Evaluating a Canadian regional air quality model using ground-based observations in north-eastern Canada and United States
Ray J.
Yang
,
Adam G.
Xia
,
Diane V.
Michelangeli
*,
David A.
Plummer
,
Lori
Neary
,
Jacek W.
Kaminski
and
John C.
McConnell
Department of Earth and Atmospheric Science, York University, Toronto, ON, Canada M3J 1P3. E-mail: dvm@yorku.ca; Fax: +1-416-736-5817
First published on 26th September 2002
Abstract
The simulated concentrations from a numerical 3-dimensional regional air quality model (MC2AQ) are compared to those of ground-based observations in north-eastern Canada and the United States. The model has oxidant chemistry for both inorganic and organic species and deposition routines driven online by a mesoscale compressible community meteorological model (MC2). A standard emission inventory of anthropogenic, natural and biogenic sources for the year 1990 for 21 atmospheric trace species was used in the simulation. The model was run for July 1999, because of the occurrence of a high ozone episode and the availability of the monitoring data for surface O3, SO2, NO, NO2 and NOx. The comparisons during the episode show that the model performs quite well for predicting concentrations and diurnal variations of the surface ozone. The predictions for other gaseous species show some discrepancies with observations, but they are consistent with the results from other models evaluated in the literature. The uncertainties in the emission inventory for these species might be the main causes of the discrepancies. Further studies are needed to improve the predictability of SO2 and NOx, especially as the model is developed to include particulate matter formation as a result of these gaseous precursors.
Introduction
The impact of ground level pollution on the environment and human health is of concern to the local and national governments of Canada and the United States, as well as many other countries worldwide.1 High concentrations of ozone in the lower troposphere are considered an important issue of air quality and tropospheric chemistry.2 Surface ozone concentrations during summertime in many regions of north-eastern Canada and the United States have been measured to exceed standard levels.3Usually it is difficult to quantify the direct contribution of precursor species to the pollutant concentrations. Chemical species, after being directly emitted into the atmosphere from anthropogenic and natural sources, undergo complex non-linear physical-chemical transformations, in which new species can be formed from their precursors.4 Moreover, meteorology can also play important roles in the pollution formation processes. Biswas and Rao5 and Hogrefe et al.6 pointed out that photochemical models can be very sensitive to meteorological inputs after they examined the variability in the predictions of the Urban Airshed Model (UAM-V) when the meteorological input was derived from two different meteorological models, namely, RAMS (or RAMS3b) and MM5. Hogrefe et al.6 found that such a variability can be decomposed into intra-day, diurnal, synoptic and long-term meteorological effects. The observed concentrations at a given site are therefore the comprehensive result of emissions, chemical transformations, microphysical processes and meteorological actions.
Many regional air quality models have been developed so far to study ozone, and more recently particulate matter (PM). Jacobson et al.7 developed the model GATOR for modelling ozone and PM in the Los Angeles metropolitan area.8–10 Other air quality models include CIT,11 URM12 UAM-V,13 CAMx,14 SAQM,15 MAQSIP,16,17 SMRAQ18 and Models-3.19 Comparison studies for many air quality models can be found in Russell and Dennis.20 In Canada, two air quality models have been developed: ADOM (Acid Deposition and Oxidant Model)21 and CHRONOS (Canadian Hemispheric and Regional Ozone and NOx system).22
Until now, pre-calculated meteorological fields were used to drive the transport of chemical species in most of the regional air quality models mentioned above. Such “off-line” models often include differences between the vertical and horizontal grids and sub-grid scale parameterizations can be inconsistent for temporal resolution.23 An online chemistry modeling system, in which the meteorological fields are computed concurrently with the chemistry, will have significant advantages. In this paper, we present comparisons of results obtained from the online regional air quality modeling system MC2AQ to observation data from the routine ground-based monitoring network in Canada and the United States.
Model description
The air quality model MC2AQ was described by Plummer in his Ph.D. thesis,24 and in Plummer et al.25 as well as McConnell et al.26 Here only some of the important features and new developments of the model are summarized.Meteorological model
MC2AQ is based on the non-hydrostatic 3-dimensional Mesoscale Compressible Community model (MC2), developed by the Meteorological Service of Canada (MSC) in collaboration with the University of Quebec in Montreal.27 It uses a semi-implicit treatment for gravity and acoustic waves, and a semi-Lagrangian treatment for advection. This combination enables long simulation timesteps in comparison with the Eulerian formulation and results in efficient and accurate solutions of the primitive equations.28Chemistry module
The governing equation for chemical trace species can be written as | (1) |
Forty-seven inorganic and organic chemical species are involved in 98 chemical reactions and 16 photolysis reactions. The gas phase chemistry mechanism is that of Version II of ADOM.21 An operator-splitting approach is used to solve the system. Initial and boundary conditions for the chemical fields in the simulation were derived from global Chemical Transport Model (CTM) results.24 Those species, which are only middle products of chemical reactions and do not appear in the CTM chemistry, were assigned near-zero values for their initial and boundary conditions.
Emissions
For this study, the 1990 emission inventory for 21 atmospheric trace species was used.29 Anthropogenic emissions and emissions from natural sources such as forest fires were taken from standard pre-processed emission files developed for the ADOM speciation and the MC2 grid scheme (21.2 × 21.2 km2 on a polar stereographic projection).29 Biogenic emissions use BEIS2 in which emissions from 120 surface vegetation types24,25,26 are driven by the model meteorology, whereas the anthropogenic emissions are prescribed. Annual emissions are partitioned by seasons and further divided into hourly emission rates for each of three day-of-the-week categories: weekday, Saturday and Sunday.The emissions of 1985 and 1990 have been extensively tested and proven to be a reliable dataset for regional modelling studies within the Canadian modelling community.25 Whilst the new emission inventory is now under construction, this 1990 inventory is up to now the best available data for this application.
Dry deposition
The dry deposition rates of species depend not only on meteorological conditions in the boundary layer and chemical properties of the species, but also on land surface types. Eight land use categories are used in MC2AQ: inland waters, deciduous broadleaf trees, evergreen needle-leaf trees, wetland with plants, crops and mixed farming, grass, urban and desert. The land surface type at each grid is composed of these eight categories. Further details on the treatment of deposition in the model can be found in Plummer.24Observation data
Data from the standard ground-based monitoring networks in Canada and the United States were used. The observation data for July 1999 were selected for this model scenario, because an extensive Canadian field campaign in southern Ontario took place during this time.30 The goal of the field campaign was to obtain detailed hourly data for the comparison with PM estimated by regional models. The measured pollutants include O3, SO2, NO, NO2, NOx, PM2.5 and PM10. The current work focuses on the gas phase measurements. The Canadian data were obtained from the Analysis and Air Quality Division of Environment Canada. The hourly meteorological data for Ontario and Quebec were obtained from the Ontario Climate Centre of Environment Canada. The measured hourly pollutant concentrations and the meteorology data for the north-east United States in the model domain were extracted from the AIRS database in the United States Environmental Protection Agency's (US EPA). The number of monitoring sites for the domain in both Canada and the United States are given in Table 1, and their geographical locations are shown in Fig. 5.| United States | Canada | |
|---|---|---|
| O3 | 194 | 70 |
| NO2 | 62 | 46 |
| NO | 35 | 46 |
| NOx | 43 | 28 |
| SO2 | 148 | 38 |
| Wind | 40 | 78 |
Another reason we chose observation data for July 1999 is that an occurrence of high ozone concentration at ground level is found in this month. Ozone episode formation is strongly influenced by meteorology.1 Light surface winds, a strong subsidence inversion, sunny and warm weather associated with a high pressure system will minimize the dispersion of pollutants and provide favourable conditions for the photochemical generation of ozone.1 Ozone episodes in southern Ontario occur predominately when a stationary high-pressure ridge lies to the east of the Great Lakes region. Under this condition, synoptic flow is from the south or southwest direction, placing southern Ontario downwind from regions with significant anthropogenic emissions in the American Midwest and Ohio valley. The ozone episode case in July 1999 was formed under similar conditions.
Modelling scenario
The model domain is an 83 × 83 horizontal grid with 21.2 km horizontal resolution and 25 vertical layers up to 20 km. The model has previously been run at a horizontal resolution of 5.3 km, 21 km, and 42 km over the domain covering most of eastern North America for resolution sensitivity tests, and the results can be found in Plummer.24 Initial and boundary conditions for the chemical fields for 21 km resolution simulations were derived from the global CTM developed by McConnell et al.26 More details about chemical and meteorological initialization can be found in Plummer.24The model was run for the entire month of July 1999 during a high ozone episode in this month, with concentrations above the national standards in both Canada and the United States. The results for the period from July 10 to 22 are presented to cover three four-days states: ‘before’, ‘during’ and ‘after’ the high ozone episode. The simulated ozone concentration was obtained by using bilinear interpolation. The comparison between observations and simulations in this period will help evaluate the model performance under pre-episodic, episodic and post-episodic conditions.
Comparisons and discussions
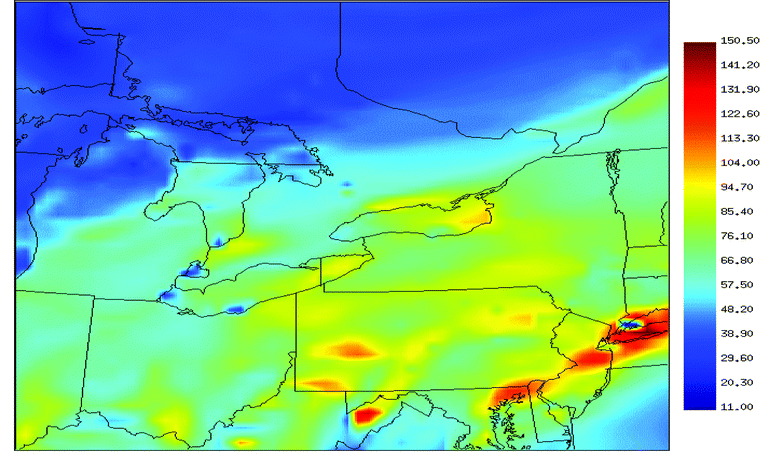
Our primary focus is on a comparison of simulated ozone with ground-based measurements since ozone is one of the main species of interest.2 For the industrialized areas in the model domain for this work, anthropogenic emissions dominate in the emission inventories, especially for organic species that greatly contribute to ozone production. So it can be expected that the anthropogenic emissions have a major contribution to ozone concentrations.Fig. 1 shows simulated ozone mixing ratios (in ppbv) at the peak of the episode, which corresponds to 2 pm Eastern Daylight Time (EDT) in eastern North America on July 17, 1999. The factors driving this ozone episode have been mentioned above. The maximum ozone level can be found along the eastern seaboard, in western Pennsylvania and in West Virginia. Ozone concentrations in the areas east of Sarnia and north of Lake Ontario exceed the 82 ppbv national standard in Canada. Fig. 2 shows the wind direction over the model domain corresponding to the conditions for Fig. 1. The prevailing winds are from the west over most of the model domain, from the southwest in southern Ontario and along the Saint Lawrence River, and from the south over the eastern seaboard. A stable high-pressure system off the eastern coast was responsible for this episode. These winds bring ozone and other chemical species into far-distance transport, which can contribute to the ‘base’ loadings of these species in downwind areas as shown in Fig. 3.
 | ||
| Fig. 2 Simulated surface wind vectors (in Knots) at 2 pm (EDT) on July 17, 1999. | ||
 | ||
| Fig. 3 Time series of ozone mixing ratios from the ground-based observations (black curve) and simulations with emissions of 1985 (red curve) and 1990 (blue curve) for selected sites in Canada and the United States in July 1999 (EDT). The vertical lines correspond to midnight every second day of the simulation. | ||
Fig. 3 shows the time series of ozone mixing ratios predicted with the 1985 and 1990 emission inventories and observed for eight sites among all those in the model domain. Four of them are in Canada. They are London, Toronto, Peterborough and Montreal, extending from the south-west to the north-east, representing the airshed upwind, within and downwind of major metropolitan areas. The other four sites are in the United States: New York City, Ohio Warren, Pennsylvania Centre and Pennsylvania Lawrence. These sites were selected due to their spread-out geographical extent and their high ozone levels. The comparison between the predicted and the measured mixing ratios shows that the qualitative agreements in all cases are quite good. During afternoons, ozone reaches its maximum concentration because of high photochemical production resulting from the emissions of NOx and volatile organic compounds (VOCs) in urban air. The titration process of ozone by NOx results in the decrease of ozone concentration within the areas of high NOx emissions. This is illustrated by the lower ozone concentrations in the urban regions, as shown in Fig. 1. During nights, high O3 is seldom observed because of low ozone production, net deposition, and the titration of ozone in the urban plume. The diurnal variations are well captured by the model, which suggests the ozone production and removal processes in the boundary layer are well characterized by the model.
By comparing two model predictions associated with two emission inventories, we can find that ozone levels predicted by using the emission inventory of 1985 (red curves) are in slightly better agreement with observations than those of the 1990 emission inventory (blue curve) at Canadian monitoring sites. While at monitoring sites in the United States, the 1990 emission inventory predicts better results than the 1985 emission. The old emissions lead to higher ozone predictions at high ozone levels. It should be noted that different emission processing methods are used in Canada and the United States. Although results with the two inventories underestimate ozone at peak levels, the general predicted geographical distributions and patterns for ozone agree well.
As mentioned above, mesoscale long-range transport can play an important role in determining the level of surface ozone.5,6 Such transport effects are clearly indicated at the four Canadian sites, as shown in Fig. 3. During nights, high ozone is seldom observed because of few ozone productions, net deposition, and the titration of ozone in urban plume. The only contribution to high ozone at night is from transport. Ozone levels don’t drop to low levels at night in Canadian sites compared with the four sites in the United States, especially for the period between July 16 and July 18. It is also evident that ozone transport effects at Montreal and Peterborough are greater than at Toronto and London. The north-eastward wind, as shown in Fig. 2, brings pollutants from high concentration areas in the American Midwest and Ohio valley, and therefore, increases the ‘base’ level in downwind areas during this period.
Fig. 4 shows the correlation coefficients between the simulated and the observed ozone on an-hourly basis for the eight sites of Fig. 3. The coefficients were computed for four days ‘before’, ‘during’ and ‘after’ the episode, as well as for the total period, at each location. The overall correlations are quite good for the eight sites, with total period correlations between 0.85 and 0.92 for the United States and between 0.79 and 0.86 for Canada (Fig. 4). In the United States, the best correlation seems to be during the peak in the episode when the maximum ozone concentration occurs. The correlations for the sites in the United States are in general better than those of the four Canadian locations. There is also a trend of decreasing correlation as time progresses ‘before’, ‘during’ and ‘after’ the episode in Canada. Fig. 5 shows the correlation coefficient for each site in the model domain over the total period. Most of the sites have satisfied values larger than 0.70, and many even larger than 0.8, which suggests the model has dynamically and chemically captured the main features of the oxidant chemistry. It is also interesting to note that the further a site is from the source region, for example in Quebec at the north-eastern region of the model domain, the worse the correlation. This is probably due to the meteorological uncertainties in the long-range transport of trace species as indicated by Biswas and Rao5 and Hogrefe et al.6 The wind and other meteorological fields are usually not predicted as well in the surface layer as in upper layers of the model.
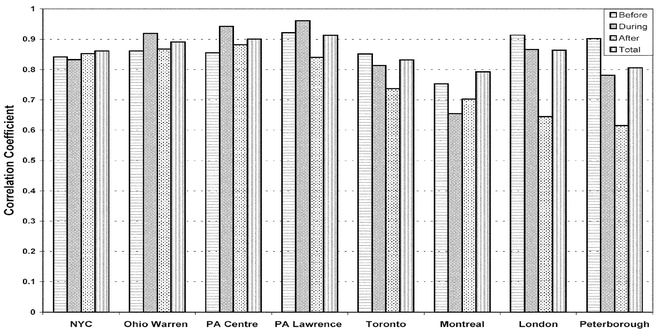 | ||
| Fig. 4 Correlation coefficients calculated during four-day periods ‘before’, ‘during’ and ‘after’ the ozone episode, and for the total period in July 1999 for the eight selected sites of Fig. 3. | ||
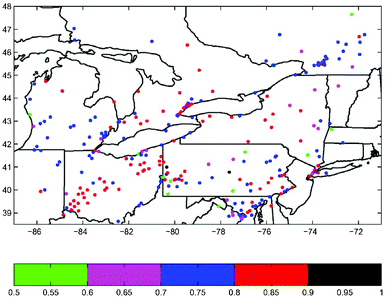 | ||
| Fig. 5 Correlation coefficient for O3 from July 10 to 21, 1999. | ||
In addition to the correlation coefficient, the index of agreement is also introduced complementarily to evaluate the model performance.34 It is expressed as:
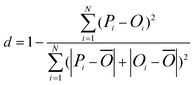 | (2) |
To statistically evaluate model performance, two other parameters are calculated: the normalized gross error G and the normalized bias B. They are defined as
 | (3) |
 | (4) |
Fig. 6 shows the geographical distribution of G in the entire domain for ozone evaluated with a cut-off of 60 ppbv. Observation–prediction pairs are often excluded if the observations are below a cut-off, typically chosen to be 60 ppbv for ozone20 based on the US EPA-recommended criteria for evaluating ozone performance.33 All sites but one have values smaller than 35%. When the cut-off is reduced to 30 ppbv, which is much lower than usually selected, there are only 9 sites with G larger than 35%, but still smaller than 40%.
 | ||
| Fig. 6 Mean normalized gross error G for O3 from July 10 to 21, 1999. | ||
Fig. 7 gives the normalized bias B for ozone over the model domain. Most sites have B within ±15%. Russell and Dennis20 presented overall bias and gross error from seven air quality models. The biases ranged from −24% to +7.4%, and the gross errors ranged from 15% to 36%. The California Air Resources Board31 and the US EPA have established acceptable ranges for G and B for model performance evaluation, which are <35% for G and within ±15% for B. Over the entire MC2AQ domain, the values of 18.5% for G and −10.9% for B are within the acceptable ranges. It is clear that the MC2AQ performs quite well for surface ozone, especially in the regions of southern Ontario, Ohio, Pennsylvania and the New York City area.
 | ||
| Fig. 7 Mean normalized bias B for O3 from July 10 to 21, 1999. | ||
While the primary focus of this paper is to investigate the model’s performance with respect to ozone, it is interesting to look at the predictions of other gaseous concentrations. Fig. 8 presents a summary of the correlation coefficients for NO, NO2, NOx, and SO2 over the model domain. NOx was evaluated separately from NO and NO2, because certain monitoring locations only record total NOx, and because there are possible uncertainties in the ratio of NO/NO2 in the emissions from each source. The correlation extends up to 0.8 for NO, 0.65 for NO2, 0.7 for NOx and 0.6 for SO2. The best correlations occur in the Greater Toronto Area, northern Virginia and Pennsylvania for NO2, in eastern Pennsylvania and central New York for SO2, in Toronto and New York City for NO.
 | ||
| Fig. 8 Histograms of correlation coefficients for NO, NO2, NOx, SO2, wind speed and wind angle agreement within 30 degrees between the observations and the model results from July 10 to 21, 1999. | ||
While the correlations for these gases seem not as good compared to those for ozone, they nevertheless are within the ranges obtained by others. For example, Biswas et al.32 obtained a correlation of about 0.2 with a maximum of 0.4 for NOx in their unfiltered data in the RAMS/UAM-V model evaluation. Russell and Dennis20 showed a systematic under-prediction of NOx by the models and indicated that there seems to be a tendency of the models to convert NOx too rapidly to NOy. The correlation value of this work is quantitatively similar to their results at most of the sites, except for the over-predictions of NOx for the sites around New York City. Compared with the 1985 emission inventory, predictions of NOx have been improved around the Great Toronto Area, and the SO2 predictions have been improved around Montreal.
The reason that modelled NOx deviates from observations is that measurements are made near the sources where the model emission schemes will never be able to reproduce small-scale fluctuations observed. Diurnal temporal variations of NOx and SO2 emissions can also account for the poor predication of these species.32 Sub-grid scale variability in emissions will have a major impact on the comparison between the model and observations. When only observations between 2 and 6 pm are considered, the correlation coefficients for NOx improve, with 18 stations having values between 0.5 and 0.7, compared to only 8 when the entire day is analyzed. This is a reflection of the fact that in the afternoon, the lower atmosphere is well mixed, which minimises the uncertainties in sub-grid scale variability. Further work to improve the predication for these species is necessary in order to model accurately the secondary formation of nitrate and sulfate with the regional air quality model. If the precursor emissions are not adequately represented in the model, or the precursor chemistry is not correctly simulated, the predictive capability of the model for PM will be reduced significantly.
The meteorological prediction of the model has been extensively studied by the original MC2 group.27 The purpose here is to provide a level of confidence in the predictions of wind speed and direction as they relate to the gas phase concentrations predicted by the model. Thus Fig. 8 presents a histogram of the correlation coefficient of the wind speed over the entire domain. The correlation extends to 0.75, with a maximum between 0.2 and 0.6. Fig. 8 also presents a histogram of the fraction of hours at each site where the difference between the observed wind direction and that predicted by the model is less than 30 degrees. The poorest correlations for winds occur around the Great Lakes. This is likely due to poorly resolved lake breeze circulation in that region, which would require much higher horizontal spatial resolution (e.g. Plummer24). The study of the effects of the lake breeze circulation at high resolution with MC2AQ is underway.
Conclusions
An air quality modelling system MC2AQ with a coupled online gas chemistry module and tracer transport algorithms, integrated with an hourly-based emission inventory, was evaluated. The simulation results were statistically and graphically compared against observations for surface O3, NO, NO2, NOx and SO2 as well as wind speed and direction, in eastern Canada and the United States. The time series comparisons for ozone on the surface demonstrate that the simulated results provide a good representation of the diurnal variation and peak concentration. The mean normalized gross error and the normalized bias for the surface ozone are within 35% and ±15%, respectively, which are acceptable for air quality models. The study indicates that the model can capture many main features of ozone incidents. The estimations for other species are not as good as ozone, but are in good agreement with other air quality models evaluated.20 Although further improvements of precursor predictions are necessary, the present analysis shows that MC2AQ can be applied to the studies of surface ozone and other pollutants. The model can also serve as a basis for further developments to include the formation, microphysical evolution and transport of PM so that it can be used for regulatory purposes in Canada.Acknowledgements
The authors would like to thank Dr. Virginia Ambrose of US EPA’s AIRS for providing the valuable monitoring datasets in the north-east US domain, Dr. Tom Dann of the Analysis and Air Quality Division of Environment Canada for the air quality datasets in Canada, and Dr. Bryan Smith of Ontario Climate Centre of Environment Canada for the meteorology datasets. The authors also acknowledge the Centre for Research in the Earth and Space Technology (CRESTech), the Natural Science and Engineering Research Council (NSERC), and the Canadian Petroleum Products Institute (CPPI) for their generous funding of this research.References
- National Research Council, Rethinking the ozone problem in urban and regional air pollution, National Academy Press, Washington DC, 1991 Search PubMed.
- P. J. Crutzen, in Tropospheric Ozone, ed. I. S. A. Isaksen, Reidel Publishing Co., 1988 Search PubMed.
- Fed. Regist., 1996, 61 (241), Dec. 13, 65763 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, J. Wiley and Sons, New York, 1998 Search PubMed.
- J. Biswas and S. T. Rao, J. Appl. Meteorol., 2001, 40, 117 Search PubMed.
- C. Hogrefe, S. T. Rao, P. Kasibhatla, G. Kallos, C. T. Tremback, W. Hao, D. Olerud, A. Xiu, J. McHenry and K. Alapaty, Atmos. Environ., 2001, 35, 4159 CrossRef CAS.
- M. Z. Jacobson, R. Lu, R. P. Turco and O. B. Toon, Atmos. Environ., 1996, 30, 1939 CrossRef CAS.
- M. Z. Jacobson, J. Geophys. Res., 2001, 106, 5385 Search PubMed.
- R. Lu, R. P. Turco and M. Z. Jacobson, J. Geophys. Res., 1997, 102, 6063 CrossRef CAS.
- R. Lu, R. P. Turco and M. Z. Jacobson, J. Geophys. Res., 1997, 102, 6081 CrossRef CAS.
- Z. Meng, J. Geophys. Res., 1998, 103, 3419 CrossRef CAS.
- N. Kumar, M. T. Odman and A. G. Russell, J. Geophys. Res., 1994, 99, 5385 CrossRef CAS.
- Systems Applications International (SAI), Users Guide to the variable Grid Urban Airshed Model (UAM-V), Systems Applications International, San Rafael, CA, 1995p. 131 Search PubMed.
- ENVIRON, User's Guide to the Comprehensive Air 'Quality Model with Extensions (CAMx), ENVIRON International Corporation, Novato, CA, 1997 Search PubMed.
- J. S. Chang, S. Jin, Y. Li, M. Beauharnois, C. H. Lu, H. C. Huang, S. Tanrikulu and J. DaMassa, The SARMAP Air Quality Model, California Environmental Protection Agency, Sacramento, CA, Final Report, 1997 Search PubMed.
- M. T. Odman and C. L. Ingram, Multiscale Air Quality Simulation Platform (MAQSIP): source code documentation and validation, Research Triangle Park, 1996, Technical Report, ENV-96TR002-v1.0. MCNC Search PubMed.
- N. Wheeler, Proceedings of the Air and Waste Management Association’s 1998 Annual Meeting, Paper 98-A739, A&WMA, Pittsburgh, PA Search PubMed.
- P. Kasibhatla and W. L. Chameides, Geophys. Res. Lett., 2000, 27, 1415 CrossRef CAS.
- D. W. Byun and J. K. S. Ching, Science Algorithms of EPA models-3 Community Multiscale Air Quality (CMAQ) Modeling System, 1999, EPA/600/R-99/030 Search PubMed.
- A. Russell and R. Dennis, Atmos. Environ., 2000, 34, 2283 CrossRef CAS.
- A. Venkatram, P. Karamchanchandani and P.K. Misra, Atmos. Environ., 1988, 24, 737 CrossRef.
- J. A. Pudykiewicz, A. Kallaur and P. K. Smolarkiewicz, Tellus Ser. B, 1997, 49, 231 Search PubMed.
- R. L. Dennis, D. W. Byun, J. H. Novak, K. J. Galluppi, C. J. Coats and M. A. Vouk, Atmos. Environ., 1996, 30, 1925 CrossRef CAS.
- D. A. Plummer, PhD Thesis, York University, 1999.
- D. A. Plummer, J. C. McConnell, L. Neary, J. Kaminski, R. Benoit, J. Drummond, J. Narayan, V. Young and D. R. Hastie, Atmos. Environ., 2001, 35, 6453 CrossRef CAS.
- J. C. McConnell, D. A. Plummer, J. W. Kaminski and L. Neary, The Ozone Monitoring Network in SW Ontario: Assessment using MC2AQ, Report to the Ontario Ministry of Environment, Sept. 28, 1999 Search PubMed.
- R. Benoit, M. Desgagne, P. Pellerin, S. Pellerin, Y. Chartier and S. Desjardins, Mon. Weather Rev., 1997, 125, 2382 Search PubMed.
- M. Tanguay, A. Robert and R. Laprise, Mon. Weather Rev., 1990, 118, 1970 Search PubMed.
- T. Scholtz, A. Taylor and A. Ivanoff, Preparation of 1990 North American emissions inventory modelling files for AES regional air quality models, Report for Environment Canada, Jan. 21, 2000 Search PubMed.
- CRESTech, Application of the Models-3 Framework for a Canadian City Using an Advanced Aerosol Module and Ambient Monitoring Techniques, Report to CRESTech, Aug. 22, 2001 Search PubMed.
- CARB (California Air Resources Board), Technical guidance document: Photochemical modeling, Sacramento, 1992 Search PubMed.
- J. Biswas, C. Hogrefe, S. T. Rao, W. Hao and G. Sistla, Atmos. Environ., 2001, 35, 6129 CrossRef CAS.
- US Environmental Protection Agency, Guideline for Regulatory Applications of the Urban Airshed Model, Research Triangle Park, NC 27711, 1991, EPA-450/4-91-013 Search PubMed.
- C. Willmott, Bull. Am. Meteorol. Soc., 1982, 63, 1309 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |