The unique nucleophilic reactivity of arylaminochlorocarbenes†
Ying
Cheng
*a,
Hua
Yang
a and
Otto
Meth-Cohn
*b
aDepartment of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: yincheng@95777.com; Tel: +861062205558
bChemistry Department, Sunderland University, Sunderland, UK SR1 3SD. E-mail: otto.meth-cohn@sunderland.ac.uk
First published on 26th November 2002
Abstract
4-Methyl and 4-methoxyphenylaminochlorocarbene (readily formed by deprotonation of the Vilsmeier reagent derived from the corresponding N-methylformanilide with Hünig’s base) reacted with diethyl acetylenedicarboxylate to give 1∶2 quinoline adducts, while p-halophenylaminochlorocarbenes yielded benzoazepine derivatives from 2∶1 interaction of the carbene with oxalyl chloride under the same reaction conditions.
Carbenes can be divided into electrophilic, nucleophilic and ambiphilic types according to their chemical behaviour. Whilst dichlorocarbene is a typical electrophilic carbene, which readily adds to alkenes, diaminocarbenes behave as nucleophiles in their reactions with, for example, the N-phenylmaleimide,1 isocyanates and isothiocyanates.2 Several years ago,3 we isolated a range of products derived from arylaminochlorocarbenes, which were readily generated by deprotonation of a Vilsmeier reagent with a tertiary amine. All of these products derived from the aminochlorocarbene reacting firstly with the parent Vilsmeier reagent to give the corresponding dimer in high yields.3,4 We envisaged the reactivity of the aminochlorocarbenes to lie between that of a dichlorocarbene and a diaminocarbene. They could be nucleophilic or ambiphilic. In order to clarify the reactivity of aminochlorocarbenes, we herein report our study of their reactions. They showed no reactivity whatever towards simple alkenes or, for example, benzoyl chloride but did react with diethyl acetylenedicarboxylate and with oxalyl chloride.
Diethyl acetylenedicarboxylate is reported to react with nucleophilic carbenes, such as aminooxycarbenes,5 dioxycarbenes6 and aminothiocarbene.7 Initially, the Vilsmeier reagent, derived from a 4-substituted N-methylformanilide 1 and oxalyl chloride 2, was deprotonated by Hünig’s base 3 in dry chloroform or THF at −10 °C to 0 °C under a nitrogen atmosphere. Addition of diethyl acetylenedicarboxylate 4 in the same solvent, was followed by stirring at room temperature for a period of time. However, under these conditions solely the HCl adduct of diethyl acetylenedicarboxylate 5, together with the carbene dimers 6 were recovered, indicating that the ‘dimerisation’ of the carbenes was much faster than its possible reaction with 4. The hydrochloride salt of Hünig’s base (formed in the reaction) was shown to react readily with 4 to give 5 in high yield.
In order to minimise the dimerisation, a non-polar solvent was used which would minimise the solubility of the Vilsmeier reagent, and a much lower initial reaction temperature was employed. Thus, the Vilsmeier reagent prepared by reaction of a 4-substituted N-methylformanilide 1a–e and oxalyl chloride at 0 °C, was cooled to −78 °C. To this solid Vilsmeier reagent, a solution of Hünig’s base in xylene was added with stirring to form a suspension. After a ½ h, diethyl acetylenedicarboxylate 4 in xylene was added and the temperature increased from −78 °C to room temperature during 2 h. The reaction mixture was stirred at room temperature for 1 h and then at 50 °C for another 2 h, when after chromatography, red products were isolated along with compound 5 and dimers 6.
Surprisingly, the structure of the red products derived from 4-methoxy- or 4-methyl-N-methylformanilide (1a and 1b respectively) and diethyl acetylenedicarboxylate were totally different from those derived from 4-halo-N-methylformanilides 1c–e. The NMR and MS spectroscopic data showed that the former contained one arylaminochlorocarbene moiety and two diethyl acetylenedicarboxylate units, while the latter required two formanilide units but no diethyl acetylenedicarboxylate at all. This was corroborated by the observation that the same products were formed in the absence of diethyl acetylenedicarboxylate in the case of the haloformanilides. The structures of these red products 7a (X = MeO) and 8c (X = F) were put beyond doubt by X-ray crystallography‡ (Scheme 1).
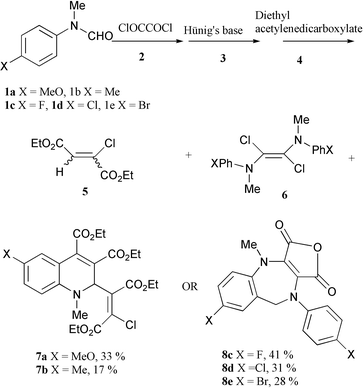 | ||
| Scheme 1 | ||
The formation of the quinoline derivatives 7 could be explained by a mechanism involving nucleophilic addition of the aminochlorocarbene 10 to diethyl acetylenedicarboxylate 4, cyclisation of the dipolar intermediate 11, a second nucleophilic addition to 4 and finally rearrangement of the chloro-substituent (Scheme 2).
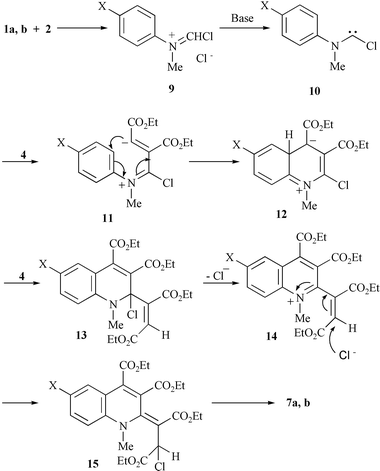 | ||
| Scheme 2 | ||
The structure of the benzo[1,4]diazepine dicarboxylic anhydrides 8 is not sufficiently trivial to be explained by being formed solely from the formanilide 1 and oxalyl chloride. As with the formation of the quinolines 7, the formation of 8 appears to involve bis-addition of the carbene 10 to the oxalyl chloride to form an iminium salt 16 and thence 17. The methyl group adjacent to the iminium function is essentially similar to one attached to a carbonyl function and can thus be easily deprotonated allowing the new iminium ion 19 to cyclise to a diazepine. The enamine 21 can then undergo nucleophilic addition to oxalyl chloride at the less hindered carbon atom of the double bond. The well-known8 decarbonylation of glyoxalyl chlorides 23 would yield the bis-acid chloride 24, which could form the anhydrides 8 with traces of water or on work-up (Scheme 3).
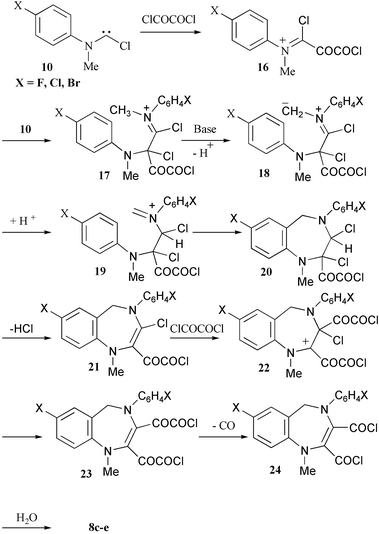 | ||
| Scheme 3 | ||
It is evident that the aminochlorocarbenes are weakly nucleophilic with a high reactivity towards Vilsmeier reagents as electrophiles. Unlike the 4-methylphenyl- and 4-methoxyphenyl substituted carbenes, the more weakly nucleophilic 4-halophenyl-substituted carbenes are insufficiently reactive towards diethyl acetylenedicarboxylate but do react with electron-deficient acid chlorides such as oxalyl chloride when the interaction with the Vilsmeier reagent is suppressed. It is noteworthy that an azepine 8 (24–26%) also formed from the 4-methyl- or from the 4-methoxy-N-methylformanilide 1, in the absence of diethyl acetylenedicarboxylate.
This work was supported by the National Natural Science Foundation of China and the Excellent Young Teachers Program of MOE, P. R. C.
Notes and references
- S. E. Kondrat’eva and S. I. Burmistrov, Zh. Org. Khim., 1975, 11, 197 Search PubMed.
- R. W. Hoffmann, B. Hagenbruch and D. M. Smith, Chem. Ber., 1977, 110, 23 Search PubMed.
- Y. Cheng, S. Goon and O. Meth-Cohn, Chem. Commun., 1996, 1395 RSC; Y. Cheng, S. Goon and O. Meth-Cohn, J. Chem. Soc., Perkin Trans. 1, 1998, 1619 RSC.
- Y. Cheng, Y.-H. Zhan, H.-X. Guan, H. Yang and O. Meth-Cohn, Synthesis, 2002 in press.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561 CrossRef CAS.
- R. W. Hoffmann, W. Lilienblum and B. Dittrich, Chem. Ber., 1974, 107, 3395 Search PubMed.
- G. Scherowsky, K. Dünnbier and G. Höfle, Tetrahedron Lett., 1977, 2095 CrossRef CAS.
- See for example: (a) H. Staudinger and M. Stockmann, Ber. Dtsch. Chem. Ges., 1909, 42, 3492 Search PubMed; (b) L. F. Yietze, C. Schneider and M. Pretor, Synthesis, 1993, 1079 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details, characterization data for compounds 7 and 8. See http://www.rsc.org/suppdata/cc/b2/b210098c/ |
| ‡ All the products have been identified by IR, 1H NMR, 13C NMR, MS, HRMS or elemental analysis. The X-ray crystallographic data will be reported elsewhere. |
| This journal is © The Royal Society of Chemistry 2003 |