The application of proton transfer reaction-mass spectrometry (PTR-MS) to the monitoring and analysis of volatile organic compounds in the atmosphere
C. N.
Hewitt
,
S.
Hayward
and
A.
Tani
Department of Environmental Science, Institute of Environmental and Natural Sciences, Lancaster University, Lancaster, UK LA1 4YQ
First published on 13th September 2002
Abstract
Proton transfer reaction-mass spectrometry (PTR-MS) is a new and emerging technique for the measurement and monitoring of volatile organic compounds (VOCs) at low concentrations in gaseous samples in more-or-less real time. Utilising chemical ionisation, it combines the desirable attributes of high sensitivity and short integration times with good precision and accuracy. Recently it has been exploited in applications related to atmospheric science. Here, the principles of operation of the PTR-MS are described, its advantages and disadvantages discussed, its inherent uncertainties highlighted, some of its uses in atmospheric sciences reviewed, and some suggestions made on its future application to atmospheric chemistry.
1. Introduction
Proton transfer reaction-mass spectrometry (PTR-MS) is a new and emerging technique for the measurement and monitoring of volatile organic compounds (VOCs) at low concentrations in gaseous samples in more-or-less real time. Utilising chemical ionisation, it combines the desirable attributes of high sensitivity and short integration times with good precision and accuracy. Although not readily portable in its present configurations, it has been utilised in a range of field locations and deployed on research aircraft and ships. It is rapidly becoming an essential tool in the arsenal of all environmental chemists involved in the measurement and monitoring of VOCs in the atmosphere.Since its introduction, PTR-MS has been used in a number of differing applications. These have included human breath analysis in health-related studies,1–3 the monitoring of VOCs resulting from the decay of food,4 the analysis of VOC emissions from decaying vegetation5,6 and a variety of applications in atmospheric chemistry. These have included such diverse applications as the measurement of VOC fluxes from vegetation to the atmosphere,7–11 urban air quality monitoring (e.g. monitoring of benzene concentrations in a city street12), regional scale air quality monitoring13–15 and measurement of VOCs in the troposphere in remote regions of the world.16–18
Here, the principles of operation of the PTR-MS are described, its advantages and disadvantages discussed, its inherent uncertainties highlighted, some of its uses in atmospheric sciences reviewed, and some suggestions made on its future application to atmospheric chemistry.
2. PTR-MS method description
PTR-MS is an on-line gas analysis tool utilising low-energy chemical ionisation to simultaneously monitor a suite of volatile organic compounds in air. The technique utilises proton transfer reactions from protonated water molecules (H3O+) to volatile organic compounds which are subsequently separated according to their mass/charge ratio (usually their molecular mass plus 1) and detected quantitatively downstream. A detailed discription of the development and principles of operation of a commercially-available PTR-MS (Ionicon GmbH, Innsbruck) instrument have been described in detail elsewhere.4,19,20 Therefore only a brief description of the salient features will be given here. The instrument comprises four key components; an ion source, a flow drift tube, a quadrupole mass analyser and an ion detection system. Here we shall discuss in detail only the ion source and flow drift tube since components of the ion filtration and detection system are commonplace in existing commercially available mass spectrometers.Under normal operating conditions, reagent ions consisting largely of H3O+ are produced at a high density from pure water vapour within a hollow cathode ion source. Under normal operating conditions, approximately (2.5–3.5) × 106 reagent ions are detected downstream of the reaction region per second. Reagent ions are passed via a Venturi type inlet21 into a flow drift tube (the reaction region), which is under the influence of an electric field E (typically 62.5 V cm−1 under normal operating conditions, but operator-variable) and vacuum pump. Within the drift tube, a proportion of reagent ions become hydrated to form cluster ions of the form H3O+(H2O)n (n = 1, 2, 3….), which can often, but not always, act as reagent ions (proton donors). The abundance and distribution of such cluster ions within the drift tube is strongly dependent upon the PTR-MS operating conditions (drift tube electric field and pressure, which affect ion kinetic energy) and the humidity of the sample gas stream, but under normal operating conditions is typically <5% of the total reagent ion abundance. Suppression of reagent ion clustering in PTR-MS is desirable since the reaction of neutrals with reagent clusters can confuse the interpretation of the instrument spectrum. A good understanding of the reagent and hydrated reagent ion distribution is essential for the interpretation of PTR-MS output, and will be discussed in section 4.
Under normal operating conditions, the sample air is introduced into the drift tube at a flow rate of ∼11 mL min−1, maintaining a drift tube pressure in the region of 1.950–2.050 mbar. The electric field, pressure and temperature of the drift tube under normal operating conditions are such that the ratio E/N (where N is the number density of the drift tube buffer gas molecules, i.e. the major components of air) is in the range 120–130 Td (1 Td = 1 Townsend = 10−17 cm2 V molecule−1). The significance of this range is discussed in section 4.
Due to their relatively low proton affinities (approximately 176–594 kJ mol−1), the major components of air undergo non-reactive collisions with H3O+ ions, and therefore act as a buffer gas. These components are subsequently removed by a turbomolecular vacuum pump. However, any collisions of H3O+ ions with gaseous constituents, R, possessing a proton affinity greater than that of water (∼697 kJ mol−1), will result in a proton transfer reaction (eqn. (1)):
| H3O+ + R → RH+ + H2O | (1) |
| [RH+] = [H3O+]0(1 − e−k[R]t) ≈ [H3O+]0[R]kt | (2) |
3. PTR-MS operation
Since PTR-MS utilises a low energy ‘soft’ ion source, many (although not all) proton-transfer processes are non-dissociative. As a result, many constituents in the atmosphere, for example the chemically reactive C5 compound isoprene (protonated mass = 69 amu), produce only one product ion which is measured as the molecular mass, plus one. This is useful when measuring compounds in air containing simple or qualitatively known gaseous mixes. However, problems can and do arise during the measurement of complex and qualitatively unknown gaseous mixtures. Since PTR-MS does not utilise compound separation prior to analysis, the likelihood of more than one VOC (or indeed VOC fragment ions) contributing to the observed signal for a particular ion mass cannot be discounted. As a result, PTR-MS is not a truly species-specific quantitative method of analysis and parallel compound identification by a complimentary technique is highly desirable, and indeed is often essential. Alternatively, tentative compound identification can be achieved by an assessment of theoretical versus measured isotopic abundances. For example, the theoretical abundance of the mono-substituted 13C analogue of isoprene is 5.3%, and an analysis of the ratio of the instrument signal at masses 69 and 70 can provide some evidence of whether the instrument signal originates from a C5 compound. This technique is not absolute however since other compounds or fragment ions may cause interference at these particular masses, but has proven useful in a number of cases.12,20 Due to these limitations, PTR-MS should be regarded as an on-line monitoring tool. However, a manipulation of instrumental operating parameters can yield further information on compound identification, and can shed light on whether an observed ion signal originates from a molecular ion, fragment ion, or both. Details of this technique are given in section 5.Collisions with neutrals in the drift tube do in a number of circumstances lead to molecular ion fragmentation, with certain groups of compounds seemingly more susceptible than others.8,22 Other than compound specificity, molecular fragmentation occurs largely as a function of mean collision energy (which itself is a function of drift tube E/N). The significance of this for on-line identification and quantification is discussed further in section 5.
Ion count rates, measured in counts per second (cps) for selected user defined molecular and fragment ions, are successively measured using pre-determined integration periods for each, enabling longer integration periods to be applied where expected concentrations are low and vice versa. For example, an integration period of 20 s or more may be appropriate where an ambient volume mixing ratio of only a few tens to a few hundred pptv (10−12) is expected, or when rapid concentration changes on the order of a few seconds are not expected, or not of interest. Instrumental precision for a given ionic mass is governed by ion signal strength (cps) and integration period, and has been shown to be well described by Poisson statistics. As a result, instrument noise is proportional to the square-root of the signal, and genuine ion-signal changes can easily be distinguished from instrument noise.12 Experimental precision can be increased for VOCs at low concentrations by increasing the integration period, although temporal resolution between successive measurements of the same mass may be compromised, particularly when a large number of ions are being analysed. Additionally, lengthy integration periods may mask any concentration changes that occur on short time scales. It can therefore be seen that the time taken between successive measurements of the same mass (i.e. temporal resolution) is dictated by the number of masses under consideration and the integration period for each.
Instrumental accuracy is largely determined by uncertainties in values assigned for k (the proton-transfer reaction rate coefficients) and in the value used for t at different drift tube E/N ratios. Accuracy is currently assessed as better than 30%, and is likely to be improved further by suppressing reagent ion clustering.4 However, the use of specific gas standards for calibration can improve accuracy to better than 15%.
4. Drift tube ion chemistry and molecular fragmentation
The basic concept behind PTR-MS is that the ion chemistry is kept as simple as possible, minimising fragmentation and allowing the concentration measurement to be based on the relative concentrations of product and primary ions (eqn. (2)) (which in turn eliminates the impact of any instrument drift). Drift tube E/N (and thus the mean collisional energy between reactant ions and neutrals) has a profound effect on both reagent and product ion distribution and abundance. The range 120–130 Td that is utilised under ‘normal’ operating conditions minimises many of the uncertainties associated with PTR-MS, since many of the parameters necessary for interpretation of the ion signals (for example reaction time and reagent ion distribution) are well characterised within this range. Outside of this range, accuracy is less well defined and the use of calibration standards is desirable.The 120–130 Td range utilised under ‘normal’ PTR-MS operating conditions represents a compromise between reagent ion hydration (often termed reagent or water clustering) on the one hand and molecular (product) ion fragmentation on the other. Drift tube E/N ratios greater than 130 Td, which are achieved by decreasing the drift tube pressure or increasing the electric field, suppress reagent ion clustering (thus increasing the density and count rate of H3O+) by increasing ion kinetic energy, but often at the expense of increased product ion fragmentation. Fig. 1 shows the PTR-MS fragmentation spectrum for the C10 monoterpene compound limonene across a wide E/N range. Above 120–130 Td, considerable molecular fragmentation is observed, and the abundance of the molecular ion (mass 137) shows significant decline. Excessive fragmentation such as this may obscure the resultant spectrometry since any one VOC can potentially yield many product ions. In complex mixtures, this is particularly problematic with regards to compound identification since any number of molecules may contribute, to some extent, to the observed signal for a particular mass.
 | ||
| Fig. 1 Percentage limonene fragment ion distribution (counts per second, cps) as a function of E/N at a vapour pressure of 1.25 kPa (RH = 40%). Note how the molecular ion (mass 137) dominates at low E/N and how molecular fragmentation increases as E/N increases, a phenomenon typical of many other VOCs. The abundance of mass 155, the protonated limonene–water cluster, only becomes significant at low E/N. The ‘normal’ operating range of 120–130 Td represents a compromise between fragmentation suppression and reagent ion clustering (see Fig. 2). | ||
Conversely, drift tube E/N ratios less than 120 Td, which are achieved by decreasing the electric field surrounding the drift tube or increasing the drift tube pressure, tend to inhibit molecular fragmentation but promote reagent ion clustering, initially as the monohydrate ion, and, as an E/N ratio of 80 Td is approached, increasingly as the dihydrate ion H3O+(H2O)2. This is illustrated in Fig. 2 which presents the abundance of the reagent ion H3O+ (mass 19) and reagent ion water clusters, H3O+(H2O) (mass 37) and H3O+(H2O)2 (mass 55) as a function of drift tube E/N. Reagent ion hydration is exacerbated when sampling humid air since the abundance of H2O in the drift tube is increased. Indeed, the instrument can be calibrated with a dew point generator to measure the vapour pressure of the sampled air stream at any E/N by monitoring the ion counts at masses 37 and 55. This has been demonstrated elsewhere.23
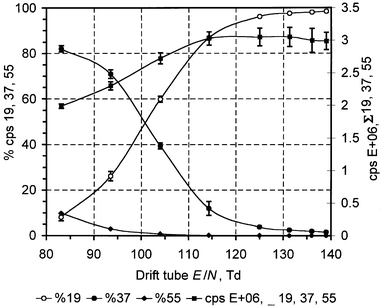 | ||
| Fig. 2 Reagent ion distribution as a function of drift tube E/N at a vapour pressure of 1.25 kPa (RH = 40%). Note how the relative abundance of hydrated reagent ions increase as E/N decreases. The total reagent ion count (19 + 37 + 55) decreases as E/N decreases due to the lower general ion mobility of hydrated reagent ions, the decreased abundance of the unhydrated reagent ion, and a general decrease in ion mobility that is observed for ions at lower E/N. Error bars represent 1 × standard deviation based upon n observations where 20 < n < 40. | ||
The uncertainties associated with PTR-MS operation at low E/N are manifold. First, in eqn. (2), the reagent cluster ion signals at masses 37 and 55 need to be accounted for, since in many cases these clusters are able to protonate neutrals possessing a large enough proton affinity. However, the proton transfer reaction rate coefficients of these clusters are lower than those of the unhydrated reagent ion (but in many cases are undocumented), and consequently the rate coefficient k for each cluster needs to weighted towards cluster abundance. However, Warneke et al.23 have shown that at low E/N the measured reagent ion distribution is markedly different to that of the theoretical distribution, calculated according to the procedure of Lau et al.24 Warneke et al. found that at low E/N, the measured reagent cluster abundance was significantly underestimated. Using measurements with standard atmospheres of benzene and toluene (which have both low proton affinities and dipole moments, thus preventing reaction with H3O+(H2O) and higher water clusters) they showed that the theoretical distribution was a more accurate representation than the measured distribution. The reason for this disparity was thought to be collision-induced dissociation of hydrated reagent clusters in the region between the drift tube and the filtration/detection system. In addition to this uncertainty, it is also known that some VOCs (including benzene and toluene, as mentioned above), due to their low proton affinities and/or dipole moments, do not undergo proton transfer reaction with mono-, di- and higher hydrate water clusters.23 This phenomenon requires consideration when such compounds are being measured at low E/N, especially in humid air when the cluster concentration increases. This also has some impact on instrumental sensitivity for such compounds since the number of available reagent ions is effectively decreased. However, under such conditions reaction time is also increased which can negate this latter effect. This is detailed below.
Secondly, at low E/N, the number of cluster ions of the form R·H+(H2O)n increases (e.g.Fig. 1; mass 155). At higher E/N these are suppressed by switching reactions in which one or more water molecules are ejected to leave the unhydrated protonated form. Again, the presence of considerable amounts of such clusters can significantly increase the complexity of the resultant mass spectrometry. For example, the protonated acetaldehyde–water cluster has the same mass as protonated unhydrated dimethyl sulfide (a naturally occurring species of considerable importance in the atmospheric cycling of sulfur and in the formation of cloud condensation nuclei), leading to the possibility of erroneous attribution of observed ions.
Thirdly, varying E/N alters reagent ion mobility and thus the drift velocity of reagent ions.25 This ultimately impacts upon reaction time t in the drift tube, and thus instrument sensitivity, as will be discussed below. Reaction time t is difficult to measure directly without dedicated equipment, although it can be calculated if proton transfer reaction rate coefficients k are accurately known, and if the true distribution of reagent and reagent cluster ions is understood. However, measured reaction time will always be an average of the differing reaction times of the reagent ions and hydrated reagent ion clusters. In any case, it is thought that while traversing the length of the drift tube, the reagent ions undergo numerous switching reactions.23 Alternatively, average reaction times can be calculated using the ‘reduced’ ion mobility data of Dotan et al.,25 who report direct measurements of protonated water and associated hydrated cluster ion mobilities in nitrogen at drift tube pressures of ∼0.2 mbar. However, because the true reagent distribution in the drift tube is difficult to ascertain,23 further uncertainty may be introduced.
5. PTR-MS operation at non-standard conditions
Operation at non-standard conditions can offer many benefits. For example, decreasing drift tube E/N yields greater instrument sensitivity (cps ppbv−1) since the reagent ion mobility is decreased resulting in an increased reaction time. However, as discussed in the previous section, this introduces additional uncertainty since the average reagent ion reaction time t requires reassessment, and the resultant elevated hydrated reagent ion cluster abundance needs consideration. Such an exercise has been conducted by Warneke et al.23 who successfully measured benzene and toluene at an E/N lower than that normally used, obtaining comparable results between PTR-MS and whole air sampling with GC-FID analysis. Furthermore, at low E/N, VOCs with proton affinities only slightly greater than that of water, e.g. formaldehyde, may undergo the reverse reaction to eqn. (1), hence depressing sensitivity. Operation at drift tube E/N greater than normal conditions may be beneficial when the suppression of hydrated reagent cluster ions is of importance, yet loss of sensitivity is not so important. Such a situation may arise, for example, when monitoring benzene and toluene in urban air.Another use of PTR-MS operation at E/N greater than standard conditions is the suppression of cluster ions of the form R·H+(H2O)n. The abundance of clusters ions of this form is usually low under normal conditions since switching reactions result in the ejection of a water molecule to leave the protonated molecule. However, in humid air, the abundance of such clusters can be significant. These clusters can cause some confusion in the resultant mass spectrometry, but by temporarily increasing drift tube E/N these can normally be identified.
A further benefit of varying drift tube E/N is the information that is often yielded on compound structure. The on-line analysis of VOC emissions from living vegetation, the coniferous tree species Sitka spruce in this instance, serves to illustrate this point well. Tani et al.22 found that by switching drift tube E/N between the normal operating conditions of ∼120 Td and 80 Td, the measured ion signal at mass 137 increased, while the signal for masses 81 and 95 simultaneously decreased. This result suggested that the ion signal for mass 137 most likely originated from a molecular ion, while the signals measured for masses 81 and 95 were more likely to represent fragments, since a reduction of E/N resulted in a decline in their signal strength. This experiment, which operated in selected ion mode, was unable to confirm conclusively whether the fragment ions of mass 81 and 95 originated from the molecular ion of mass 137. However, a literature examination revealed that the signal measured at mass 137 was most likely monoterpenoid in origin, since Sitka spruce is a known monoterpene emitter.26 As a result, parallel laboratory analyses with authentic monoterpene standards revealed that many monoterpenes do indeed fragment during PTR-MS analysis to yield ions dominated by masses 81 and 95. As a result, Tani et al. were able to report with some confidence emissions of monoterpenes from Sitka spruce. However, comprehensive verification of individual monoterpene species (most of which have a protonated molecular mass of 137) proved more difficult since identification of fragment ions unique to each monoterpene is required. Unique ions do not always exist, and in some cases the fragment ions of other non-monoterpenoids may interfere.
6. Other uncertainties
Perhaps the most significant source of uncertainty in PTR-MS arises in the allocation of proton transfer reaction rate coefficients, k. The Langevin rate coefficient kL, and the ion capture rate coefficient, kc can provide estimates for k in the case of non-polar and polar compounds respectively. In the former case, the proton transfer reaction is assumed to proceed at the collisional limiting Langevin rate (capture rate), while in the latter case the capture rate exceeds the Langevin rate.27 The two rate coefficients can be calculated using the parametrized trajectory formulae given by Su and Chesnavich, provided that information on neutral reactant masses, polarisability and permanent dipole moments are known.28 In many cases, these data are not readily available and estimations are required.Alternative to estimations, proton transfer reaction rate coefficients can be measured directly, provided that reaction time t is accurately known. Tani et al.22 carried out such measurements for a range of monoterpenes and related compounds. In their measurements, reaction time t was first accurately determined using proton transfer reaction determinations of standard atmospheres of toluene, for which k has been clearly established.29 Parallel GC-FID analysis was used to determine the standard atmosphere concentration, and t calculated according to eqn. (2). Next, PTR-MS measurements of nominal standard monoterpene atmospheres were made, and k determined using eqn. (2), after consideration of simultaneous GC-FID determinations of the standard atmospheres. In cases where it was possible to estimate kL and kc, the measured rate coefficients of Tani et al. were usually within ±10% of the estimated rate coefficients, comparing favourably with the 15% uncertainty range given by Lindinger et al.4
Further uncertainty in k is introduced when proton transfer reactions involving reagent ion other than H3O+ occur. For example, the PTR-MS rate coefficient between the monoterpenes and the monohydrate reagent ion cluster has been calculated as being ∼24% less than the rate coefficient with the unhydrated reagent ion. This may be of some importance when significant amounts of reagent ion clusters are present, such as at low E/N or when sampling humid air. However, under normal operating conditions, over 95% of the reagent ions are present in the unhydrated form, and as a result the average PTR-MS rate coefficient (for both unhydrated and monohydrated reagent ions) has been calculated to be only ∼1.2% lower than the rate coefficient for the unhydrated reagent ion alone.22 This value is within the instrumental uncertainty range and as a result can be discounted.
7. Application of PTR-MS to atmospheric measurement and monitoring
Since 1999, PTR-MS has been used for several distinct applications of relevance to atmospheric science by a small number of research groups, principally in Europe. Broadly, these applications have been (a) studies of the emissions of VOCs from the biosphere to the atmosphere; (b) studies of the occurrence and behaviour of VOCs in the atmosphere (urban, rural and remote); (c) laboratory experiments on VOC oxidation (a very limited application to date) and (d) development of the PTR-MS method for the analysis of selected organic nitrates and other exotic species (another limited application to date). In addition, papers have been published on the performance characteristics of the PTR-MS in ambient air applications,12 and on the fragmentation behaviour of an important class of biogenic VOCs, the monoterpenes, during PTR-MS analysis.22 Here, selected applications of PTR-MS to atmospheric science are briefly discussed, but what follows is not a comprehensive review of every use of the method.It is important to recognise that some of the limitations of the PTR-MS method, discussed above, may not have been adequately recognised in these early applications. There is no doubt that the fantastic capabilities of the method engendered an early enthusiasm for its application that may have lead to some of the limitations being overlooked. For example, the notion that the H3O+-mediated ionisation of a VOC molecule is a “soft” process, resulting in no fragmentation of the VOC but the sole formation of a protonated molecular ion, may have mislead some researchers into erroneous interpretations of the PTR-MS signal. As discussed above, a good example of this is the fragmentation of some C10 monoterpene compounds at high concentrations to give mass fragments which may be confused with toluene, isoprene and other lower molecular mass species.
8. Emissions of VOCs from the biosphere to the atmosphere
Probably the most successful environmental application of PTR-MS to date has been in the study of the emissions of biogenic VOCs to the atmosphere. Boschetti et al.6 monitored changes in the emission rates of selected VOCs (including alcohols, aldehydes, ketones, esters and acids) from soft fruit during post-harvest ripening and the on-set of decay. However, this study was aimed at monitoring the quality and preservation of fruit, rather than the emissions of the VOCs to the atmosphere. Of greater relevance to atmospheric science was the laboratory study of Warneke et al.5 who observed emissions of acetone, methanol, acetaldehyde and other VOCs from decaying vegetation. They studied the effects of varying temperature and of wetting on the emissions and concluded that the abiotic production of these partially oxidised VOCs following “roasting” at elevated temperatures and subsequent wetting could be responsible for the emission of 6–8 Tg of acetone and 18–40 Tg of methanol to the global atmosphere per annum.Fall et al.8 and de Gouw et al.30 used PTR-MS to monitor the emissions of the hexenal family and other partially oxygenated VOCs following leaf wounding and drying. In the first of these studies they found that the deliberate wounding of Populus leaves lead to the very rapid production and emission of (Z)-3-hexenal, which then disappeared, followed by the emission of its metabolites including (E)-2-hexanal, hexenols and hexenyl acetates. They found that the amount of emitted VOCs was proportional to the degree of wounding, was not light dependent, occurred in attached and detached leaves and was enhanced as detached leaves became dry. In the second study they showed that the cutting and drying of crops caused the emission of these same classes of compounds as well as alcohols, aldehydes, ketones, esters and acids. Karl et al.31 monitored the release of VOCs during the mowing of grass. The same hexenal family of compounds was observed, with enhanced acetone emissions following the wetting of the cut grass by rainfall. Using the micro-meteorological eddy covariance method above a hay pasture during the cutting of grass, emission flux rates of 0.1–8 mg m−2 h−1 of a range of partially oxygenated VOCs were measured by Karl et al.10 Fall et al.9 found that the wounding of plants by repeated freezing and thawing also produced partially oxygenated VOCs and the occurrence of these compounds in ambient air was attributed to the freeze–thawing of vegetation (see also Karl et al.32). However, no attempt was made at inverse modelling to show the plausibility of this contention.
From all these studies, it may be concluded that the grazing of grassland, harvesting of crops, freezing and thawing of plant tissue and other large scale “wounding” events may cause the emission of substantial quantities of partially oxygenated VOCs to the atmosphere, with possible effects on atmospheric composition and chemistry. However, it should be noted that few of the above studies used a species specific analytical method (e.g. GC-MS or HPLC) to verify compound identification. One example of the use of parallel analysis was de Gouw et al.30 Fall et al.9 used chromatographic separation of the VOCs prior to entry into the PTR-MS, allowing them to separate compounds producing the same fragment ions. For example, a number of C5 and C6 compound fragments give an ion of mass 69 (e.g. 2- and 3-methylbutanal, 2- and 3-methyl-2-butenol, and similar pentene-related compounds). Mass 69 is also the molecular ion mass of isoprene. However, given the variable fragmentation patterns of similar VOCs in the PTR-MS (see above) the ability of the coupled GC-PTR-MS method to give unequivocal identification of mixed analytes is not yet fully developed and certainly warrants further work.
Several studies have applied PTR-MS to the measurement of emissions of VOCs from plants. Holzinger et al.7 used it to study VOC emissions from the Mediterranean holm oak (Quercus ilex) under different environmental conditions. They attributed mass 93 to the emissions of toluene, although Tani et al.22 have since shown that mass 93 is an ion produced by the fragmentation of the monoterpene p-cymene. At high concentrations (e.g. as found in chamber used to measure emissions from plants) the attribution of minor ions observed in the mass spectrum can potentially lead to misidentification. Hayward et al.33 used PTR-MS in a study of VOC emissions from Sitka spruce (Picea sitchensis) and obtained very detailed information on how variations in light and temperature affect isoprene emissions from this species.
9. Monitoring concentrations of VOCs in the ambient atmosphere
PTR-MS has been used on a number of occasions for monitoring VOC concentration changes in ambient air. Hayward et al.12 presented benzene concentrations in urban air at a site in the city of Lancaster, UK, with a 4 minute time resolution, and a mean concentration of 790 pptv. They showed close correlation with the concentrations of NO, consistent with a common traffic-related source of these species.Karl et al.34 obtained a large set of data on VOC concentrations at the Sonnblick Observatory, located at 3106 m in the Austrian Alps. They also used GC-PTR-MS for species verification for some sampling periods. The data were used to establish the variability-lifetime relationship, allowing the calculation of an average OH concentration of (1.5 ± 0.4) × 105 radicals cm−3 around midday. They were also able to indirectly calculate peak OH concentrations of around (1.3 ± 0.5) × 106 radicals cm−3. By continuously monitoring VOC concentrations over a period of months under a wide range of meteorological conditions they were able to distinguish a number of VOC source-receptor relationships that may be helpful in understanding atmospheric transport and chemistry processes. However they did not provide detailed GC-MS confirmation of their interpretation of the ion counts obtained, and some of the attribution of observed protonated ions to parent VOC compounds must be viewed with caution.
Warneke and de Gouw35 used PTR-MS aboard a research vessel to measure dimethyl sulfide (DMS) and acetonitrile every 40 s in surface air on a cruise in the north-west Indian Ocean. Other VOCs were measured using gas chromatography. An average acetonitrile concentration of 120 pptv was found, with elevated concentrations attributed to the long-range transport of air polluted by biomass burning in Asia. A diurnal cycle in DMS concentration was observed, and the data used to estimate a DMS flux from the ocean surface of (1.5 ± 0.7) × 1013 molecules m−2 s−1 and a 24 h average OH concentration of (1.4 ± 0.7) × 106 molecules m−3. A more pronounced diurnal cycle in DMS concentrations in maritime air was observed by Hayward et al.12 at Mace Head in Ireland, with clear night-time maxima and daytime minima, consistent with the daytime removal of DMS by OH.
An early and ambitious application of PTR-MS was its deployment on a research aircraft over the northern Amazonian rain forest in Surinam. These results have been summarised by Crutzen et al.16 and are discussed in more detail by Williams et al.18 (who considered the interpretation of the mass scans obtained), Warnecke et al.36 (isoprene and its oxidation products) and Poschl et al.17 (acetone). The concentrations of isoprene observed (by assuming its unique contribution to mass 69) were within the range expected from an estimation of the strength of the forest as a source. Similarly the concentrations of the isoprene oxidation products (by assuming the unique contribution of methyl vinyl ketone plus methacrolein to mass 71 and of C5 hydroperoxides to mass 101) were as expected from modelling the oxidation of isoprene. In addition to these compounds, methanol (mass 33), acetonitrile (mass 42) and acetone (mass 59) were identified and monitored with some confidence. Many other masses were detected but without unambiguous attribution to particular VOC species, highlighting the absolute necessity of making simultaneous determinations of air composition by a species specific method, such as GC-MS.
The applicability of PTR-MS has clearly been demonstrated for a number of compounds in the ambient atmosphere, including benzene, isoprene, methanol, acetonitrile, acetone, dimethyl sulfide. It has also been used for a number of other species, but without possibly the same degree of confidence in mass attributions. Additionally, it obviously has the potential to be used for monitoring many other species. For example, Hansel and Wisthaler37 have shown that PTR-MS can be used to monitor peroxyacetic nitric anhydride (PAN), peroxypropionic nitric anhydride (PPN) and peroxymethacrylic nitric anhydride (MPAN) at masses 77, 91 and 103 respectively, with detection limits of ~70 pptv with integration times of 15 s. Compared with the usual GC method of analysis of PAN and other organic nitrates this represents a significant improvement in sampling frequency and sensitivity, although the applicability of the method to ambient air monitoring remains to be further developed.
10. The use of PTR-MS in atmospheric chemistry laboratory experiments
To date, PTR-MS has not been widely applied to laboratory experiments. Wisthaler et al.38 used it to study the formation of acetone and other gas phase products from the OH-mediated oxidation of monoterpenes in a 0.5 m3 Teflon reaction chamber. They utilised the fast response time of the instrument to study the yields of a wide range of intermediate and more stable products, and used FTIR for unequivocal product identification. They demonstrated the utility of the method to laboratory experiments in atmospheric chemistry, and it is only a matter of time before it is applied to sophisticated chamber experiments such as those conducted at the EUROPHORE reactor in Spain.11. Conclusions
PTR-MS is an analytical method still in its infancy, yet has already been utilised for a range of applications in atmospheric science. Its ability to monitor concentration changes over small time intervals, its high sensitivity, and its ability to detect oxygenated and ‘exotic’ VOCs has already been exploited to good use. A flush of early enthusiasm for the method may have lead some researchers to draw unwarranted conclusions from the PTR-MS data obtained without supporting species-specific evidence, but nevertheless the power of the method has been convincingly demonstrated.In the future, a better understanding of the fragmentation behaviour of the VOC species present in ambient air, backed up by adequate complementary GC-MS analyses, will allow PTR-MS to become an essential tool for atmospheric chemists. GC-PTR-MS and ion-trap PTR-MS are both undergoing development and may become of increased importance. Many other applications of the PTR-MS method, for example in isotopic labelling experiments, in isotopic discrimination in ambient air sampling, in laboratory gas kinetics and reaction products experiments, and in routine VOC monitoring, are already coming in to view.
Acknowledgements
We are grateful to Ionicon GmbH for their support and service, and to A. Jordan and A. Hansel for helpful discussions. This work was supported by the Natural Environment Research Council, UK, under grant number GR3/12750.References
- A. Jordan, A. Hansel, R. Holzinger and W. Lindinger, Int. J. Mass Spectrom. Ion Processes, 1995, 148, L1 CrossRef CAS.
- T. Karl, P. Prazeller, D. Mayr, A. Jordan, J. Rieder, R. Fall and W. Lindinger, J. Appl. Physiol., 2001, 91, 762 Search PubMed.
- C. Warneke, J. Kuczynski, A. Hansel, A. Jordan, W. Vogel and W. Lindinger, Int. J. Mass Spectrom. Ion Proceses., 1996, 154, 61 Search PubMed.
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrom. Ion Processes, 1998, 173, 191 CrossRef CAS.
- C. Warneke, T. Karl, H. Judmaier, A. Hansel, A. Jordan, W. Lindinger and P. J. Crutzen, Global Biogeochem. Cycles, 1999, 13, 9 CrossRef CAS.
- A. Boschetti, F. Biasioli, M. van Opbergen, C. Warneke, A. Jordan, R. Holzinger, P. Prazeller, T. Karl, A. Hansel, W. Lindinger and S. Iannotta, Postharvest Biol. Technol., 1999, 17, 143 CrossRef CAS.
- R. Holzinger, L. Sandoval-Soto, S. Rottenberger, P. J. Crutzen and J. Kesselmeier, J. Geophys. Res. [Atmos.], 2000, 105, 20573 Search PubMed.
- R. Fall, T. Karl, A. Hansel, A. Jordan and W. Lindinger, J. Geophys. Res. [Atmos.], 1999, 104, 15963 Search PubMed.
- R. Fall, T. Karl, A. Jordon and W. Lindinger, Atmos. Environ., 2001, 35, 3905 CrossRef CAS.
- T. Karl, A. Guenther, A. Jordan, R. Fall and W. Lindinger, Atmos. Environ., 2001, 35, 491 CrossRef CAS.
- J. A. de Gouw, C. J. Howard, T. G. Custer and R. Fall, Geophys. Res. Lett., 1999, 26, 811 CrossRef CAS.
- S. Hayward, C. N. Hewitt, J. H. Sartin and S. M. Owen, Environ. Sci. Technol., 2002, 36, 1554 CrossRef CAS.
- R. Holzinger, A. Jordan, A. Hansel and W. Lindinger, J. Atmos. Chem., 2001, 38, 187 CrossRef CAS.
- R. Holzinger, A. Jordan, A. Hansel and W. Lindinger, Atmos. Environ., 2001, 35, 2525 CrossRef CAS.
- R. Holzinger, B. Kleiss, L. Donoso and E. Sanhueza, Atmos. Environ., 2001, 35, 4917 CrossRef CAS.
- P. J. Crutzen, J. Williams, U. Poschl, P. Hoor, H. Fischer, C. Warneke, R. Holzinger, A. Hansel, W. Lindinger, B. Scheeren and J. Lelieveld, Atmos. Environ., 2000, 34, 1161 CrossRef CAS.
- U. Poschl, J. Williams, P. Hoor, H. Fischer, P. J. Crutzen, C. Warneke, R. Holzinger, A. Hansel, A. Jordan, W. Lindinger, H. A. Scheeren, W. Peters and J. Lelieveld, J. Atmos. Chem., 2001, 38, 115 CrossRef CAS.
- J. Williams, U. Poschl, P. J. Crutzen, A. Hansel, R. Holzinger, C. Warneke, W. Lindinger and J. Lelieveld, J. Atmos. Chem., 2001, 38, 133 CrossRef CAS.
- A. Hansel, A. Jordan, R. Holzinger, P. Prazeller, W. Vogel and W. Lindinger, Int. J. Mass Spectrom. Ion Processes, 1995, 150, 609 CrossRef.
- A. Hansel, A. Jordan, C. Warneke, R. Holzinger and W. Lindinger, Rapid Commun. Mass Spectrom., 1998, 12, 871 CrossRef CAS.
- W. Lindinger, in Gaseous Ion Chemistry and Mass Spectrometry, ed. J. H. Futrell, John Wiley and Sons, New York, 1986, pp. 141–154 Search PubMed.
- A. Tani, S. Hayward and C. N. Hewitt, Int. J. Mass Spectrom. Ion Processes, 2002, in press Search PubMed.
- C. Warneke, C. van der Veen, S. Luxembourg, J. A. de Gouw and A. Kok, Int. J. Mass Spectrom. Ion Processes, 2001, 207, 167 Search PubMed.
- Y. K. Lau, S. Ikuta and P. Kebarle, J. Am. Chem. Soc., 1982, 104, 1462 CrossRef CAS.
- I. Dotan, D. L. Albritton, W. Lindinger and M. Pahl, J. Chem. Phys., 1976, 65, 5028 CrossRef CAS.
- R. A. Street, S. G. Duckham and C. N. Hewitt, J. Geophys. Res. [Atmos.], 1996, 101, 22799 Search PubMed.
- W. Lindinger, in Gaseous Ion Chemistry and Mass Spectrometry, ed. J. H. Futrell, John Wiley and Sons, New York, 1986, pp. 237–257 Search PubMed.
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183 CrossRef CAS.
- P. Spanel and D. Smith, Int. J. Mass. Spectrom. Ion Processes, 1998, 181, 1 CrossRef CAS.
- J. A. De Gouw, C. J. Howard, T. G. Custer, B. M. Baker and R. Fall, Environ. Sci. Technol., 2000, 34, 2640 CrossRef CAS.
- T. Karl, R. Fall, A. Jordan and W. Lindinger, Environ. Sci. Technol., 2001, 35, 2926 CrossRef CAS.
- T. Karl, R. Fall, P. J. Crutzen, A. Jordan and W. Lindinger, Geophys. Res. Lett., 2001, 28, 507 Search PubMed.
- S. Hayward, A. Tani, S. M. Owen and C. N. Hewitt, Tree Physiol., 2002, submitted Search PubMed.
- T. Karl, P. J. Crutzen, M. Mandl, M. Staudinger, A. Guenther, A. Jordan, R. Fall and W. Lindinger, Atmos. Environ., 2001, 35, 5287 CrossRef CAS.
- C. Warneke and J. A. de Gouw, Atmos. Environ., 2001, 35, 5923 CrossRef CAS.
- C. Warneke, R. Holzinger, A. Hansel, A. Jordan, W. Lindinger, U. Poschl, J. Williams, P. Hoor, H. Fischer, P. J. Crutzen, H. A. Scheeren and J. Lelieveld, J. Atmos. Chem., 2001, 38, 167 CrossRef CAS.
- A. Hansel and A. Wisthaler, Geophys. Res. Lett., 2000, 27, 895 CrossRef.
- A. Wisthaler, N. R. Jensen, R. Winterhalter, W. Lindinger and J. Hjorth, Atmos. Environ., 2001, 35, 6181 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |