
Acknowledgements
This work was funded by the UK Food Standards Agency.References
- CRC Handbook of Chemistry and Physics, Chapman and Hall, 2001 Search PubMed.
- EU regulation 3954/87 (Euratom).
- ASTM method C1001-00 Standard Test Method for Radiochemical Determination of Plutonium in Soil by Alpha Spectroscopy Search PubMed.
- J. Truscott, P. Jones, B. Fairman and E. Evans, J. Chromatogr., A, 2001, 91–98 CrossRef CAS.
- J. Truscott, P. Jones, B. Fairman and E. Evans, Anal. Chim. Acta., 2001, 433(2), 245–253 CrossRef CAS.
- U. Giessmann and U. Greb, Fresenius’ J. Anal. Chem., 1994, 350, 186–193 CrossRef CAS.
- Inductively Coupled Plasma Mass Spectrometry, ed. A. Montaser, Wiley, Chichester, 1999 Search PubMed.
- MAFF Proficiency Testing Scheme for the Measurement of Radionuclides in Foods RP0319 1999 Search PubMed.
Footnote |
| † © Copyright LGC Limited 2003. |
| This journal is © The Royal Society of Chemistry 2003 |