Investigation of the molecular structure of the radical anions of some pyrimidine-type bases in aqueous solution by comparison of calculated hyperfine coupling constants with EPR results†
S.
Naumov
a,
J.
Reinhold
b and
D.
Beckert
c
aInstitute of Surface Modification, Permoserstrasse 15, D-04318, Leipzig, Germany
bUniversity of Leipzig, Faculty of Chemistry and Mineralogy, Wilhelm Ostwald Institute of Physical and Theoretical Chemistry, Johannisallee 29, D-04103, Leipzig, Germany
cUniversity of Leipzig, Faculty of Chemistry and Mineralogy, Interdisciplinary Group for Time Resolved Spectroscopy, Permoserstrasse 15, D-04318, Leipzig, Germany
First published on 22nd November 2002
Abstract
New results of DFT B3LYP calculations in aqueous solution are presented for the radical anions of uracil, thymine, 1-methylthymine, 1-methyluracil and 1,3-dimethyluracil. The most relevant molecular structure of the radical anions in water optimised with extended basis sets using either the Onsager or the CPCM self-consistent reaction field model is the boat conformation. The structure shows pyramidality at the radical centre C6 connected with a deviation of the C6–H atom from the molecular plane up to around 12°. Gas-phase structures, even optimised with extended basis sets, are not able to reproduce the large values of the hyperfine coupling (hfc) constant of the C6–H atom known from the experiments (about 35 MHz). Reliable values for this coupling require optimisations involving the solvent. The CPCM model appears to be superior to the Onsager model. Optimisations with inclusion of up to 10 water molecules, thus modelling hydrogen bonding with the solvent, confirm the results obtained with the continuum models.
Introduction
The radical ions and free radicals of pyrimidine-type bases are important intermediate species in DNA chemistry. Therefore, many experimental and theoretical investigations of the radical ions and free radicals have been reported.1–6 To facilitate the interpretation of the complex EPR spectra in the solid phase, quantum chemical calculations at various levels of theory were carried out and summarized in a review by Colson and Sevilla.5 Wetmore et al.6 have calculated structures, energies and hyperfine coupling (hfc) constants of dehydrogenated, hydrogenated and hydroxylated radiation products of thymine, 1-methylthymine and uracil applying density functional theory (DFT). The highly resolved EPR spectra of pyrimidine-type base radicals in aqueous solution allow to determine the hfc constants of most of the hfc-active nuclei, and well-defined radical structures can be obtained. Time-resolved FT EPR7,8 experiments with laser photolysis excitation are appropriate to study the primary radical anions of pyrimidine-type bases.9 The comparison of these highly resolved FT EPR spectra with the parameters calculated in vacuum by Wetmore et al.6 for the radical anions of uracil and thymine shows a strong deviation from the experimental values of the C6–H hfc constants. The observed couplings are much larger than the calculated ones. The molecular structure of the radical anions of the pyrimidine-type bases is point of the discussion.10 Whereas Wetmore et al.11 have obtained a puckered ring structure for the uracil and thymine radical anions, Close10 explains the C6–H couplings observed in water (35.0 MHz for the uracil radical anion9) with a planar structure, though the calculated value at the DFT B3LYP level with a large basis set is overestimated (ca. 41 MHz). Otherwise, it has been found that consideration of solvent effects by a polarized continuum model leads to a remarkable agreement between computed and experimental data for 5,6-dihydro-6-thymyl and 5,6-dihydro-5-thymyl radicals.12 It could be shown that also the molecular structure and the spin density distribution of the radical cations and their deprotonated successor radicals of pyrimidine-type bases are influenced by a surrounding dielectric solvent and that the hfc constants calculated for an aqueous medium are in good agreement with the experimental FT EPR results.13 The polarized continuum model was also very useful for the identification of the transients and the reliable calculation of the isotropic hfc constants of N1-substituted cytosines in aqueous solution.14 Otherwise, it could be shown15 that the hydrogen bonding of the p-benzosemiquinone anion radical with explicit water molecules leads to better numerical agreement of the calculated hfc constants with experiment.The questions, whether the radical anions of pyrimidine-type bases have planar or nonplanar structure in water and which computation scheme is most reliable for the interpretation of EPR data are of continuous interest in the framework of our investigations. In this paper, we present the results of extensive DFT calculations in aqueous solution for the radical anions of uracil, thymine, 1-methylthymine, 1-methyluracil and 1,3-dimethyluracil (see Scheme 1). The solvent is considered as dielectric continuum as well as by a supermolecule model with an increasing number of water molecules to take hydrogen bonding explicitly into account.
 | ||
| Scheme 1 | ||
Computational details
The calculations were done using the Gaussian 98 package.16 For the systems under study, geometry optimisations were performed applying the density functional theory (DFT) approach with B3LYP hybrid functionals.17–19 For comparison, sample systems were also calculated at the conventional ab initio Hartree-Fock (UHF) and the Möller-Plesset perturbation theory (MP2)20 levels. Frequency calculations were used to characterise the stationary points.To investigate the influence of a solvent on the molecular structure of the radical anions, geometry optimisations were carried out in water (ε![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =78) using two different self-consistent reaction field (SCRF) models: the Onsager21
(SCRF=Dipole) and the CPCM (COSMO)22,23 model (SCRF=CPCM). For the investigation of the solvent effect on the spin distribution and the hfc constants, additionally, the polarized continuum model24,25
(SCRF=PCM) and the isodensity polarized continuum model26
(SCRF=IPCM) were considered.
=78) using two different self-consistent reaction field (SCRF) models: the Onsager21
(SCRF=Dipole) and the CPCM (COSMO)22,23 model (SCRF=CPCM). For the investigation of the solvent effect on the spin distribution and the hfc constants, additionally, the polarized continuum model24,25
(SCRF=PCM) and the isodensity polarized continuum model26
(SCRF=IPCM) were considered.
A large variety of different basis sets has been involved. For the geometry optimisations, the standard 6-31G(d), 6-31G(d,p), 6-311G(d,p), 6-31+G(d,p), 6-311+G(d,p) and 6-311+G(2df,p) basis sets were used. To obtain relative energies, spin densities and isotropic hfc constants, single-point calculations were performed including also the highly extended 6-311++G(2df,p) basis set and, additionally, the EPR-2 and EPR-3 basis sets of Barone and Cossi,27 which have been optimised for the computation of hfc constants in solution by DFT methods (particularly B3LYP).
The influence of hydrogen bonding with the solvent on the molecular structure and the hfc constants of the uracil radical anion was investigated at the B3LYP/6-31G(d), B3LYP/6-31G(d,p) and B3LYP/6-31+G(d,p) levels of theory. Hydrogen-bonded complexes with up to 10 water molecules could be successfully optimised. For the complexes with up to 5 water molecules, it is possible to locate the most stable structures. This corresponds to the formation of a first rather stable water shell around the uracil radical anion (see below Fig. 7). It is practically impossible to locate the most favourable structures for the complexes with more than 5 water molecules. In these cases, there are several possibilities to form additional hydrogen bonds leading to complexes which differ only slightly in energy and structure. However, these small structural deviations have a remarkable influence on the calculated hfc constants. So, we consider the results obtained for the complexes with up to 5 water molecules to be trustworthy, whereas those with a larger number of water molecules have a more qualitative character to test the further trends.
Optimisation of molecular structures
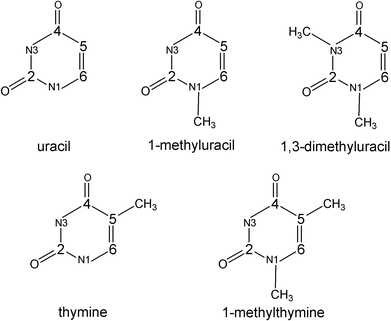
We first focus on the uracil radical anion. To investigate the dependence of the calculated molecular structure on the basis set involved, complete DFT optimisations were performed using different basis sets in both vacuum and water. A few calculations with UHF and MP2 methods have been added for comparison. Whereas the equilibrium structure of the uracil singlet ground state is known to be characterized by a planar ring,6 the optimisations for the radical anion yield two different stationary points on the potential surface, shown in Fig. 1, with relatively small differences in energy. | ||
| Fig. 1 Stable structures and spin density distribution (isospin=0.035) of the uracil radical anion calculated with B3LYP and Onsager model in water. (a) Chair conformation at 6-31G(d) basis set, (b) boat conformation at 6-311+G(d,p) basis set and (c) planar conformation (saddle point) at 6-311+G(d,p) basis set. The energy difference between the latter two is from 4.2 to 7.3 kJ mol−1, depending on the level of theory. | ||
One structure (Fig. 1c) has a planar ring in vacuum as well as in water independent of the basis set (as calculated with DFT and both UHF and MP2 methods). The frequency analysis for this structure yields two imaginary frequencies (e.g. in vacuum, 459i and 88i at B3LYP/6-311+G(d,p) or 603i and 29i at UHF/6-311+G(d,p)) indicating that this structure is a second-order saddle point.
The second structure is characterised by a remarkably deformed ring (Fig. 1a and 1b). This structure appears to be a minimum on the potential surface. Selected structural parameters are collected in Table 1. If the two nearly parallel C4–C5 and C2–N1 bonds of the ring are adopted to describe a reference plane, then the distortion of the ring structure from planarity can be characterised by the dihedral C4–C5–C6–N1 and C2–N3–C4–C5 angles. Following the arguments of Wetmore et al.,11 the ring puckering arises as a result of the localization of the radical centre at one of the doubly bound carbon atoms (C6 in uracil). Consequently, the inclusion of diffuse functions should reduce the degree of puckering due to stronger delocalisation of the unpaired electron. However, in the vacuum optimisations, the influence of diffuse functions on the C4–C5–C6–N1 angle is rather small which is in line with the results of Wetmore et al.6 Otherwise, the C2–N3–C4–C5 angle, which characterises the distortion on the opposite side of the molecular ring, is significantly influenced. This dihedral angle changes not only its absolute value, but also even the sign. In fact, in vacuum, the lack of diffuse functions leads to a chair conformation of the six-membered ring (Fig. 1a). If diffuse functions are involved, which is generally important in the case of anions, the boat conformation appears to be most stable (Fig. 1b).
=Dipole) and the COSMO (SCRF=CPCM) models. MP2/6-31G(d) and UHF/6-311+G(d,p) methods are shown for comparison
| Basis set | 6-31G(d) | MP2/6-31G(d) | 6-31G(d,p) | 6-311G(d,p) | 6-31+G(d,p) | 6-311+G(d,p) | 6-311+G(2df,p) | UHF/6-311+G(d,p) |
|---|---|---|---|---|---|---|---|---|
| Vacuum | ||||||||
| C4C5C6N1 | 7.0 | 12.6 | 6.7 | 8.2 | 12.9 | 12.6 | 11.1 | 13.3 |
| C2N3C4C5 | 9.7 | 9.4 | 9.0 | 2.0 | −7.2 | −7.6 | −5.8 | −4.2 |
| C4C5C6–H | 149.6 | 150.9 | 151.3 | 152.0 | 157.5 | 158.4 | 158.2 | 155.7 |
| C4C5,N1–H | 145.9 | 144.7 | 147.7 | 154.0 | 159.8 | 161.1 | 163.0 | 157.5 |
| C5C4N3–H | 171.7 | 172.2 | 172.3 | 176..9 | 177.3 | 177.1 | 178.0 | 180.0 |
| Onsager | ||||||||
| C4C5C6N1 | 5.4 | 6.0 | 12.0 | 11.0 | 11.4 | 10.1 | 13.1 | |
| C2N3C4C5 | 12.1 | 9.7 | 2.7 | −11.5 | −13.3 | −9.2 | −6.3 | |
| C4C5C6–H | 152.3 | 152.4 | 156.1 | 162.1 | 165.4 | 163.7 | 157.9 | |
| C4C5,N1–H | 151.5 | 152.5 | 164.6 | 171.9 | 178.8 | 175.2 | 165.4 | |
| C5C4N3–H | 169.4 | 171.4 | 177.7 | 174.6 | 169.9 | 169.9 | 177.4 | |
| COSMO | ||||||||
| C4C5C6N1 | 9.3 | 8.8 | 10.5 | 10.3 | 9.0 | 8.9 | ||
| C2N3C4C5 | −2.9 | −2.0 | −9.9 | −10.2 | −10.0 | −8.3 | ||
| C4C5C6–H | 157.3 | 158.8 | 165.4 | 166.0 | 167.2 | 167.3 | ||
| C4C5,N1–H | 177.0 | 176.9 | 174.0 | 174.1 | 174.6 | 175.9 | ||
| C5C4N3–H | 172.0 | 173.1 | 175.1 | 174.5 | 174.9 | 176.4 | ||
Solvent effects often significantly influence the equilibrium structure and the electron distribution in molecules.12,13 The Onsager model has been considerably successful in estimating changes of the molecular structure due to solvation, although it is appropriate for only relatively compact molecules. The CPCM model is able to simulate the dielectric environment also for larger molecules. We have used these two self-consistent reaction field models for the geometry optimisation of the uracil radical anion in water. The comparison of the resulting vacuum and water structures (see Table 1) shows strong influences of the dielectric environment on the molecular structure, especially on the dihedral angles. While these angles essentially agree for the vacuum case and the Onsager model, the CPCM optimisations always lead to the boat conformation, independent of whether diffuse functions are included or not.
A second structural peculiarity is the significant out-of-plane deviation of the hydrogens bound to the ring atoms. This feature is independent of the computational procedure (basis set as well as level of theory, i.e. DFT vs. UHF or MP2) and also of the dielectric environment. In Table 1, the dihedral C4–C5–C6–H, C4–C5,N1–H and C5–C4–N3–H angles are given. It appears that a certain pyramidality around the N1, N3 and C6 atoms is predicted, which is generally quite large at the radical centre C6 (compare Figs. 1a and 1b). These dihedral angles are rather sensitive to the basis set, while the bond lengths are hardly influenced (not shown).
We focus on the deviation φ of the hydrogen atom bound to C6 from the molecular plane, with φ being determined as the difference between 180° and the dihedral C4–C5–C6–H angle. φ strongly depends on both the basis set and the dielectric continuum. In vacuum, it changes from 30.4° (for the 6-31G(d) basis set) to 21.6° (for 6-311+(d,p)). This basis set effect is even more pronounced in the solvent and also depends on the self-consistent reaction field model used. With the Onsager model we get the angles 27.7° and 14.6°, and with the CPCM model—the angles 22.7° and 12.8° for the 6-31G(d) and 6-311+G(d,p) basis sets, respectively. The B3LYP/6-311+G(d,p) level of theory yields the smallest deviation from planarity in water; further extension of the basis set has only minor effect.
It is easy to understand the remarkable deviation of the C6 position from planarity in the uracil radical anion. It appears from the calculations that the most amount of the spin density (up to 70%) is located at this centre (see Fig. 1). As a consequence, a certain change of the hybridisation at the carbon atom C6 has to be assumed. In the planar case (H in the N1–C6–C5 plane, compare Fig. 1c), for which the three bond angles involving the C6 atom (see Scheme 2) sum up to 360°, we have pure sp2 hybridisation with the unpaired electron in a pπ orbital. Otherwise, in the case of pure sp3 hybridisation (with a dihedral N1,C5–C6–H angle ϕ of approximately 120°), the three bond angles should have values around 109° summing up to approximately 327°. The respective angles resulting from the optimisations for both the planar and the nonplanar structures are given in Table 2.
 | ||
| Scheme 2 | ||
=360°−σ and Δϕ=180°−ϕ are differences between planar and nonplanar structures indicating the extent of deviation from planarity (pyramidality) on the radical center C6
| Structure | Basis set | 6-31G(d) | 6-311+G(d,p) | ||||
|---|---|---|---|---|---|---|---|
| Vacuum |
Water
Onsager |
Water
COSMO |
Vacuum |
Water
Onsager |
Water
COSMO |
||
| a UHF/6-311+G(d,p) structure is shown for comparison. | |||||||
| Planar | α(C5–C6–H) | 126.1 | 125.9 | 125.8 | 125.7 | ||
| β(N1–C6–H) | 116.1 | 116.0 | 116.4 | 116.3 | |||
| γ(N1–C6–C5) | 117.8 | 118.1 | 117.8 | 118.0 | |||
| Nonplanar | α(C5–C6–H) | 120.6 | 121.7 | 122.1 | 121.3 | 123.2 | 123.5 |
| β(N1–C6–H) | 112.9 | 113.6 | 114.0 | 113.7 | 114.8 | 115.3 | |
| γ(N1–C6–C5) | 116.3 | 116.7 | 116.4 | 116.4 | 117.2 | 117.1 | |
|
σ=α+β+γ |
349.8 | 352.0 | 352.5 | 351.9 | 355.5 | 355.9 | |
| ϕ(N1,C5–C6–H) | 142.7 | 146.9 | 148.0 | 145.8(142.1)a | 154.0(144.9)a | 156.2 | |
| Δσ | 10.2 | 8.0 | 7.5 | 9.1 | 4.5 | 4.1 | |
| Δϕ | 37.3 | 33.1 | 32.0 | 34.2(37.9a | 26.0(35.1)a | 23.8 | |
It follows for the nonplanar structures that, independent of the basis set and of the dielectric continuum, the hybridisation at the carbon atom C6 clearly changes from sp2 to some sp3 character. The conclusions concerning the basis set and solvent effects on the amount of pyramidality at the radical centre correspond to those derived above for the dihedral angles. Whereas the basis set effect on the pyramidality, i.e. on the angles σ and ϕ, is relatively small in vacuum, it is somewhat stronger in water. The influence of the solvent is similar for the Onsager and CPCM models, being more pronounced for the large 3-611+G(d,p) basis set.
Calculation of hyperfine coupling constants
Dependence of the hyperfine coupling constants on the molecular structure
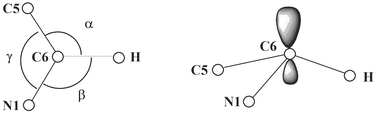
As previously noted by Wetmore et al.,6 the planar structure of the uracil radical anion is not a minimum energy configuration. This is in line with our results identifying the planar structure as a second-order saddle point. Additional information concerning the uracil radical anion structure may be derived from our calculations of the isotropic hfc constants of the system. The calculated coupling constant a(H,6) for the planar structure is too large (the experimental values is 35.0 MHz).9 This results in values between −42 and −49 MHz in vacuum, depending on the basis set. This agrees with the value −41.7 MHz calculated for the planar structure by Close10 at the B3LYP/6-311++G(3df,3dp)//B3LYP/6-311+G(d,p) level. Moreover, in the case of the planar structure, the comparison of the calculated coupling constants with the experimental values for all other atoms shows a relatively poor agreement. Thus, there are several reasons (unstable structure, poor fit for all couplings and possible solvent effects on the molecular and electronic structure), which make us suspicious about the planar structure. Therefore, in our opinion, it is reasonable to pay more attention to the nonplanar minimum structure of the uracil radical anion. In this case, the main question is, why the calculated major C6–H coupling is too small. To find out the reasons for this discrepancy, a large variety of comparative calculations were performed.It is well known that the hyperfine coupling is very sensitive to the molecular structure. As the calculations show (see Table 1), there are two strong influences on the structure of the uracil radical anion—first, the basis set used for the geometry optimisation and, second, the solvent effect of water—the latter being more pronounced. Thus, it is reasonable to study the dependence of the electronic structure, i.e. the calculated isotropic hfc constants, on the level of the geometry optimisation. The resulting a(H,6) constants depending on the molecular structure in vacuum as well as in water resulting from optimisations applying various basis sets, are shown in Fig. 2.
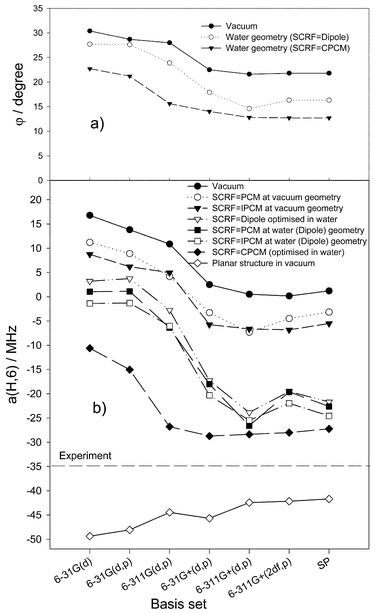 | ||
| Fig. 2 Dependence of DFT B3LYP calculated parameters of the uracil radical anion on the basis set for various levels of geometry optimisation in the vacuum case and in water involving different SCRF models. (a) Deviation of the C6–H atom from the molecular plane and (b) the hfc constant a(H,6). SP indicates a single-point calculation at the B3LYP/6-311++G(2df,p)//6-311+G(d,p) level. | ||
It appears that in the vacuum case, the agreement with the experiment is rather poor for all basis sets used. In particular, the calculated major C6–H coupling is completely wrong (Fig. 2b). However, there is a significant trend in the value of the C6–H coupling, which correlates with the deviation (angle φ) of the C6–H atom from the molecular plane (determined as the difference between 180° and the dihedral C4–C5–C6–H angle, see above)
(Fig. 2a). a(H,6) varies from +16.79 MHz at φ=30.4°
(6-31G(d) basis set) to +0.16 MHz at φ=21.6°
(large 6-311+G(2df,p) basis). Remarkably, in the vacuum case, even large basis sets are not suitable to calculate correct C6–H coupling constants.
Otherwise, in the case of the solvent, the effect of the basis set on the calculated hfc constants is more pronounced, especially on the major C6–H coupling. The a(H,6) values show a similar trend as in vacuum and, again, correlate with the deviation (angle φ) of the C6–H atom from the molecular plane. With the Onsager model, a(H,6) drops to +3.2 MHz at φ=27.7° for the 6-31G(d) basis set and then reaches a large negative value of −23.87 MHz at φ=14.6° for the 6-311+G(d,p) basis. Application of the CPCM model for consideration of the solvent effect significantly reduces the resulting major C6–H coupling constant. The a(H,6) value drops to −10.61 MHz at φ=22.7° for the 6-31G(d) basis set and reaches the large negative value of −28.38 MHz at φ=12.8° for the 6-311+G(d,p) basis. It appears that geometry optimisations using relatively large basis sets and involving the solvent, lead to reasonable absolute values of the major C6–H hfc constant in comparison with the experiment. Clearly, the inclusion of diffuse functions is essential. Otherwise, additional f-functions do not improve the agreement.
To separate the influence of the molecular structure from the computational procedure for the evaluation of the hfc constants, we have compared different self-consistent reaction field models for the dielectric continuum (Dipole, CPCM, PCM and IPCM, see the section Computational details) at vacuum and water structures, depending on the basis set. This is also included in Fig. 2.
The figure clearly shows that gas-phase structures, even those using large basis sets, are not reliable to reproduce the large negative experimental value of the C6–H hfc constant. Consideration of the dielectric continuum at the gas-phase structures, as shown for the PCM and IPCM models, makes some improvement, but is not sufficient to get reliable values. However, consideration of the solvent also for the geometry optimisation significantly reduces the calculated a(H,6) values. It follows that a rather good agreement with the experimental value is achieved for structures optimised in water with relatively large basis sets involving diffuse functions. The structures resulting for the CPCM model with such basis sets can be adopted as the most relevant ones. Thus, a distinct deviation of the C6–H atom from the molecular plane is indicated, which amounts to around 13° from the calculations (Fig. 2a).
Basis set dependence of hyperfine coupling constants
The choice of the reliable basis set for the calculation of hfc constants of pyrimidine-type base radicals at fixed molecular structure by DFT is not evident. The basis set dependence has been examined both through ab initio and DFT methods.6,28–34 A pleasant aspect of DFT is that, unlike the case of ab initio methods concerning the inclusion of electron correlation, the accuracy of predicting spin densities does not seem to be much improved with increasing the size of the basis. The basis set dependence is small for hydrogen hfc constants.32 For the nitrogen atom, the calculated hfc constant is relatively insensitive to the chosen basis set and is fortuitously close to the experiment.33 However, the inclusion of diffuse functions for heavy atoms is found to be particularly important for the prediction of hfc constants for anionic systems.34We have examined, for the case of the uracil radical anion, the basis set dependence of the coupling constants calculated by the DFT B3LYP method using nine different basis sets, i.e. the standard 6-31G(d), 6-31G(d,p), 6-311G(d,p), 6-31+G(d,p), 6-311+G(d,p), 6-311+G(2df,p), 6-311++G(2df,p) basis sets and the EPR-2 and EPR-3 basis sets of Barone.27 With these basis sets, series of single-point calculations were performed at fixed molecular structures. The most reliable structures optimised in water for the Onsager and the CPCM model with the 6-311+G(d,p) basis set were chosen. Fig. 3 shows the effect of the basis set on the major hfc coupling constant a(H,6).
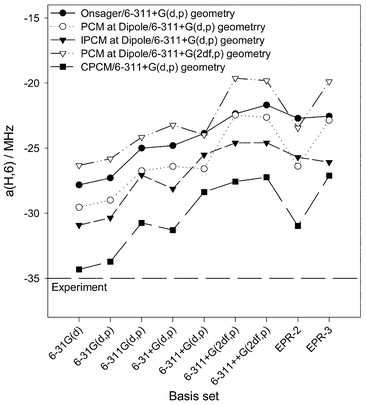 | ||
| Fig. 3 Dependence of the hfc constant a(H,6) of the uracil radical anion (DFT B3LYP) on the basis set in water and different SCRF models at fixed molecular structure. | ||
It can be seen that the basis set dependence follows the same trend for all self-consistent reaction field models. Independent of the molecular structure, the isotropic hfc constant of the C6–H atom shows only a relatively small sensitivity to the basis set. For both structures, the best agreement with the experimental value is calculated with the medium-sized 6-31G(d) and 6-31G(d,p) basis sets.
The results collected in Figs. 2 and 3 show that two of the solvent approximations—the Onsager and the CPCM models—provide similar solvent effects, although, in the case of the uracil radical anion, the CPCM coupling constants are in a slightly better agreement with the experiment, probably because of a more reliable cavity.
We have examined the dependence of the major C6–H coupling on the extent of pyramidality at the radical centre C6. Based on a fixed molecular structure (the nonplanar one optimised for the Onsager model with the 6-311+G(d,p) basis set) single-point calculations using the polarised continuum model with the 6-31G(d) basis set were performed varying the dihedral C4–C5–C6–H angle (and, simultaneously, the N1,C5–C6–H angle). The resulting values of a(H,6) constant are presented in Fig. 4. In addition, we look at the character of the spin density distribution at the radical centre C6, as described by the singly occupied MO (SOMO) (actually the highest occupied α-MO without β-counterpart). The SOMO is mainly of pπ-character, but has, on the radical centre C6, also some s-character. The amount of the latter depends strongly on the extent of pyramidality, i.e. on the dihedral N1,C5–C6–H angle. It ranges from 0% for pure sp2 hybridisation (180°) to around 12% at almost pure sp3 hybridisation (120°).
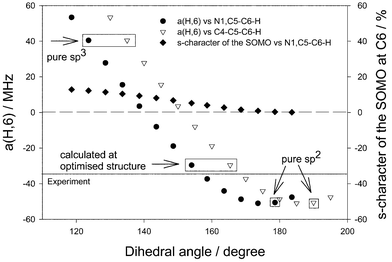 | ||
| Fig. 4 Dependence of the hfc constant a(H,6) of the uracil radical anion from the extent of pyramidality on the radical centre C6 and deviation of C6–H atom from the molecular plane calculated with B3LYP/6-31G(d)/SCRF=PCM in water at fixed water geometry (B3LYP/6-311+G(d,p)/SCRF=Dipole). | ||
It is important to note that, due to the deformation of the molecular ring, even if the C6–H atom is located in the molecular plane (with the C4–C5–C6–H angle being 180°), there is certain pyramidality at the C6 position, characterised by the N1,C5–C6–H angle, which amounts to 168.6° in the chosen structure. It follows from Fig. 4 that the calculated a(H,6) coupling constant matches the experimental value (35 MHz) for a deviation of the C6–H atom from the molecular plane by about 11° (169° for the C4–C5–C6–H angle and 158° for the N1,C5–C6–H angle). At such an extent of pyramidality, there is about 5% s-character of the SOMO at the radical centre C6. If the deviation of the C6–H atom from the molecular plane decreases, the hfc constant changes in negative direction and achieves a maximum negative value of about −51 MHz if the deviation vanishes (180° for the N1,C6–C6–H angle and 0% s-character of the SOMO at the C6 position). If the deviation of the C6–H atom increases, the hfc constant changes in positive direction up to +41 MHz for a N1,C5–C6–H angle of 120° (pure sp3 hybridisation). The latter is connected with an increase of the amount of the respective s-character up to approximately 13%. From Fig. 4 it follows that in the case of pure sp2 hybridisation at the C6 atom (dihedral N1,C5–C6–H angle of 180°) a strongly overestimated value of the hfc constant a(H,6) follows (−49 MHz).
Other pyrimidine-type bases
To test the suitability of the considered computation procedures for the evaluation of the hyperfine structure of the radical anions of other pyrimidine-type bases, we have involved thymine, 1-methylthymine, 1-methyluracil and 1,3-dimethyluracil. To base the calculations on reliable molecular structures, which have shown to be very important for the uracil system, geometry optimisations of the respective radical anions were performed in vacuum and in aqueous solution both with the Onsager and CPCM models and various basis sets. In agreement with what has been found for the uracil radical anion, a nonplanar ring results for the most stable structure of all radical anions considered. In each case, there is certain pyramidality at the radical centre C6. As before, there is a strong dependence of the major a(H,6) coupling on the level of geometry optimisation. Vacuum structures, even optimised with the large 6-311+G(2df,p) basis set, are not suitable to calculate reliable a(H,6) coupling constants. Optimisations including solvent effects, however, lead to significant improvements of the absolute values of the constants. Fig. 5 shows the calculated a(H,6) coupling constants of the thymine radical anion in vacuum and in water, depending on the level of the geometry optimisation. It appears that the molecular structure optimised in water with the CPCM model is superior to that resulting with the Onsager model. The most reasonable structures are obtained with the large 6-311+G(d,p) and 6-311+G(2df,p) basis sets.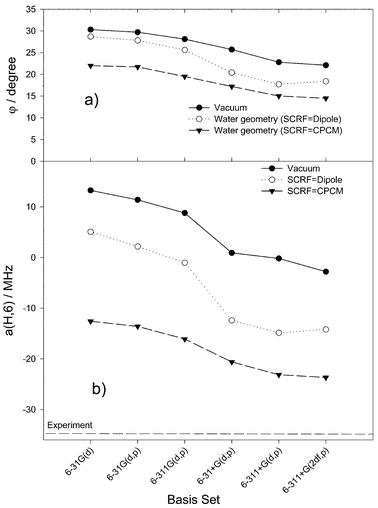 | ||
| Fig. 5 Dependence of DFT B3LYP calculated parameters of the thymine radical anion on the basis set for various levels of geometry optimisation in the vacuum case and in water involving both Onsager and CPCM models. (a) Deviation of the C6–H atom from the molecular plane; and (b) the hfc constant a(H,6). | ||
With two fixed structures (optimised with the Onsager and the CPCM model using the 6-311+G(d,p) basis set), the hfc coupling constants for the variety of radical anions were calculated with various basis sets using the four different self-consistent reaction field models involved in this investigation. In Table 3 are presented the calculated coupling constants and the deviation of the C6–H atom from the molecular plane (angle φ). It follows that by selecting appropriate computational procedures, hfc constants for pyrimidine-type bases can be calculated which are in reasonable agreement with the experiment (where available).
=Dipole (Onsager) and B3LYP/6-311+G(d,p)/SCRF=CPCM (COSMO). Single point calculations with SCRF=Dipole, SCRF=PCM and SCRF=CPCM models (at water geometry), φ-dihedral angle of deviation of C6–H atom from the molecular plane
|
Geometry Optimization
SCRF Basis set |
Vacuum/6-311+G(d,p)
Vacuum 6-311+G(d,p) |
6-311+G(d,p)/Dipole | 6-311+G(d,p)/CPCM | Exp | |||
|---|---|---|---|---|---|---|---|
|
Dipole
6-311+G(d,p) |
PCM
6-31G(d) |
CPCM
6-311+G(d,p) |
CPCM
6-31G(d) |
||||
|
a
Ref. 9.
b Unpublished results by D. Beckert,
c At B3LYP/6-311+G(2df,p)/SCRF=CPCM geometry.
|
|||||||
| Uracil | a(N,1) | −0.95 | −1.18 | 0.13 | −1.48 | −0.02 | ∼0a |
| a(N,3) | 0.91 | 1.57 | 4.17 | 1.65 | 4.08 | 4.1 | |
| a(H,1) | −1.49 | −4.76 | −5.49 | −4.77 | −5.34 | 2.4 | |
| a(H,3) | −5.00 | −2.32 | −2.71 | −5.39 | −5.06 | 2.4 | |
| a(H,5) | −7.21 | −3.33 | −6.19 | −2.85 | −5.25 | 2.5 | |
| a(H,6) | 0.51 | −23.87 | −27.83 | −28.38 | −34.00 | 35.0 | |
| φ | 21.6 | 14.6 | 14.6 | 12.8 | 12.8 | ||
| Thymine | a(N,1) | 1.34 | −1.09 | 0.25 | −1.23 | 0.10 | ∼0a |
| a(N,3) | 1.00 | 1.01 | 3.57 | 1.54 | 3.80 | 0.6 | |
| a(H,1) | 0.92 | −3.41 | −4.37 | −4.35 | −4.84 | 2.4 | |
| a(H,3) | 5.47 | −4.14 | −5.03 | −4.66 | −4.99 | 2.4 | |
| a(CH3,5) | 4.06 | 2.81 | 3.82 | 1.34 | 3.34 | 3.8 | |
| a(H,6) | 0.02 | −14.48 | −21.36 | −23.14 | −29.49 | 35 | |
| φ | 23.1 | 17.9° | 17.9° | 15.0° | 15.0° | ||
| 1-Methyl thymine | a(N,1) | −1.10 | −1.04 | 0.51 | −0.84 | 0.49 | |
| a(N,3) | 0.55 | 0.80 | 3.23 | 1.17 | 3.15 | ||
| a(H,3) | −4.73 | −3.95 | −5.52 | −5.29 | −5.67 | ||
| a(CH3,1) | 1.75 | 2.16 | 4.12 | 2.56 | 3.54 | ||
| a(CH3,5) | 4.32 | 2.83 | 3.73 | 1.97 | 3.94 | ||
| a(H,6) | −12.18 | −21.99 | −29.33 | −25.69 | −32.80 | ||
| φ | 18.8 | 12.2° | 12.2° | 12.7° | 12.7° | ||
| 1,3-Dimethyl uracil | a(N,1) | −1.02 | −0.98 | 0.48 | −0.79 | 0.47 | |
| a(N,3) | 1.58 | 1.84 | 3.79 | 2.43 | 4.08 | ||
| a(H,5) | −6.50 | −4.36 | −5.19 | −3.64 | −5.39 | ||
| a(CH3,1) | 1.90 | 2.41 | 3.70 | 2.12 | 2.60 | ||
| a(CH3,3) | 6.16 | 6.68 | 7.46 | 7.47 | 7.38 | ||
| a(H,6) | −9.42 | −16.72 | −24.08 | −22.37 | −29.44 | ||
| φ | 18.1 | 17.0° | 17.0° | 14.2° | 14.2° | ||
As mentioned above, the calculated stable structure of the radical anions has a puckered ring. Similar to the neutral radicals derived from DNA bases,12 there are two minima on the potential surface corresponding to the two enantiomeric forms of the boat conformation. In this case, vibrational averaging could be discussed. The potential energy barrier separating the two minima (between 4.2 and 7.3 kJ mol−1 in the case of the uracil radical anion) corresponds to a planar six-membered ring (transition state). The first deformational vibration level (80 cm−1=0.96 kJ mol−1) is localized inside the potential well. Consequently, equilibration between the two energy minima, which leads to an average planar structure of the radical anions, can occur or not, depending on the temperature. At room temperature, a complete averaging seems to have not been achieved, because the calculated hfc constants for the C6–H atom at the planar structure are strongly overestimated (ca. 41 MHz in the case of the uracil radical anion).
Effect of hydrogen bonding
To estimate the influence of hydrogen bonding on the molecular and electronic structure of the uracil radical anion we consider a supermolecule model with an increasing number of water molecules. The results obtained are collected in Figs. 6 and 7.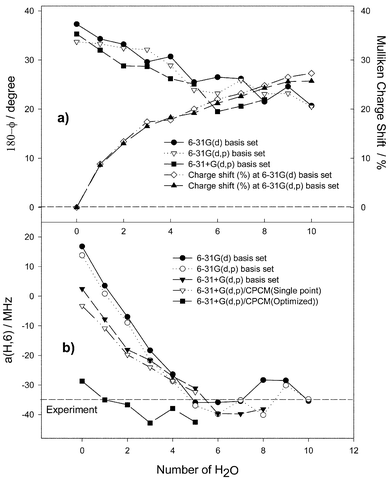 | ||
| Fig. 6 Dependence of B3LYP calculated parameters of the uracil radical anion on the number of hydrogen-bonded water molecules for various levels of geometry optimisation. (a) Here (●), (▽) and (■) are the extent of the pyramidality on the radical centre calculated as the difference 180° − dihedral N1,C5–C6–H angle; and (⋄) and (▲) are the negative Mulliken charge shift (%) from the radical anion to hydrogen-bonded water molecules. (b) The hfc constant a(H,6). | ||
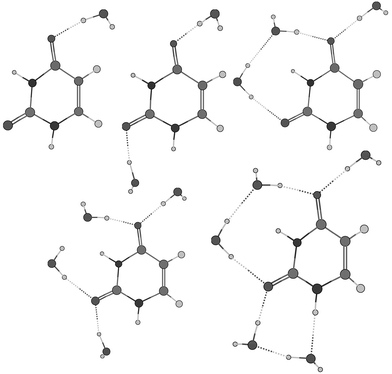 | ||
| Fig. 7 The most stable structures of the supermolecules with up to five water molecules calculated with 6-31+G(d,p) basis set. | ||
First, we consider the supermolecules without an additional reaction field (vacuum case). It appears that explicit water molecules lead to the boat structure of the uracil radical anion independent of the basis set. There is a similar trend for the calculated values depending on the number of the water molecules for all basis sets used. The value of the C6–H coupling constant drops down from positive to large negative values and achieves saturation near the experimental value of 35 MHz (Fig. 6b). This trend correlates with the extent of pyramidality at the radical centre C6 (Fig. 6a). The dihedral N1,C5–C6–H angle φ decreases continuously from 142.7° for the isolated uracil radical anion to 159.3° for the complex with 8 water molecules using the 6-31G(d) basis set (from 144.7° to 159.4° with 6 water molecules using 6-31+G(d,p)) and achieves saturation as well. Moreover, the analysis of the Mulliken charges depending on the number of water molecules shows that each oxygen-bound water withdraws up to 0.05 e. The charge shift from the radical anion to the water molecules reduces the negative charge of the former by almost 30% for a larger number of water molecules (Fig. 6a) and, consequently, reduces the distortion of the molecular ring and the extent of pyramidality at the radical centre. It follows from Fig. 6 that, clearly, only the first 5 or 6 water molecules are essential, a larger number seems to have no significant influence on both the molecular structure and the calculated values of a(H,6).
Now, we consider the supermolecules with an additional reaction field (SCRF=CPCM). Single-point calculations for the vacuum structures yield only a minor improvement of the a(H,6) value. Complete optimisations with consideration of the reaction field, however, strongly influence both the radical anion structure and the a(H,6) value. The latter is somewhat overestimated when more than 2 water molecules are involved.
A comparison of the different models for the simulation of the water environment is given in Table 4. The vacuum optimisation of the complexes with 4 or 5 water molecules leads to structural parameters at the radical centre and to an a(H,6) value, which are very similar to those obtained for the single radical anion molecule optimised in the dielectric continuum. So, both of these computational alternatives seem to be suitable for describing appropriately the molecular structure at the radical centre in water. The subsequent consideration of the reaction field by single-point calculations for the vacuum-optimised supermolecules leads to only a small improvement of the a(H,6) value. It seems that the optimization of a complex including a few water molecules with consideration of the reaction field yields the most reliable results. This strategy, however, is restricted to sample systems because of the considerable computational effort.
=CPCM), (2) radical anion with explicit water molecules in gas phase, (3) radical anion with explicit water molecules optimised in dielectric continuum (SCRF=CPCM)
|
Structure
Model |
Uracil
CPCM Optimized in water |
Uracil+4H2O
Optimized in vacuum |
Uracil+5H2O
Optimized in vacuum |
Uracil+2H2O
CPCM Optimized in water |
Uracil+4H2O
CPCM Optimized in water |
Uracil 5H2O
CPCM Optimized in water |
|---|---|---|---|---|---|---|
|
a Radical anion with explicit water molecules in dielectric continuum (SCRF=CPCM) at fixed vacuum geometry.
|
||||||
| C4C5C6N1 | 10.3 | 10.6 | 11.3 | 7.1 | 10.2 | 9.5 |
| C4C5C6–H | 166.0 | 164.4 | 166.2 | 168.2 | 169.1 | 175.2 |
| N1,C5C6–H | 155.7 | 153.8 | 154.9 | 161.1 | 158.9 | 165.7 |
| a(H,6) | −28.73 | −27.10(−28.69)a | −31.11(−32.39)a | −36.66 | −37.94 | −44.74 |
Conclusions
The present DFT study of the radical anions of pyrimidine-type bases has shown that the calculated values of the hyperfine coupling constants are extremely sensitive to the molecular structure. Gas-phase structures even optimised with extended basis sets are not able to reproduce the large negative hfc constant of the C6–H atom known from the experiments. Reliable structures result only from optimisations in water using large basis sets. The inclusion of diffuse functions is essential for the anions. Features of the resulting structures are a noticeably deformed molecular ring and a significant pyramidality at the radical centre C6 connected with a deviation of the C6–H atom from the molecular plane up to around 12°. The molecular structure optimised in water using the CPCM model with the 6-311+G(d,p) basis set seems to be most relevant. Obviously, the Onsager approach becomes less accurate than the CPCM one when the molecules have a more irregular shape, as it is the case for the uracil derivatives involved.For a fixed optimised molecular structure, the calculated values of the isotropic hfc constant of the C6–H atom show only a relatively small sensitivity to the basis sets used. Inclusion of diffuse functions leads only to small improvements of the values. It was concluded that the moderate 6-31G(d) and 6-31G(d,p) basis sets lead to the best agreement of the a(H,6) coupling constant with the experiments. The special EPR-2 and EPR-3 basis sets give rather good hfc constants for all hfc-active nuclei of the radical anions.
Explicit consideration of water molecules in a supermolecule model leads to a boat structure for the uracil radical anion independent of the basis set. The vacuum optimisation of the complexes with 4 or 5 water molecules leads to structural parameters at the radical centre and to an a(H,6) value, which are very similar to those obtained for the single radical anion molecule optimised in the dielectric continuum. So, both of these computational alternatives seem to be suitable to describe appropriately the molecular structure at the radical centres in water.
Acknowledgements
Financial support of this work was kindly provided by the Deutsche Forschungsgemeinschaft.References
- C. von Sonntag, The Chemical Basis of Radiation Biology, Taylor and Francis, London, 1987 Search PubMed.
- J. Hüttermann, in Radical Ionic Systems, ed. A. Lund and M. Shiotani, Kluwer, Dordrecht, 1991, p. 435 Search PubMed.
- Effects of Ionizing Radiation on DNA, ed. J. Hüttermann, ed. W. Köhnlein, R. Teoule and A. J. Bertinchamps, Springer, Berlin, 1978 Search PubMed.
- M. G. Simic, L. Grossman and A. C. Upton, Mechanisms of DNA Damage and Repair, Plenum Press, New York, 1986 Search PubMed.
- A.-O. Colson and M. D. Sevilla, Int. J. Radiat. Biol., 1995, 99, 3867 CAS.
- S. D. Wetmore, R. J. Boyd and L. A. Eriksson, J. Phys. Chem. B, 1998, 102, 5269 CrossRef.
- J. Säuberlich and D. Beckert, J. Phys. Chem., 1995, 99, 12520.
- T. Kausche, J. Säuberlich, E. Trobitzsch and D. Beckert, Chem. Phys., 1996, 208, 375 CrossRef CAS.
- J. M. Lü, J. Geimer, S. Naumov and D. Beckert, Phys. Chem. Chem. Phys., 2001, 3, 952 RSC.
- D. M. Close, Phys. Chem. Chem. Phys., 2002, 4, 43 RSC.
- S. D. Wetmore, R. J. Boyd and L. A. Eriksson, Chem. Phys. Lett., 2000, 322, 129 CrossRef CAS.
- F. Jolibois, J. Cadet, A. Grand, R. Subra, N. Rega and V. Barone, J. Am. Chem. Soc., 1998, 120, 1864 CrossRef CAS.
- S. Naumov, A. Barthel, J. Reinhold, F. Dietz, J. Geimer and D. Beckert, Phys. Chem. Chem. Phys., 2000, 2, 4207 RSC.
- S. Naumov, K. Hildenbrand and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 2001, 1648 RSC.
- P. J. O'Malley, J. Phys. Chem., 1997, 101, 6334 CrossRef CAS.
- GAUSSIAN 98 (Revision A.11), M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and A. J. Pople, Chem. Phys. Lett., 1990, 166, 281 CrossRef CAS.
- M. W. Wong, M. J. Frisch and K. B. Wiberg, J. Am. Chem. Soc., 1991, 113, 4776 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- N. Rega, M. Cossi and V. Barone, Chem. Phys., 1996, 105, 11060 CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- J. B. Foresman, T. A. Keith, K. B. Wiberg, J. Snoonian and M. J. Frisch, J. Phys. Chem., 1996, 100, 16098 CrossRef CAS.
- N. Rega, M. Cossi and V. Barone, J. Chem. Phys., 1996, 105, 11060 CrossRef CAS.
- B. Engels, L. A. Eriksson and S. Lunell, Adv. Quantum Chem., 1996, 27, 298 Search PubMed.
- R. Batra, B. Giese, M. Spichty, G. Gescheidt and K. N. Houk, J. Phys. Chem., 1996, 100, 18371 CrossRef CAS.
- S. D. Wetmore, R. J. Boyd and L. A. Eriksson, J. Chem. Phys., 1997, 106, 7738 CrossRef CAS.
- M. J. Raiti and M. D. Sevilla, J. Phys. Chem. A, 1999, 103, 1619 CrossRef CAS.
- Y. Inadomi, K. Morihashi and O. Kikuchi, J. Molec. Struct. (THEOCHEM), 1998, 428, 143 Search PubMed.
- I. Carmichael, J. Phys. Chem. A, 1997, 101(25), 4633 CrossRef CAS.
- J. W. Gauld, L. A. Ericsson and L. Radom, J. Phys. Chem. A, 1997, 101(7), 1352 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: calculated hfc constants depending on the level of geometry optimisation and structure of the hydrogen bonded complex of a uracil radical anion with 5 water molecules. See http://www.rsc.org/suppdata/cp/b2/b207732a/ |
| This journal is © the Owner Societies 2003 |