Application of a compact all solid-state laser system to the in situ detection of atmospheric OH, HO2, NO and IO by laser-induced fluorescence
William J.
Bloss
,
Thomas J.
Gravestock
,
Dwayne E.
Heard
*,
Trevor
Ingham
,
Gavin P.
Johnson
and
James D.
Lee
Department of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK LS2 9JT. E-mail: dwayneh@chem.leeds.ac.uk
First published on 29th November 2002
Abstract
A tuneable, high pulse-repetition-frequency, solid state Nd:YAG pumped titanium sapphire laser capable of generating radiation for the detection of OH, HO2, NO and IO radicals in the atmosphere by laser induced fluorescence (LIF) has been developed. The integration of the laser system operating at 308 nm into a field measurement apparatus for the simultaneous detection of hydroxyl and hydroperoxy radicals is described, with detection limits of 3.1 × 105 molecule cm−3 (0.012 pptv in the boundary layer) and 2.6 × 106 molecule cm−3 (0.09 pptv) achieved for OH and HO2 respectively (30 s signal integration, 30 s background integration, signal-to-noise ratio = 1). The system has been field tested and offers several advantages over copper vapour laser pumped dye laser systems for the detection of atmospheric OH and HO2 radicals by LIF, with benefits of greater tuning range and ease of use coupled with reduced power consumption, instrument footprint and warm-up time. NO has been detected in the atmosphere at ∼ 1 ppbv by single photon LIF using the A 2Σ+ ← X 2Π1/2 (0,0) transition at 226 nm, with absolute concentrations in good agreement with simultaneous measurements made using a chemiluminescence analyser. With some improvements in performance, particularly with regard to laser power, the theoretical detection limit for NO is projected to be ∼ 2 × 106 molecule cm−3 (0.08 pptv). Whilst operating at 445 nm, the laser system has been used to readily detect the IO radical in the laboratory, and although it is difficult to project the sensitivity in the field, an estimate of the detection limit is < 1 × 105 molecule cm−3 (< 0.004 pptv), well below previously measured atmospheric concentrations of IO.
1. Introduction
The composition, physical and chemical properties of the atmosphere are controlled by reactions of atmospheric species present at trace concentrations through their participation in catalytic reaction cycles. Assessment of the impact of such reactions requires measurement of the abundance and distribution of trace constituents, achieved through a variety of remote sensing and in situ techniques. One such constituent is the hydroxyl radical, OH, which is the principal oxidising species in the troposphere,1 driving the removal and hence abundance of many species emitted into the atmosphere through natural and anthropogenic processes. OH is highly reactive, with a chemical lifetime in the boundary layer ranging from ∼ 0.1–1 s, and typical maximum boundary layer concentrations only reaching (1–5) × 106 molecule cm−3 (0.04–0.2 pptv (parts per trillion by volume)). Quantitative detection of OH radicals at these levels is essential to understand the chemistry of the troposphere and to validate models of (for example) hydrocarbon oxidation and urban ozone production.2Measurements of in situ OH concentrations have been reported using mass-spectrometric,3 radiocarbon oxidation,4 long-path optical absorption5 and wet chemical titration6 methods, however the most widely used technique is laser-induced fluorescence (LIF), in particular the FAGE (fluorescence assay by gas expansion) methodology.7–11 In such experiments, the on-resonance fluorescence resulting from laser excitation of the (0,0) band of the OH A 2Σ+ ← X 2Πi transition near 308 nm is measured to determine [OH]. A low-pressure gas expansion is used to extend the OH fluorescence lifetime beyond the duration of the excitation laser pulse, allowing the detector to be switched off through electronic gating during the laser pulse, thus minimising collection of scattered laser light.12 An alternative fluorescence scheme is to employ excitation of the (1,0) band of the OH A 2Σ+ ← X 2Πi transition near 282 nm, with collection of red-shifted fluorescence near 308 nm. Although successfully employed in the stratosphere,13 such a scheme is not used in the troposphere due to the generation of artefact OH in the instrument, arising from the 282 nm photolysis of ambient ozone and subsequent reaction of O(1D) with ambient water vapour.14 Even at 308 nm the laser pulse energy has to be low to minimise O3 photolysis and to avoid optical saturation of the OH transition. A high pulse-repetition-frequency (PRF), of the order of several kHz, is therefore required to obtain sufficient signal to detect the extremely low atmospheric OH concentrations on a suitably short timescale. Although all-solid-state laser systems have been developed in the laboratory for the detection of OH,15,16 the 308 nm radiation required for field measurement of atmospheric OH by LIF has hitherto been generated via frequency doubled dye laser systems, with the high PRF provided by pumping the dye with copper-vapour lasers (CVL)7–10 or solid state Nd:YAG (neodymium:yttrium-aluminium-garnet) lasers.11,13,17 The CVL/dye laser systems exhibit disadvantages of complexity and wet chemical requirement (dyes) and large bulk (∼ 3 × 2 × 0.5 m instrument footprint), high power consumption (5 kW with the requirement of three-phase power), and long warm-up time (∼ 1 h).
In this paper we describe the implementation of an alternative all-solid-state Nd:YAG-pumped frequency tripled Ti:Al2O3 (Ti:Sapphire) laser system (section 2), and the integration of this laser into a FAGE instrument for the detection of atmospheric OH and HO2 (section 3). The wavelength tuning range accessible with such a system also facilitates the detection of other molecular species by LIF. This possibility has been investigated for two further atmospheric trace species: IO and NO.
Iodine oxide species may have a significant impact upon the atmospheric chemistry of the marine boundary layer (MBL), through their participation in catalytic ozone destruction cycles,18 their involvement in autocatalytic halogen release19 from the condensed phase and their potential role in forming aerosol particles which may be a significant source of cloud condensation nuclei.20 IO radicals have been observed in the MBL using differential optical absorption spectroscopy (DOAS) at concentrations of up to 6 pptv;21,22 however as DOAS absorption paths typically cover several km of both ocean and shoreline environments, and the intertidal zone is known to be a major source area for short-lived iodocarbons, the importance of open ocean versus shoreline iodine chemistry is the subject of debate. Local, in situ measurement of IO concentrations, via LIF, should assist in resolving the importance of iodine chemistry throughout the MBL.
The nitrogen oxides NO and NO2, collectively termed NOx, play a major role in atmospheric chemistry, notably in the oxidation of volatile organic compounds (VOCs) in the troposphere.23 Under very “clean” (low NOx) conditions, tropospheric ozone is destroyed by photolysis, whereas at higher levels of NOx, ozone production arising from VOC oxidation can occur. The point at which chemical production and destruction of ozone is balanced (the so called compensation point) occurs at a NOx mixing ratio of approximately 15–30 pptv in remote marine air.24,25 Accurate measurement of NOx levels at such low concentrations is of importance for modelling of ozone levels and hence calculation of the oxidising capacity of the troposphere. NO and NO2 can be readily interconverted, by photolysis or reaction with O3, thus a method for detection of either species can be used to determine atmospheric concentrations of both constituents, and hence [NOx].
Atmospheric NO has hitherto almost exclusively been detected by chemiluminescence techniques26,27 in which the fluorescence from electronically excited NO2 formed following the NO + O3 reaction is detected. State-of-the-art chemiluminescence analysers can achieve a detection limit of around 1 pptv, but can be subject to artefact interference at low concentrations (< 20 pptv). A two-photon LIF technique28 has been used for the detection of NO and related nitrogen oxide species (e.g. NO2 and organic nitrates). Laser radiation at 226 nm first excites NO to the A 2Σ state, and a second photon from another laser system operating at 1.1 µm excites NO further to the D 2Σ state. Fluorescence accompanying relaxation back to the X 2Π state is then observed from 187 to 220 nm. The two-photon method offers good signal-to-noise performance as the fluorescence is blue-shifted relative to both laser-excitation wavelengths, minimising the level of scattered laser light detected. However, this system suffers from large bulk, weight and complexity. In this work single photon excitation through the A 2Σ←X 2Π1/2 γ-band at 226 nm is used to generate NO LIF.
Section 4 of this paper describes the application of the Ti:Sapphire laser system to the measurement of IO and NO by laser induced fluorescence. Laboratory detection of each species has been demonstrated, and preliminary measurements of atmospheric NO in the urban boundary layer are reported.
2. The Nd:YAG–Ti:Sapphire laser system
The pump laser beam is provided by a diode-pumped, Q-switched, intra-cavity frequency doubled Nd:YAG laser (Photonics Industries DS 20-532), capable of producing ca. 12 W of 532 nm radiation when operated at a pulse repetition frequency of 5 kHz, with a typical pulse length of 25 ns and a beam diameter of approximately 1 mm. The diodes are fibre-coupled to the Nd:YAG cavity facilitating field replacement. The Nd:YAG second harmonic at 532 nm is used to pump a Ti:Sapphire laser, producing broadband near-IR radiation in the range 690–1000 nm (Photonics Industries TU-UV 308). The desired Ti:Sapphire oscillator wavelength is selected using a diffraction grating whose incident angle is computer controlled with a pico-motor driver (New Focus 8732). The grating and laser cavity design give a TEM00 beam profile with the number of longitudinal modes (defining the laser linewidth) such that following harmonic generation there is efficient overlap with the spectral profile of the OH A 2Σ+ ← X 2Πi (0,0) transition at 308 nm under the FAGE measurement conditions. The near-IR fundamental output of the Ti:Sapphire laser is passed through a flexible arrangement of two non-linear harmonic generation stages, consisting of two cerium lithium borate (CLBO) crystals separated by a half wave plate for intermediate control of the laser polarisation. In addition to the fundamental Ti:Sapphire wavelength range (690 – 1000 nm) the following wavelengths can also be generated: (a) 440–500 nm via second harmonic generation, (b) 230–330 nm through third harmonic generation via sum frequency mixing of the fundamental and second harmonic, and (c) 200–250 nm via fourth harmonic generation. The ranges are determined by the wavelength dependence of the Ti:Sapphire output power, controlled by the choice of grating and the coatings on the Ti:Sapphire cavity optics, and the particular cut-angles of the non-linear crystals. For fourth harmonic generation a non-linear crystal specifically designed for the Ti:Sapphire laser was not available and so a generic type-II β-barium borate (BBO) crystal was used. During scanning of the fundamental laser wavelength (via rotation of the grating) the laser is specified to automatically control the phase-matching angles of the non-linear crystals to ensure optimum output at the wavelengths of the higher harmonics. Approximate (± 0.001 nm) wavelength calibration is performed by splitting a small fraction of the second harmonic (blue) radiation into a wavemeter (Coherent Wavemaster). A more precise wavelength calibration is obtained through the use of a reference cell containing the particular species being measured, and using tabulated line positions of individual transitions. The overall dimensions of the YAG laser, Ti:Sapphire laser and the power supply units are approximately 600 × 200 × 150, 650 × 360 × 190 and 480 × 470 × 180 mm, respectively (L × W × H), with a total weight of 50 kg. In addition to these, a small chiller is used to cool the housings of the solid state optical units and maintain a constant temperature (21 °C). The total power consumption (including chiller) is approximately 1 kW, single phase.For OH excitation at 308 nm, the Ti:Sapphire oscillator is operated at 924 nm. The 924 nm radiation is frequency tripled via two non-linear stages, using the first CLBO crystal to generate the second harmonic at 462 nm, and the second to perform sum-frequency mixing of this wavelength with the fundamental (924 nm) to obtain the desired 308 nm radiation. The system can produce up to 150 mW of UV radiation at 308 nm, when operated at a PRF of 5 kHz, equivalent to 30 µJ per pulse. The UV beam is approximately 3 mm in diameter, with a typical pulse length of 35 ns, and a spectral bandwidth of 0.065 cm−1. The laser bandwidth is well matched with the lineshape of a single OH rotational transition that has a Doppler-broadened profile of 0.08 cm−1 under normal FAGE measurement conditions. All average laser power measurements were performed with a calibrated joulemeter (Molectron PM10).
3. Detection of atmospheric OH and HO2
The Leeds FAGE instrument has been described in detail previously10,12,29 therefore only a brief description is given here. The system consists of an evacuated fluorescence chamber into which ambient air is drawn through a 1 mm diameter flat nozzle where it undergoes a supersonic expansion. The chamber pressure is maintained at 1 Torr using a pumping system consisting of a Roots blower backed by a rotary pump. The laser excitation and perpendicular fluorescence detection axes intersect the gas expansion approximately 80 mm below the nozzle in the subsonic region. LIF from OH is collimated by a pair of anti-reflection (AR) coated plano-convex lenses, passed through an interference filter (transmission > 50% at 308 nm with very high rejection (transmission < 10−6) at other wavelengths) and focused using another pair of AR coated plano-convex lenses onto the cathode (∼ 8 mm diameter) of a channeltron photomultiplier tube (PMT) (Perkin Elmer C943P). This PMT is much smaller than the 14-dynode end-window device previously used in the FAGE instrument.10,12 The solid angle of fluorescence collection is approximately doubled with a spherical mirror mounted opposite the collimating optics. The PMT is switched off immediately prior to and during the laser pulse using a modification of a previously reported gating circuit developed at Leeds12 to apply a 100 V positive voltage to the cathode relative to the channeltron body. OH fluorescence is recorded by photon-counting during a 500 ns wide integration window, commencing 100 ns after the start of the laser pulse. The delay accounts for the laser pulse width (ca. 35 ns) and the PMT gating circuit time response (ca. 50 ns). A second integration window, delayed 50 µs after the laser pulse, is used to measure the signal arising due to scattered solar radiation through the nozzle, for subsequent subtraction.In the original configuration of the FAGE instrument laser radiation was delivered to a single fluorescence cell using a series of laser steering mirrors and a focusing lens.10 In a recent modification the laser radiation is now delivered to two fluorescence cells through a fibre optic system. Beamsplitters are used to divide the 308 nm radiation emerging from the laser into three fractions: 60%, 35% and 5%, for the OH, HO2 and reference cells respectively. The first two fractions are focused into fibre optic cables (Oz Optics QMMJ-55-UVVIS) using micrometer positioned coupling mounts (Elliot Scientific MDE511-SPEC) for transmission to the ambient measurement cells. Fibre transmission is approximately 60% for the 5 m lengths used, with the majority of losses occuring at the entrance and exit stages. Light emerging from the fibres is collimated and directed into each cell through a baffled side arm, giving a 10 mm beam diameter at the fluorescence excitation region. The magnification ratio of the fluorescence collection optics (0.75) provides a good match with the PMT cathode diameter, and minimises the collection of laser scattered light that originates away from the overlap of the laser and molecular beams. After traversing the cell, the laser beam exits through a second baffled side arm, via a Brewster window, and is directed onto a UV photodiode, to permit subsequent normalisation of the LIF signal for fluctuations in laser power. The third fraction (5%) of the 308 nm radiation from the laser is passed through a reference cell in which a relatively high concentration of OH is generated in a microwave discharge through humidified air at reduced pressure. The signal from this cell provides a wavelength calibration and is used to lock the laser wavelength to the centre of a particular OH transition.
Whilst measuring [OH] in ambient air, the data acquisition sequence begins by locating the peak of the OH Q1(2) rotational line via the reference cell signal. The laser is then held at this wavelength for a period of (typically) 30 s to 5 min, during which the OH fluorescence signal (with the solar-scattered signal subtracted) is recorded. The wavelength is then stepped off the centre of the OH line to a closely adjacent value where there is no absorption by OH, and the signal (arising solely from scattered laser light) measured for subsequent subtraction from the OH LIF signal.
A second LIF cell is used to simultaneously measure [HO2], detected as OH following its chemical conversion by the addition of NO through an injection ring positioned concentrically around the expansion jet in the subsonic region:
| HO2 + NO → OH + NO2 | (1) |
The instrument is calibrated through the 184.9 nm photolysis of humidified air, leading to the formation of OH and HO2 in equal concentrations, quantified relative to the concomitant formation of O3 from O2 photolysis:
| H2O + hν (184.9 nm) → H + OH | (2) |
| H + O2 + M → HO2 + M | (3) |
| O2 + hν → O + O | (4) |
| O + O2 + M → O3 + M | (5) |
| S = CXP [X] | (i) |
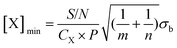 | (ii) |
 | (iii) |
If the experimental parameters of a particular instrument are known for detection of a given species, then the theoretical LOD can be determined by calculation of CX using:
 | (iv) |
 | ||
| Fig. 1 A laser-excitation spectrum of OH in the A 2Σ+ (v′ = 0) ← X 2Π3/2(v″ = 0) band showing a single rotational transition (Q1(1) and its satellite Q1′ (1)), obtained by scanning the output of the Nd:YAG-pumped Ti:Sapphire laser system. The wavelength increment was 0.0002 nm, and at each wavelength point the signal was averaged for 1 s (5000 laser shots). The 308 nm radiation was generated by frequency doubling the 924 nm Ti:Sapphire fundamental wavelength to 462 nm, followed by frequency mixing of the 462 nm radiation with residual 924 nm. Comparison of the ratio of the relative peak intensities with the corresponding ratio of Einstein B coefficients shows that at the laser power used (∼ 30 mW) the transitions are not subject to significant optical saturation. The measured full-width at half-maximum of each peak and the calculated Doppler-broadened linewidth for OH give a laser-linewidth at 308 nm of 0.065 cm−1. | ||
 | ||
| Fig. 2 A diurnal profile of the OH concentration (points) and rate of production of O(1D) from ozone photolysis, J(O1D), (solid line) recorded at the Mace Head Atmospheric Research Station (53.19° N, 9.54° W) on 30th July 2002 during the NAMBLEX field campaign. Each OH point represents an average of all data recorded in a given 240 s (4 min) time period. The sequence of data acquisition for each of these periods is: (i) 40 s peak search, (ii) 5 × 20 s OH data and (iii) 5× 20 s laser-background. The gap in the data around 06:00 UT corresponds to an instrument calibration, from which a detection limit of ∼ 3 × 105 molecule cm−3 was obtained. The laser power in the centre of the fluorescence cell was 20 mW, and each point has been normalised for laser power, as measured after the cell using a photodiode. The J(O1D) measurements were made every 60 s using a filter radiometer. | ||
| Factor | Units | OH | IO | NO |
|---|---|---|---|---|
| a For IO B, N(v”,J”)/N and ϕ are considered together. Using measured absorption cross-sections reported by Atkinson et al.40 the product B × (N(v′′, J′′)/N) × ϕ is calculated to be 2.5 × 1020 cm3 J−1 s−2. b counts s−1 mW−1 molecule−1 cm3. c For a total integration time of 60 s, i.e.m = n = 1, t = 30 s. Background signals: Slb = 30 (OH), 1000 (IO), 20 (NO) counts s−1; Ssb = 50 (OH) 500 (IO) and 20 (NO) counts s−1; Sdb =0 counts s−1 (all species) | ||||
| Electronic transition | A 2Σ+ ← X 2Π3/2 (0,0) band Q1(2) | A 2Π3/2 ← X 2Π3/2 (2,0) R1 bandhead | A 2Σ+ ← X 2Π1/2 (0,0) band Q1(5) | |
| Approx. wavelength | nm | 308.00 | 444.89 | 226.18 |
| Fluorescence cell pressure | Torr | 1 | 400 | 4 |
| Einstein B coefficient | cm3J−1s−2 | 9.09 × 1023 | a | 1.905 × 1023 |
| Sum of laser and Doppler linewidths (ΔνD2 + ΔνL2)1/2 | cm−1 | 0.16 | 0.07 | 0.13 |
| Population of laser excited level N(v”,J”)/N | 0.15 | a | 0.032 | |
| Length of laser and molecular beam overlap, l | cm | 0.35 | 0.35 | 0.35 |
| Fluorescence collection efficiency, ε | 0.125 | 0.125 | 0.125 | |
| PMT quantum efficiency, η | 0.18 | 0.15 | 0.12 | |
| Transmission efficiency of optics, T | 0.49 | 0.25 | 0.22 | |
| Fraction of fluorescence collected, fgate | 0.70 | 1 | 0.54 | |
| Fluorescence quantum yield, ϕ | 0.403 | a | 0.40 | |
| Ratio of air densities inside and outside cell, (ρin/ρout) | 9.2 × 10−4 | 0.53 | 5.26 × 10−3 | |
| Sampling efficiency, γsampling | 1 | 1 | 1 | |
| Calibration factor, CX | b | 8.9 × 10−7 | 3.2 × 10−6 | 6.4 × 10−8 |
| Laser power, P | mW | 30 | 50 | 20 |
| Calculated detection limit, [X]minc | cm−3 (pptv) | 8.6 × 104 (0.004) | 6.2 × 104 (0.0025) | 1.7 × 106 (0.07) |
4. Application to atmospheric measurements of other trace constituents
The broad wavelength tunability of the Ti:Sapphire crystal, coupled with a flexible choice of non-linear harmonic generation stages, enables the laser system to be readily re-configured to produce a variety of different wavelengths for the detection of trace atmospheric constituents other than OH and HO2 using the LIF technique. Such an ability has been explored for the detection of IO and NO radicals.(a) Detection of IO radicals near 445 nm
LIF from IO has been observed previously following excitation in the (0,0), (2,0), (2,1) and (3,0) bands of the A 2Π3/2 ← X 2Π3/2 transition between 435 and 468 nm.30 In this work the (2,0) band near 445 nm was selected, following consideration of the wavelength dependent laser power, IO absorption cross-section and the excited state fluorescence lifetime. The A 2Π3/2 state of IO is highly predissociative, with many of the (v′,0) bands exhibiting no structure in their absorption spectra due to the extremely short lifetime of the A 2Π3/2v′ state. The lifetime of the v′ = 2 state is relatively long (up to 100 ps, the value being rotational level dependent),31 and the absorption spectrum of the (2,0) band is fully rotationally resolved, and is therefore suitable for the detection of IO by LIF. The required 445 nm radiation was readily obtained by operating the Ti:Sapphire oscillator at 890 nm, followed by a single second harmonic generation stage using the same CLBO crystal as for operation at 924 nm. As 890 nm is towards the end of the useful tuning range of the Ti:Sapphire crystal, only approximately 150 mW of blue light was obtained at 5kHz PRF. The laser linewidth at 445 nm was estimated to be 0.07 cm−1 from measured IO spectra, compared to an IO Doppler linewidth of 0.023 cm−1 at 295 K. IO experiences very little Doppler broadening as a result of its high molecular mass but the laser linewidth is still sufficiently narrow to achieve good spectral resolution with negligible background arising from overlap with multiple lines.IO radicals were produced in the reference cell through the reaction of O atoms, generated from a microwave discharge through synthetic air, with either CF3I (taken direct from a pressurised cylinder and diluted in nitrogen) or CH3I (entrained from the liquid in a flow of nitrogen). The reference cell pressure was maintained at approximately 6 Torr. IO was excited in the (2,0) band and fluorescence was detected off resonance at ca. 500–600 nm from several vibrational bands of the A 2Π3/2 → X 2Π3/2 electronic transition. The fluorescence signal was distinguished from laser and solar scatter using a Schott glass GG-495 filter with a long-pass cut-on wavelength of ∼ 500 nm. A typical laser excitation spectrum of the IO (2,0) band LIF in the region near the bandhead is shown in Fig. 3, together with rotational assignments. As the excited state lifetime is shorter than the laser-pulse, the photon counting gate was set to directly overlap temporally with the laser pulse. The LIF signal from IO is not appreciably quenched by air, and the fluorescence quantum yield is not expected to show any marked pressure dependence. The IO radical concentration in the reference cell was very uncertain, but was estimated to be approximately 1012 molecule cm−3 for the laser excitation scan shown in Fig. 3, that exhibits an excellent signal-to-noise ratio of > 500. Preliminary experiments using a quick modification of the FAGE fluorescence cell to detect IO instead of OH in the laboratory were unsuccessful. Attempts to generate IO at atmospheric pressure, using the photolysis of a flow of O2 at 184.9 nm to produce O(3P) atoms in the presence of organic iodide precursors, did not yield any LIF signal ascribable to IO when the radical source was sampled by the FAGE instrument. The most likely explanation is that the IO radicals were removed between generation and the laser excitation region as a result of reaction with the precursors used. Calculation of the theoretical sensitivity for IO viaeqn. (iv) (see Table 1) gives an excellent detection limit of 6 × 104 molecules cm−3 (0.003 pptv), supporting the postulate that IO radicals were lost prior to the laser-excitation region. Despite not being able to observe IO in the main FAGE cell in the laboratory, an opportunity arose during the NAMBLEX field campaign to make an attempt to measure IO in the marine boundary layer at Mace Head, Ireland, where previously IO had been detected under favourable conditions.21,22 The calculated detection limit is more than adequate to detect IO radicals at the several pptv level anticipated for the coastal MBL during sunlit conditions. However, no signal from IO could be distinguished from the laser background signal, which was much higher (> 1000 counts s−1) than in the case of OH detection. The random variation in the laser background signal was too large to enable any IO signal to be observed when the wavelength was tuned alternatively on and off an IO transition. Shading of the nozzle from sunlight resulted in a solar scattered signal that was considerably smaller than the laser scattered signal, and was not thought to be a limiting factor. Assuming that IO radicals are transmitted by the sampling nozzle, the limiting factor appears to be the high laser background, the fluctuations of which are significantly larger than any signal expected from IO. Future development of the IO detection scheme will include construction of a separate field measurement cell with fibre delivery, laser beam collimation and fluorescence collection (optimised for longer wavelengths) that is designed specifically for the detection of IO. A high quality interference filter to effectively block laser scattered light yet transmit a high fraction of the IO LIF will be employed. Finally it is possible that clustering or polymerisation of iodine oxide species could be responsible for reducing the IO radical concentration after sampling at the nozzle but prior to excitation in the LIF region of the cell. While such polymerisation has been reported in laboratory experiments20 the transit time for IO radicals from the nozzle to detection region was short (approximately 0.7 ms), thus (for example) IO self-reaction kinetics,32 proceeding with a near-collisional rate coefficient of 8.6 × 10−11 molecule−1cm3s−1, result in negligible reduction in the IO concentration at anticipated atmospheric levels of 5–10 pptv. It is envisaged that future laboratory experiments will investigate the IO LIF signal, under constant [IO]nozzle, for various cell pressure/residence time/humidity conditions to test for any such behaviour.
 | ||
| Fig. 3 Laser-excitation spectrum of IO in the A 2Π3/2(v′ = 2) ← X 2Π3/2(v″ = 0) band, together with rotational assignments in the region of the (2,0) bandhead, recorded in the reference cell at a pressure of 6 Torr, T = 298 K. Each point represents 1 s averaging (5000 laser shots), with 100 mW laser power and 0.065 cm−1 laser linewidth. The unidentified lines correspond to the weak Q branch of the Π–Π transition. The most intense peak is the R1 bandhead consisting of three blended rotational transitions, and was chosen for attempts to measure IO in the main FAGE fluorescence cell. | ||
It is important to consider any potential interference or artefacts in the measured LIF signal that may arise from the excitation of other fluorescing species, or from the photolysis of atmospheric precursors to generate additional IO that is then excited by the laser. As the gas flow rate in the excitation region of the chamber is sufficiently fast to ensure that a fresh sample of gas is present for successive laser pulses, in order to constitute an artefact signal any IO must be generated by photolysis and excited to generate fluorescence in the same laser pulse. For IO detection at 445 nm, photolysis of alkyl iodides followed by the I + O3 reaction could form IO, but CH3I (σ < 10−23 cm2 molecule−1)33 and other simple RI species do not absorb significantly at 445 nm. In addition the I + O3 reaction is slow, and any I atoms formed would be converted to IO on a very long timescale at ambient levels of ozone compared to the length of a single laser pulse. Of the potential direct IO photolytic precursors, only IONO2 and possibly I2O2 could contribute to the detected IO signal. HOI photolyses to give I + OH,34 OIO has no observed absorption below 482 nm (and is not thought to photolyse to O + IO),35 and INO2 concentrations and 445 nm absorption cross sections are too low to be significant. Even at 75 pptv IONO2, and assuming 100% yield for IO from IONO2 + hν (445 nm) and σ(IONO2) = 5 × 10−19 cm2 molecule−1 (the 445 nm cross section is (1–2) × 10−19 cm2 molecule−1)36 only 0.08 pptv IO production is calculated for the FAGE laser parameters. I2O2 concentrations in the MBL are expected to be much lower than those of IONO2 (<10 pptv) and the estimated I2O2 cross section at 445 nm is 2.5 × 10−19 cm2 molecule−1,32 thus little IO production will result from photolysis of this species. Moreover it is expected that upon photolysis of these species IO will be generated with a rovibrational distribution that is considerably more excited than the thermal distribution of sampled ambient IO, and as the number of collisions in the molecular beam is very small, only a small fraction of any photolytically generated IO will be in the rotational state excited by the laser. LIF has been observed from atmospheric NO2 at 440 nm using a YAG-pumped optical parametric oscillator,37 and could contribute to the observed signal; however the NO2 cross section varies little over the wavelength range (< 0.1 nm) between the IO “on-line” and “off-line” measurements (the off-line measurement is performed to enable subtraction of laser scatter from the fluorescence signal). Thus any contribution from NO2 will be subtracted as background.
(b) Detection of NO at 226 nm
The Ti:Sapphire laser was operated as described in section 3, with a fundamental wavelength of ∼ 904 nm selected using the diffraction grating. Frequency quadrupling was achieved via two successive frequency-doubling operations: The CLBO crystal common to all 3 radical detection schemes was used to generate 452 nm radiation, and a BBO type II crystal (additional to the core laser system and mounted externally) was used to perform the second frequency doubling operation. The resulting 226 nm radiation was separated from the other harmonics using a Pellin Broca prism arrangement. Approximate wavelength control (± 0.001 nm) was achieved using the wavemeter that sampled a small fraction of the 452 nm radiation. The BBO crystal gave inefficient conversion of the 452 nm radiation from the Ti:Sapphire laser system as it was designed for higher pulse energies typical of 10 Hz PRF excimer- or YAG-pumped dye-laser systems, and only ∼0.5 mW of 226 nm radiation was produced at 5 kHz PRF. It was extremely difficult to measure the laser power accurately, and the value of 0.5 mW must be regarded as an upper limit. The laser linewidth at 226 nm was estimated to be 0.10 cm−1 from laser-excitation spectra of single rotational transitions in NO, giving efficient overlap with the calculated Doppler-broadened NO linewidth under the FAGE experimental conditions of 0.09 cm−1. Laser radiation at 226 nm was directed to the LIF chamber using laser steering mirrors.NO was introduced into the fluorescence chamber from a cylinder (Air Products standard 440 ± 22 ppbv in N2), and further diluted in synthetic air to 50 ppbv (calculated from gas flow rates). A Schott glass UG-5 filter, with a transmission window of approximately 240–390 nm, was used to discriminate the NO LIF from scattered laser light. A low detection cell pressure reduced the effects of collisional quenching and extended the fluorescence lifetime of the NO A2Σ+ state (the natural radiative lifetime is ∼ 205 ns38) beyond the duration of the laser pulse (∼ 35 ns). Operation at such reduced pressure enabled gating of the PMT to distinguish between the fluorescence signal and red shifted scattered laser light, originating from the walls of the cell and from Raman scattering. The optimum balance between the NO number density, fluorescence lifetime and the background due to Rayleigh and Mie scattering (which scale differently with pressure) was found to be at ∼ 4 Torr. Fig. 4 shows a laser excitation spectrum of NO in the Q1 bandhead region obtained under these conditions, for 50 ppbv NO at the sampling nozzle. In addition to excitation scans in the laboratory, ambient measurements of NO were performed in the courtyard adjacent to the Department of Chemistry at the University of Leeds for 2 days during April 2002. Air was drawn into the laboratory through 6 mm i.d. PTFE tubing. Fig. 5 shows the LIF signal observed whilst sampling ambient air when the laser wavelength was repeatedly scanned over the Q1(5) line at 226.182 nm. We believe these measurements to be the first example of the detection of atmospheric NO using single-photon LIF via the A 2Σ–X 2Π transition at 226 nm. Calibration with the NO standard enabled the observed signal to be converted to an absolute NO concentration, and the peak signals in Fig. 5 correspond to 1–1.5 ppbv, in good agreement with measurements from a commercial chemiluminescence analyser (TECO 49C) sampling adjacent to the PTFE sampling inlet. The signal-to-noise level in this figure, while adequate for NO measurements in polluted air, is not sufficient for detection of NO in much cleaner air (for example at the 1–10 pptv typical of the remote marine boundary layer). However, the UV laser power was low (≤ 0.5 mW) and the beam quality was poor. Future developments using a custom-specified fourth harmonic generation crystal (CLBO) to generate 226 nm, designed for use at high PRF/low pulse power, should yield an excitation laser beam power of 50 mW or above. Further improvements, for example the use of laser-turning mirrors coated for 226 nm reflection, cell windows with an anti-reflection coating optimised for NO (226 nm) rather than for OH (308 nm) detection and an interference filter specifically designed for the collection of NO LIF will also increase the fluorescence signal whilst reducing the laser background. Calculation of the theoretical sensitivity for NO viaeqn. (iv) yields a detection limit of 1.7 × 106 molecule cm−3 (0.07 pptv) for a 60 s signal integration (30 s NO LIF, 30 s laser background) obtained for 20 mW of laser power at 226 nm. Such performance should be more than adequate for monitoring ambient NO levels in clean air at and below the ozone production/destruction compensation point. It is hoped that a future development will be an aircraft-based LIF instrument for the measurement of NO, for which (in the background troposphere) a good detection limit and short averaging time is required.
![Laser-excitation spectrum of NO in the A 2Σ+(v′
= 0)
← X 2Π1/2(v″
= 0) band, together with rotational assignments in the region of the Q1 bandhead, recorded in the FAGE cell at a pressure of 4 Torr for [NO]
= 50 ppbv. Each point represents 1 s averaging (5000 laser shots), with a laser power and linewidth of < 0.5 mW and 0.1 cm−1, respectively. A Boltzmann analysis of the line intensities yielded a rotational temperature of 225 ± 14 K.](/image/article/2003/EM/b208714f/b208714f-f4.gif) | ||
| Fig. 4 Laser-excitation spectrum of NO in the A 2Σ+(v′ = 0) ← X 2Π1/2(v″ = 0) band, together with rotational assignments in the region of the Q1 bandhead, recorded in the FAGE cell at a pressure of 4 Torr for [NO] = 50 ppbv. Each point represents 1 s averaging (5000 laser shots), with a laser power and linewidth of < 0.5 mW and 0.1 cm−1, respectively. A Boltzmann analysis of the line intensities yielded a rotational temperature of 225 ± 14 K. | ||
![Measurement of ambient NO as the laser wavelength is repeatedly scanned over the Q1(5) rotational transition at 226.18 nm for a period of about 5 min. NO was sampled using a length of PTFE tubing bringing air to the FAGE sampling nozzle from the Brotherton Courtyard on the campus of the University of Leeds. The feint line shows the raw data with 1 s averaging (5000 laser shots), whereas the heavy line shows a 6 s running average. The LIF signal corresponds to [NO] between 1 and 1.5 ppbv, in good agreement with a chemiluminescence analyser that made simultaneous measurements in the courtyard.](/image/article/2003/EM/b208714f/b208714f-f5.gif) | ||
| Fig. 5 Measurement of ambient NO as the laser wavelength is repeatedly scanned over the Q1(5) rotational transition at 226.18 nm for a period of about 5 min. NO was sampled using a length of PTFE tubing bringing air to the FAGE sampling nozzle from the Brotherton Courtyard on the campus of the University of Leeds. The feint line shows the raw data with 1 s averaging (5000 laser shots), whereas the heavy line shows a 6 s running average. The LIF signal corresponds to [NO] between 1 and 1.5 ppbv, in good agreement with a chemiluminescence analyser that made simultaneous measurements in the courtyard. | ||
A potential interference in the NO LIF signal could arise from 226 nm photolysis of atmospheric nitrogen species leading to generation of NO which could subsequently be excited in the laser pulse. Possible candidates are the UV photolysis of NO2, HONO, HNO3, alkyl nitrites (RONO) and nitrates (RONO2, e.g. PAN). Calculations of the expected interference from such species for LIF detection of atmospheric NO using, for example, laser pulse energies at 226 nm far in excess (a factor of >100) of those to be used in the Leeds system have determined that such interference effects are insignificant.39 (Any artefact NO signal with a photolytic origin will have a squared dependence upon laser-pulse energy.) Further checks for photolytic interference can be performed in the laboratory by admitting high concentrations of potentially NO forming precursors and checking for any NO LIF signal, and in the field by tuning the laser wavelength to excite a higher vibrational or rotational level that can only be populated by photolysis (i.e. having no thermal population for ambient NO). Alternatively, detuning the laser wavelength slightly from the centre of an NO line enables the detection of molecules formed with large amounts of translational energy following photolysis.
5. Conclusions
An all-solid-state, tuneable, high pulse-repetition-frequency Nd:YAG-pumped Ti:Sapphire laser system with the capability for the direct measurement of OH, NO and IO by LIF has been integrated into a field instrument. Theoretical calculations of the sensitivity for an instrument utilising this laser system give detection limits for OH, HO2 (after conversion to OH), IO and NO of 8.6 × 104 molecule cm−3 (0.006 pptv), 1 × 106 molecule cm−3 (0.04 pptv), 6.3 × 104 molecule cm−3 (0.0025 pptv) and 1.7 × 106 (0.07 pptv), respectively (mixing ratios for boundary layer conditions). The detection limit is calculated for a signal-to-noise ratio of 1, for a 60 s integration period, 30 s each of signal and background integration. Quantitative detection of very low concentrations of OH and HO2 were performed in a recent campaign at Mace Head, Ireland, with a demonstrated OH detection limit of 3.1 × 105 molecule cm−3 (0.01 pptv). Detection in the atmosphere of ∼ 1 ppb NO using the A 2Σ–X 2Π (0,0) transition at 226 nm was demonstrated in Leeds, using a very low laser power of ≤ 0.5 mW. It should be possible to detect much lower concentrations of NO, typical of the remote atmosphere, with higher laser powers using a non-linear crystal optimised for the generation of 226 nm radiation in the Ti:Sapphire laser system. Very large LIF signals were generated from the IO radical in the reference cell, but a preliminary attempt to observe IO in the field at Mace Head, Ireland, was not successful. The absolute sensitivity towards IO has yet to be experimentally determined, but the theoretical detection limit for this instrument optimised for IO is better than 0.01 pptv. Efforts are underway to design a fluorescence cell specifically for IO detection to demonstrate such a detection limit.Finally we note that the high PRF laser system could also be employed to detect atmospheric species using absorption techniques, for example via cavity ring-down spectroscopy (CRDS) in ambient air. The ring down time of the cavity can be observed with the laser tuned on and off an appropriate narrow absorption feature of the species of interest to obtain a differential absorption spectrum. CRDS is being developed as a field measurement technique, and operation in pulsed mode at high PRF offers significant advantages.
Acknowledgements
The authors wish to thank Dr. M. Blitz for his expertise in the laboratory, Dr. D. Creasey, J. MacLeod and D. McCoy of Photonics Solutions Plc., and R. Edwards of Photonics Industries Inc. for assistance in specifying the laser system, and Professor M. J. Pilling for advice and encouragement. The suggestions of two anonymous referees of this manuscript are gratefully acknowledged. This work was supported by the UK Natural Environment Research Council (Grant No. GR3/12813). DEH thanks the Royal Society for the award of a University Research Fellowship.References
- H. Levy, Science, 1971, 173, 141.
- N. Carslaw, D. J. Creasey, D. E. Heard, A. C. Lewis, J. B. McQuaid, M. J. Pilling, P. S. Monks, B. J. Bandy and S. A. Penkett, J. Geophys. Res., 1999, 104, 30241 CrossRef CAS.
- F. L. Eisele and D. J. Tanner, J. Geophys. Res., 1991, 96, 9295 CAS.
- C. C. Felton, J. C. Sheppard and M. J. Campbell, Environ. Sci. Technol., 1990, 24, 1841 CAS.
- G. Hubler, D. Perner, U. Platt, A. Toennissen and D. H. Ehhalt, J. Geophys. Res., 1984, 89, 1309.
- X. Chen and K. Mopper, J. Atmos. Chem., 2000, 36, 81 CrossRef CAS.
- T. M. Hard, C. Y. Chan, A. A. Mehrabzadeh and R. J. O′Brien, Environ. Sci. Technol., 1984, 18, 678.
- P. S. Stevens, J. H. Mather and W. H. Brune, J. Geophys. Res., 1994, 99, 3543 CrossRef CAS.
- F. Holland, M. Hessling and A. Hofzumahaus, J. Atmos. Sci., 1995, 52, 3393 Search PubMed.
- D. J. Creasey, D. E. Heard, P. A. Halford-Maw, M. J. Pilling and B. J. Whitaker, J. Chem. Soc., Faraday Trans., 1997, 93, 2907 RSC.
- Y. Kanaya, Y. Sadanaga, J. Hirokawa, Y. Kajii and H. Akimoto, J. Atmos. Chem., 2001, 38, 73 CrossRef CAS.
- D. J. Creasey, P. A. Halford-Maw, D. E. Heard, J. E. Spence and B. J. Whittaker, Rev. Sci. Instrum., 1998, 69, 4068 CrossRef CAS.
- P. O. Wennberg, R. C. Cohen, N. L. Hazen, L. B. Lapson, N. T. Allen, T. F. Hanisco, J. F. Oliver, N. W. Lanham, J. N. Demusz and J. G. Anderson, Rev. Sci. Instrum., 1994, 65, 1858 CrossRef CAS.
- G. Zeng, D. E. Heard, M. J. Pilling and S. H. Robertson, Geophys. Res. Lett., 1998, 25, 4497 CrossRef CAS.
- H. R. Barry, B. Bakowski, L. Corner, T. Freegarde, O. T. W. Hawkins, G. Hancock, R. M. J. Jacobs, R. Peverall and G. A. D. Ritchie, Chem. Phys. Lett., 2000, 319, 125 CrossRef CAS.
- D. J. Binks, P. S. Golding and T. A. King, J. Mod. Opt., 2000, 47, 1899 CrossRef CAS.
- W.H. Brune, I. C. Faloona, D. Tan, A. J. Weinheimer, T. Campos, B. A. Ridley, S. A. Vay, J. E. Collins, G. W. Sachse, L. Jaeglé and D. Jacob, Geophys. Res. Lett., 1998, 25, 1701 CrossRef CAS.
- G. McFiggans, J. M. C. Plane, B. J. Allan and L. J. Carpenter, J. Geophys. Res., 2000, 105, 14371 CrossRef CAS.
- R. Vogt, P. J. Crutzen and R. Sander, Nature, 1996, 383, 327 CrossRef CAS.
- C. D. O′Dowd, J. L. Jimenez, R. Bahreini, R. C. Flagan, J. H. Seinfeld, K. Hämeri, L. Pirjola, M. Kulmala, S. G. Jennings and T. Hoffmann, Nature, 2002, 417, 632 CrossRef.
- B. Alicke, K. Hebestreit, J. Stutz and U. Platt, Nature, 1999, 397, 572 CrossRef CAS.
- B. J. Allan, G. McFiggans and J. M. C. Plane, J. Geophys. Res., 2000, 105, 14363 CrossRef CAS.
- R. P. Wayne, Chemistry of Atmospheres, Oxford University Press, Oxford, 2000 Search PubMed.
- P. S. Monks, L. J. Carpenter, S. A. Penkett, G. P. Ayers, R. W. Gillet, I. E. Galbally and C. P. Meyer, Atmos. Environ., 1998, 32, 3647 CrossRef CAS.
- R. A. Cox, J. Geophys. Res., 1999, 104, 8047 CrossRef CAS.
- B. A. Ridley and L. C. Howlett, Rev. Sci. Instrum., 1974, 45, 742 CrossRef CAS.
- D. Kley and M. McFarland, Atmos. Technol., 1980, 12, 63 Search PubMed.
- J. D. Bradshaw, M. O. Rodgers, S. T. Sandholm, S. K. Sheng and D. D. Davis, J. Geophys. Res., 1985, 90, 12861.
- D. J. Creasey, D. E. Heard and J. D. Lee, Atmos. Environ., 2001, 35, 4713 CrossRef CAS.
- A. A. Turnipseed, M. K. Gilles, J. B. Burkholder and A. R. Ravishankara, Chem. Phys. Lett., 1995, 242, 427 CrossRef CAS.
- S. M. Newman, W. H. Howie, I. C. Lane, M. R. Upson and A. J. Orr-Ewing, J. Chem. Soc., Faraday Trans., 1998, 94, 2681 RSC.
- W. J. Bloss, D. M. Rowley, R. A. Cox and R. L. Jones, J. Phys. Chem. A, 2001, 105, 7840 CrossRef CAS.
- J. C. Mossinger, D. E. Shallcross and R. A. Cox, J. Chem. Soc., Faraday Trans., 1998, 93, 2839 Search PubMed.
- D. Bauer, T. Ingham, S. A. Carl, G. K. Moortgat and J. N. Crowley, J. Phys. Chem., 1998, 102, 2857 Search PubMed.
- R. A. Cox, W. J. Bloss, D. M. Rowley and R. L. Jones, Geophys. Res. Lett., 1999, 26, 1857 CrossRef CAS.
- J. C. Mossinger, D. M. Rowley and R. A. Cox, Atmos. Chem. Phys., 2002, 2, 227 Search PubMed.
- Y. Matsumi, S. Murakami, M. Kono, K. Takahashi, M. Koike and Y. Kondo, Anal. Chem., 2002 Search PubMed , in press.
- J. Luque and D. R. Crosley, LIFBASE: Database and Spectral Simulation Program (Version 1.5), SRI International Report MP 99-009, 1999.
- J. D. Bradshaw, M. O. Rodgers, S. T. Sandholm, S. K. Sheng and D. D. Davis, J. Geophys. Res., 1985, 90, 12861.
- D. B. Atkinson, J. W. Hudgens and A. J. Orr-Ewing, J. Phys. Chem. A, 1999, 103, 6173 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |