Organometallic sesquialkoxides of aluminium, gallium and indium
Bernhard Neumüller
Fachbereich Chemie der Universität Marburg, Hans-Meerwein-Strasse, D-35032, Marburg, Germany
First published on 20th November 2002
Abstract
Organometallic sesquihalides of aluminium are important intermediates in technical processes. However, those compounds and their homologues with gallium or indium centers have not been structurally characterized so far. On the other hand organometallic sesquialkoxides have been isolated. The major synthetic routes and the structures of the corresponding products will be discussed. Furthermore, cage-contructiveness reactions having sesquialkoxides as educts will be shown. Discussion will focus primarily on the syntheses, the spectroscopic findings and a structural comparison. Especially the structural motifs deserve attention because of the structural connection to the well-known earth metal alkoxides.
 Bernhard Neumüller Bernhard Neumüller | Professor Dr Bernhard Neumüller obtained his Diploma and his PhD at the University of Stuttgart (Germany) under Professor Dr Dr h c Ekkehard Fluck (1984–1987) after which he became a post doctoral fellow at the MIT under the supervision of Professor Dr Dietmar Seyferth (Cambridge, Massachusetts, USA, 1987–1988). He started his Habilitation at the University of Marburg (Germany) in 1988 in the group of Professor Dr Dr h c Kurt Dehnicke with a Liebig fellowship of the Fonds der Chemischen Industrie. In 2000 he was appointed as an apl professor in Marburg. Currently, he is working at the University of Stuttgart (Germany), substituting a professorship (2001–2002). |
1 Introduction
Organometallic sesqui compounds serve as explanation in a variety of technical processes and are part of many text book chapters.1–4 Nevertheless, a closer view exhibits our lack of information concerning the nature of those sesqui compounds. For example there are no structural data for any organometallic sesquihalide, even if one is convinced of their presence during reactions, e.g. the Hüls process for the synthesis of diorganoaluminiumhalides (Scheme 1).1 The assumption that the sesquihalide will have a dimeric structure with an Al2X2 four-membered ring is derived from the wellknown species R2MX and RMX2.2,3 Attempts at a well directed synthesis of organoindium sesquichlorides were not successful.5 Very recently, the existence of sesquihydrides, [{tBu2MH}2{tBuMH2}2] (M = Al, Ga), have been verified by W. Uhl et al.6 | ||
| Scheme 1 | ||
The existence of organometallic sesquialkoxides was proven during the last 10 years, mainly for aluminum compounds like [Al{(OEt)2AlMe2}3] and in two cases for the heteronuclear aluminium–gallium compounds [Al{(OEt)2GaR2}3] (R = Me, Et) (Fig. 1). The most electropositive metal occupies the central position.
 | ||
| Fig. 1 Graphical representation of the general build-up of aluminium-based sesquialkoxides. | ||
The synthesis of such sesquialkoxides is basically a ligand redistribution reaction between metallanes and earth metal alkoxides.7,8 The sesquialkoxides in Fig. 1 are also interesting in the search for unimolecular precursors to Al2O3 since they already contain the desired ratio of group 13/16 elements in their composition.7
Another useful variant for the introduction of alkoxo functions may be the treatment of metallanes with alcohols to form diorganoalkoxometallanes and organodialkoxometallanes, respectively.
Diorganoalkoxometallanes of aluminium, gallium and indium are well-known with regard to their synthesis and structures.2,3,9,10 In contrast to this organodialkoxometallanes are rare species, even if derivatives are mentioned in literature.2,3 However, the reliable data are sparse. Especially the strategies of synthesis are different from those for the first introduced class of compounds.9,10 The mostly applied syntheses for [R2M–OR′]n are the treatment of metallanes with alcohols [eqn. (1)] and the metathesis reaction of organohalogenometallanes with alkoxides of alkali metals [eqn. (2)].
| MR3 + HOR′ → 1/n [R2M–OR′]n + RH | (1) |
| R2MX + M′OR → 1/n [R2M–OR′]n + M′X M = Ga, In; M′ = alkali metal; R, R′ = alkyl, aryl | (2) |
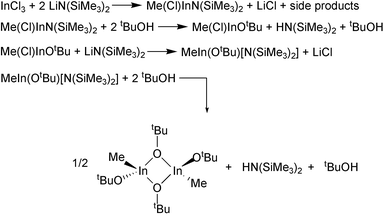 | ||
| Scheme 2 | ||
2.1 Aluminium compounds
The first mentioned organometallic sesquialkoxides were obtained by the treatment of AlMe3 or AlEt3 with alcohols derived from thiophene or furane [eqn. (3)]15a followed by the previously reported [Al{(OR′)2AlR2}3] (R = Me, R′ = 10-undecene).15b | (3) |
A second synthesis route to homonuclear aluminium sesquialkoxides exists in the combination of equimolar amounts of aluminium alkoxide and alane in toluene at 25 °C with subsequent reaction under reflux conditions [eqn. (4)].7,8
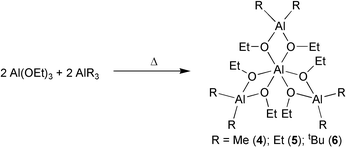 | (4) |
Compounds 1–6 may be described either as Al3+ ion coordinated with three metalate units [R2Al(OR′)2]− or, in emphasizing their sesqui character, as [{R2AlOR′}2{RAl(OR′)2}2]. According to the structural characterized compounds 1, 4 and 5, the complexes possess nearly D3 symmetry (Fig. 2 and 3).
 | ||
| Fig. 2 Structure of a sesquialkoxide with 2-thiophenemethoxide functions (1). | ||
![Result of the X-ray structure determination of [Al{(OEt)2AlMe2}3] (4).](/image/article/2003/CS/b205568f/b205568f-f3.gif) | ||
| Fig. 3 Result of the X-ray structure determination of [Al{(OEt)2AlMe2}3] (4). | ||
The methylene protons of the OCH2R′ groups must be diasterotopic because of the chirality of 1–6. This should lead to AB spin systems in the 1H NMR spectra under the assumption of slow ligand redistribution reactions in comparison to the NMR time scale.7,15a Compounds 4–6 give multiplet signals for the methylene protons and one signal for the R–Al group indicating the supposed behaviour.7 The 27Al spectra of 4 and 5 show two signals at ∼11 and ∼150 ppm for the Al atoms with coordination number (cn) six and for those with cn 4, respectively. The basic structural motif in 1, 4 and 5 and similar aluminium compounds7,8 is well-known from aluminium alkoxides.16 Spectroscopical investigations16,17 and X-ray structure determinations showed the tetrameric character of the OiPr and OCH2Ph derivative in solution and the solid state (Fig. 4).17–20
![Structure of the tetrameric aluminium alkoxide [Al(OiPr)3]4.](/image/article/2003/CS/b205568f/b205568f-f4.gif) | ||
| Fig. 4 Structure of the tetrameric aluminium alkoxide [Al(OiPr)3]4. | ||
Especially [Al(OiPr)3]4 is known to be the key compound in the Meerwein-Ponndorf-Verley reduction.21 Compound 6 undergoes an equilibrium between the tetranuclear and two trinuclear species (Scheme 3).7
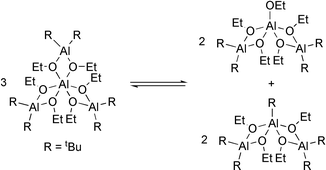 | ||
| Scheme 3 | ||
The trinuclear complex contains an Al3+ center with cn 5, as was shown for the solid state structure of Al(OcHex)3, [cHexOAl{Al(OcHex)4}2] (Fig. 5).22
![Structure of the trimeric aluminium alkoxide [Al(OcHex)3]3.](/image/article/2003/CS/b205568f/b205568f-f5.gif) | ||
| Fig. 5 Structure of the trimeric aluminium alkoxide [Al(OcHex)3]3. | ||
However, other degrees of association are known, depending mainly on the bulk of the alkoxo group, e.g. the dimeric species [Al(OtBu)3]223 and [Al(OSiMe3)3]2.24
Heteronuclear sesquialkoxides containing Al and Ga were formed in the reaction of GaR3 (R = Me, Et) with Al(OEt)3 in a molar ration of 3∶2, whereas the products [Al{(EtO)2GaR2}3] (R = Me (7), Et (8)) exhibit similar tetranuclear structures (Fig. 6), found in 1, 4 and 5. Therefore, one unit of AlR3 was eliminated in both cases, 7 and 8.7,8
![Structure of the heteronuclear aluminium–gallium sesquialkoxide [Al{(EtO)2GaMe2}3] (7).](/image/article/2003/CS/b205568f/b205568f-f6.gif) | ||
| Fig. 6 Structure of the heteronuclear aluminium–gallium sesquialkoxide [Al{(EtO)2GaMe2}3] (7). | ||
Heteronuclear alkoxides with a central lanthanide atom are also well-known from their OiPr- derivatives, [Ln{M(OiPr)4}3] (Ln = Sc, Y, La–Yb; M = Al, Ga).25 All the spectroscopic data and the cryoscopical measurements point to the same structural motif of Fig. 1. The cation, which prefers a high cn (Ln3+) is always in the center of the molecule. An X-ray structure determination of the erbium derivative confirms the proposed structure.8,26 The corresponding Cr3+ compound, [Cr{Al(OiPr)4}3], was first mentioned by R. C. Mehrotra,27a but H. Meerwein had already in 1929 synthesized salts with the general composition [M{Al(OR)4)}n].27b
2.2 Gallium compounds
Although the reaction of a gallane or indane with an equimolar amount of alcohol leads directly to a diorganoalkoxometallane species, [R2M–OR′]n, the treatment with two equivalents or an excess of alcohol gives no organodialkoxo derivative, [RM(OR′)2]n, even in boiling toluene. The result of the reaction depends on the reactivity of the corresponding metallane. However, MMe3 (M = Ga, In) give products which can be explained only by generation of methane combined with ligand redistribution.The more reactive GaMe3 gives with PhCH2OH a new type of sesquialkoxide. A homonuclear Ga complex, [Ga{MeGa(OCH2Ph)3}3] (9) was formed during the reaction of GaMe3 with an excess of PhCH2OH in boiling toluene [eqn. (5)] with evolution of methane.28
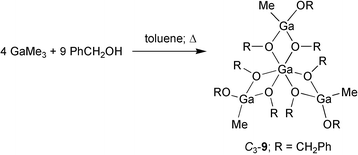 | (5) |
According to the NMR spectra two species, a C3-symmetrical and a C1-symmetrical were present in a molar ration of 1∶1. The spectroscopic findings verify the structural motif for the sesquialkoxides in solution. In all cases, were the OCH2Ph ligand is present, an AB-spin system was found because of the chirality of the complexes (diastereotopic H atoms). This result was also a crucial hint for the tetrameric character of [Al(OCH2Ph)3]4 in solution (solid state structure see ref. 20).17 The 1H NMR spectra show two AB spectra for the methylene protons in C3-9 and nine AB spectra in C1-9. An X-ray structure determination could only be performed for C3-9. The result is consistent with the structure in solution (Fig. 7).28
![Structure of the homonuclear sesquialkoxide C3-[Ga{MeGa(OCH2Ph)3}3] (C3-9).](/image/article/2003/CS/b205568f/b205568f-f7.gif) | ||
| Fig. 7 Structure of the homonuclear sesquialkoxide C3-[Ga{MeGa(OCH2Ph)3}3] (C3-9). | ||
However, according to the structure in Fig. 7, 9 may be described also as new type of a sesquialkoxide, [{MeGa(OCH2Ph)2}3{Ga(OCH2Ph)3}].
2.3 Indium compounds
The less reactive metallane InMe3 gives even with an excess of alcohol the type [{Me2InOR}2{MeIn(OR)2}2] [R = cHex (10), CH2Ph (11)] [eqn. (6)].28,29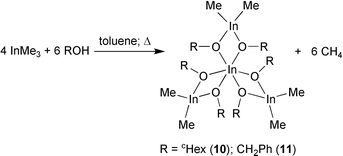 | (6) |
The reactions occur at 20 °C and in boiling toluene to give the same products. Compounds 10 and 11 possess nearly D3-symmetry in the solid state with characteristic sesquialkoxide structures (Fig. 8 and 9).
![Structure of [{Me2InOcHex}2{MeIn(OcHex)2}2] (10).](/image/article/2003/CS/b205568f/b205568f-f8.gif) | ||
| Fig. 8 Structure of [{Me2InOcHex}2{MeIn(OcHex)2}2] (10). | ||
![Structure of [{Me2InOCH2Ph}2{MeIn(OCH2Ph)2}2] (11). Compounds 10 and 11 exhibit nearly D3-symmetry.](/image/article/2003/CS/b205568f/b205568f-f9.gif) | ||
| Fig. 9 Structure of [{Me2InOCH2Ph}2{MeIn(OCH2Ph)2}2] (11). Compounds 10 and 11 exhibit nearly D3-symmetry. | ||
Cryoscopic measurements and the NMR data confirm that the structural motif is preserved in solution so that the methylene groups of the OCH2Ph functions in 11 result in an AB spin system.
3 Reactions of sesquialkoxides
So the question was, is it possible to substitute more than six methyl groups by alkoxy functions in an indium compound and will the In–O skeleton still remain? To answer the question, compound 11 or InMe3 (careful addition of PhCH2OH to InMe3 at 0 °C) were heated in pure PhCH2OH at 160 °C, where further gas evolution was observed (CH4). Nevertheless, the resulting compound, 12, was surprising. After workup the unexpected pentanuclear complex [(MeIn)5(OCH2Ph)8(O)] (12) was isolated [eqn. (7)] which can be described as an O-centered, decapitated rhombohedral dodecahedron (Fig. 10).29 | (7) |
![Structure of [(MeIn)5(OCH2Ph)8(O)] (12) with an O2−-centered decapitated rhombic dodecahedronal In5O9 skeleton.](/image/article/2003/CS/b205568f/b205568f-f10.gif) | ||
| Fig. 10 Structure of [(MeIn)5(OCH2Ph)8(O)] (12) with an O2−-centered decapitated rhombic dodecahedronal In5O9 skeleton. | ||
This structural motif was also observed in [{iPrOIn}5{OiPr}8(O)]30,31 and in rare earth metal chemistry in [Ln5(OiPr)13(O)] (Ln = Sc, Y, Yb)31 as well as in [(CpY)5(OMe)8(O)].32 Another interesting compound for comparison is the hexameric [Li2N{(H)Si[N(SiMe3)CH2CH2NSiMe3]}]6,33 which possesses a central Li9N5 skeleton, an inverse arrangement to the previous M5O9 skeletons.
Very significant are the data from vibrational spectroscopy. Because of the strong ionic character of the M–O bonds the intensities of the corresponding M–O skeletal vibrations are quite different in the IR and the RE (Raman effect) spectra. Strong and partly broad absorptions at 495, 460 (10), 368 (11), and 364, 281 (12) cm−1 in the IR spectra correspond with very weak emissions in the RE spectra at 506 (10) and 368 (11) cm−1.28,29
The four μ2-bridging oxygen atoms in 12 form a basal plane, which should be able to act like a tetradentate ligand to large cations like Cs+. Reaction of 12 with Cs metal leads not to a single electron transfer but to two of them displacing a unit of InMe under formation of a double negative charged [(RIn)4(OR)8(O)]2− unit [eqn. (8)].29 This unit coordinates two Cs+ ions to give [Cs{Cs(THF)}{MeIn(OCH2Ph)2}4(O)] (13), supported by additional cation–π-electron interactions (Fig. 11).
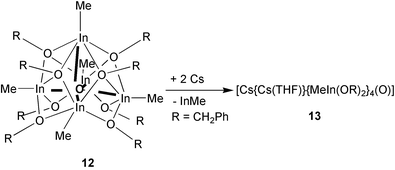 | (8) |
![Molecular structure of the rhombic dodecahedronal heteronuclear complex [Cs{Cs(THF)}{MeIn(OCH2Ph)2}4(O)] (13).](/image/article/2003/CS/b205568f/b205568f-f11.gif) | ||
| Fig. 11 Molecular structure of the rhombic dodecahedronal heteronuclear complex [Cs{Cs(THF)}{MeIn(OCH2Ph)2}4(O)] (13). | ||
A THF ligand belongs to the coordination sphere of one Cs+ ion. The C2H2–C3H2 ‘backbone’ of the THF ligand fits perfectly into the cavity of the second Cs+ ion belonging to a adjacent molecule 13 to form infinite chains along [010].
The existence of a molecule like 13 confirms previous results in synthesizing such M–O cages, because there is a totally different way to arrive at this class of compound. In this second route the triorganochloroindate Cs[(PhCH2)3InCl] was treated with dry dioxygen giving the complex [Cs2{PhCH2In(OCH2Ph)2}4(O)] (14) under insertion of oxygen atoms into In–C bonds (Fig. 12).34
![Structure of the benzyl derivative [Cs2{PhCH2In(OCH2Ph)2}4(O)] (14).](/image/article/2003/CS/b205568f/b205568f-f12.gif) | ||
| Fig. 12 Structure of the benzyl derivative [Cs2{PhCH2In(OCH2Ph)2}4(O)] (14). | ||
In both cases the O2− ion is an integral part of the In4O92− complex framework found also for a variety of other organometallic complexes and clusters.35
4 Structures
The molecular structures of 1, 4, 5, 7 and 9–11 exhibit quite similar structural features. Planar M2O2 four-membered rings with small inner ring angles at M (mean value 75°) and angles greater than 90° (mean value 105°) at O. The M–O distances can be divided into three categories. The longest M–O values were observed for the six central M–O bonds because of coordination number six at the metal ion (Al–O: 189 pm (4, 5, 7);7 Ga–O: 198 pm (9);28 In–O: 216 pm (10, 11)28,29). Shortenings were found for M(cn 4)–O bond lengths (Al–O: 180 pm (4, 5); Ga–O: 192 pm (7, 9); In–O: 216 pm (10, 11); in 10 and 11 the accumulation of six electronegative bonding partners at the central In3+ ion compensate the lower cn at the peripheral metal atoms). The terminal Ga–O bonds possess the smallest values with 181.2(2) pm.Compound 12 combines μ2- and μ3-bridging alkoxo groups as well a μ5-bridging O2− function leading to a distinct variation of the In–O bonds. The average value for the basal μ2-bridging alkoxo groups amounts to 215 pm whereas the μ3-functionality exhibits 234 pm. The central O2− function gives a mean In–O bond distance of 228 pm caused by the combination of two charges and cn five. This variation of the In–O bonding parameters was found also in [{iPrOIn}5{OiPr}8(O)].30,31
The common structural motif of 13 and 14 is an oxygen-centered hetereonuclear rhombic dodecahedron. This is built up by an anionic torus [{RIn(OCH2Ph)2}4(O)]2− acting as a metalla crown ether for the coordination of two Cs+ ions. In additon both cations are wrapped by the phenyl groups of the benzyl ligands forming weak Cs+–π-aryl contacts.29,34 All OCH2Ph groups have μ3-character if one totals In–O and Cs–O contacts leading to an average In–O value of 225 pm in 13. The Cs+–O distances amount to 305 pm. Because of the central position of the O2−-function in 13 (cn 4 + 2) this In–O bond length of 219 pm is significantly shorter than the 228 pm in 12.
5 Concluding remarks
The Meerwein-Ponndorf-Verley reagent [Al(OiPr)3]3 possesses a structural motif which dominates a part of the organometallic chemistry of group 13 metals. Especially the combination of alkoxo ligands and organic groups leads not only in ligand redistribution reactions of an exact molarity to sesquialkoxides but also all attempts to generate organodialkoxo metallanes from the simple treatment of MMe3 with excess alcohol yield sesquialkoxides as products. However, it seems to be favourable for nature to separate such sesqui compounds into two electron precise species with an electron octet at M, an M3+ ion and metalate ions [MR4]−, which form together chelate complexes. In heterometallic sesqui compounds the most electropositive metal ion occupies the center of the molecule caused by the tendency of the those metals to maximize their M–O interactions. The majority of organometallic sesquialkoxides have stable structures in solution and the solid state and do not undergo ligand redistribution reactions.Further incorporation of oxygen atoms with simultaneous generation of large polymetallic complexes is not unusual but quite common in organometallic chemistry,35 no matter whether the source is molecular oxygen or water.
Nevertheless, the successes in synthesis and characterization of sesqui compounds during the last years are only the tip of the iceberg. Efforts have be started to get a closer insight into this area, particularly concerning the organometal halides of group 13 elements. Ligand redistributions occur in intercrossing experiments, indicating at least the temporary presence of sesqui species. As known for the aluminium alkoxides, solvent effects and the sterical demand of the organic group besides electronical reasons may be responsible for the different molecular structures in solution and the solid state.
6 References
- Ch. Elschenbroich and A. Salzer, Organometallchemie, 3rd edn., B. G. Teubner, Stuttgart, 1993 Search PubMed.
- Gmelin Handbook of Inorganic Chemistry, Gallium, Organogallium Compounds, Part 1, Springer, Berlin, New York, 1987 Search PubMed.
- J. Weidlein, in, Gmelin Handbook of Inorganic Chemistry, Indium, Organoindium Compounds, Part 1, Springer, , Berlin, New York, 1991 Search PubMed.
- Chemistry of Aluminium, Gallium, Indium and Thallium, ed. A. J. Downs, Chapmann and Hall, London, 1993 Search PubMed.
- B. Neumüller, Z. Naturforsch., 1991, 46b, 753 Search PubMed.
- W. Uhl, L. Cuypers, R. Graupner, J. Molter, A. Vester and B. Neumüller, Z. Anorg. Allg. Chem., 2001, 627, 607 CrossRef CAS.
- D. A. Atwood, J. A. Jegier, S. Liu, D. Rutherford, P. Wei and R. C. Tucker, Organometallics, 1999, 18, 976 CrossRef CAS.
- M.-A. Munoz-Hernandez, P. Wei, S. Liu and D. A. Atwood, Coord. Chem. Rev., 2000, 210, 1 CrossRef CAS and references cited therein.
- Overview on [R2GaOR′]2: (a) W. M. Cleaver, A. R. Barron, A. R. McGufey and S. G. Bott, Polyhedron, 1994, 13, 2831 CrossRef CAS; (b) M. R. Kopp and B. Neumüller, Z. Anorg. Allg. Chem., 1997, 623, 796 CrossRef CAS.
- Overview on [R2InOR′]2: M. Häußlein, H.-D. Hausen, K. W. Klinkhammer, J. Weidlein and K. Merz, Z. Anorg. Allg. Chem., 1999, 625, 1608 Search PubMed (see also ref. 29).
- T. Arai, H. Sasai, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 441 CrossRef CAS.
- S. Chitsaz and B. Neumüller, Organometallics, 2001, 20, 2338 CrossRef CAS.
- M. D. Healy, D. A. Wierda and A. R. Barron, Organometallics, 1988, 7, 2543 CrossRef CAS and references cited therein.
- M. Veith, S. Hill and V. Huch, Eur. J. Inorg. Chem., 1999, 1343 CrossRef CAS.
- (a) J. de Mel, M. L. Sierra, D. G. Hendershot and J. P. Oliver, Organometallics, 1994, 13, 2079 CrossRef; (b) J. Turunen, T. T. Pakkanen and B. Löfgren, J. Mol. Catal. A, 1997, 123, 35 CrossRef CAS.
- Reviews about metal alkoxides is given in: R. C. Mehrotra, I. Rothwell, A. Singh and D. C. Bradley, Alkoxo and Aryloxo Derivatives of Metals, Academic Press, San Diego, 2002 Search PubMed and Chemistry of Metal Alkoxides, eds. N. Turova, E. Turevskaya, V. Kessler and M. Yanovskaya, Kluwer Academic Publishers, Dordrecht, 2002 Search PubMed and references cited therein.
- J. G. Oliver and I. J. Worrall, J. Chem. Soc. (A), 1970, 1389 RSC and references cited therein.
- N. Y. Turova, V. A. Kozunov, A. I. Yanovskii, N. G. Bokii, Y. T. Struchkov and B. L. Tarnopol’skii, J. Inorg. Nucl. Chem., 1979, 41, 5 CrossRef CAS.
- K. Folting, W. E. Streib, K. G. Caulton, O. Poncelet and L. G. Hubert-Pfalzgraf, Polyhedron, 1991, 10, 1639 CrossRef CAS.
- J. Pauls and B. Neumüller, Z. Anorg. Allg. Chem., 2001, 627, 2127 CrossRef CAS.
- J. March, Advanced Organic Chemistry, 3rd edn., Wiley, New York, 1985 Search PubMed.
- J. Pauls and B. Neumüller, Z. Anorg. Allg. Chem., 2000, 626, 270 CrossRef CAS.
- R. H. Cayton, M. H. Chisholm, E. R. Davidson, V. F. DiStasi, P. Du and J. C. Huffman, Inorg. Chem., 1991, 30, 1020 CrossRef CAS.
- M. H. Chisholm, J. C. Huffman and J. L. Wesemann, Polyhedron, 1991, 10, 1367 CrossRef CAS.
- R. C. Mehrotra, P. N. Kapoor and J. M. Batwara, Coord. Chem. Rev., 1980, 31, 67 CrossRef CAS.
- M. Wijk, R. Norrestam, M. Nygren and G. Westin, Inorg. Chem., 1996, 35, 1077 CrossRef CAS.
- (a) J. V. Singh, N. C. Jain and R. C. Mehrotra, Synth. React. Inorg. Met.-Org. Chem., 1979, 9, 79 CAS; (b) H. Meerwein and T. Bersin, Liebigs Ann. Chem., 1929, 476, 113 Search PubMed.
- S. Chitsaz, E. Iravani and B. Neumüller, Z. Anorg. Allg. Chem., 2002, 628, 2279 CrossRef CAS.
- S. Chitsaz and B. Neumüller, Z. Anorg. Allg. Chem., 2001, 627, 2451 CrossRef CAS.
- D. C. Bradley, H. Chudzynska, D. M. Frigo, M. B. Hursthouse and M. A. Mazid, J. Chem. Soc., Chem. Commun., 1988, 1258 RSC.
- D. C. Bradley, H. Chudzynska, D. M. Frigo, M. E. Hammond, M. B. Hursthouse and M. A. Mazid, Polyhedron, 1990, 9, 719 CrossRef CAS.
- W. J. Evans and M. S. Sollberger, J. Am. Chem. Soc., 1986, 108, 6095 CrossRef CAS.
- S. Abele, G. Becker, U. Eberle, P. Oberprantacher and W. Schwarz in: Organosilicon Chemistry IV, eds. N. Auner and J. Weis, Wiley-VCH, Weinheim, New York, 2000 Search PubMed.
- T. Kräuter and B. Neumüller, Chem. Eur. J., 1997, 3, 568 CrossRef CAS.
- A. E. H. Wheatley, Chem. Soc. Rev., 2001, 30, 265 RSC.
| This journal is © The Royal Society of Chemistry 2003 |