The radical anions and the electron affinities of perfluorinated benzene, naphthalene and anthracene†
Yaoming
Xie
a,
Henry F.
Schaefer III
*a and
F. Albert
Cotton
b
aCenter for Computational Quantum Chemistry, University of Georgia, Athens, Georgia 30602. E-mail: HFSIII@uga.edu
bLaboratory for Molecular Structure and Bonding, Department of Chemistry, Texas A & M University, College Station, Texas 77843-3255, USA
First published on 28th November 2002
Abstract
Although benzene and naphthalene do not have electron affinities in the conventional sense, perfluorobenzene and perfluoronaphthalene have nonzero electron affinities. Theoretical methods extensively calibrated with experiment for the prediction of electron affinities (EAs) predict the EAs of perfluorobenzene, perfluoronaphthalene and perfluoroanthracene as 0.69, 1.02 and 1.84 eV, respectively. A rough estimate of 2.39 eV is made for the electron affinity of perfluorotetracene. Thus the perfluoro polycyclic aromatic hydrocarbons (PAHs) are predicted to be effective electron acceptors.
Neutral molecules with positive electron affinities are not particularly common, but certain important classes are known. Fullerenes, for example, have been shown to form ionic compounds with electron donors such as alkali metals,1 and polycyano olefins such as tetracyanoethylene (TCNE) and tetracyanoquinodimethane (TCNQ) are well known to accept electrons from a variety of donors.2 A very comprehensive review of the literature on atomic and molecular electronic affinities has recently been published.3
The possibility that perfluoro aromatic molecules could form another class has been adumbrated4,5 but not much explored. The purpose of this report is to provide an opening theoretical examination of the prospects for perfluoroaromatic molecules to serve as electron acceptors which can form compounds by interaction with electron donors in general. For this purpose the three most obvious ones, C6F6, C10F8 and perfluoroanthracene, C14F10, have been chosen. In view of the fascinating results that have been obtained already with other electron acceptors, it seems reasonable to anticipate that interesting new chemistry and materials will be obtainable by using perfluoroaromatics.
Extensive calibrative quantum mechanical studies have demonstrated recently that density functional theory (DFT) is capable of reliable predictions of molecular electron affinities.3 For 91 atomic and molecular systems for which electron affinities (EAs) have been determined reliably from experiment, the average absolute error for the B3LYP method6 is 0.14 eV. In 71% of the cases the B3LYP EA lies above the experimental value. Given the great difficulty of experimental measurements of EAs, this is an important achievement, indicating that theory can be a valuable guide in situations where meaningful experiments are not currently feasible. Detailed theoretical discussions of the use of density functional methods to describe negative ions may be found elsewhere.7,8
A basis set of double zeta plus polarization plus diffuse functions (DZP++) was used in the present work. This is the same basis set used consistently in the comprehensive calibration describe above.3 Specifically, the Huzinaga–Dunning set9,10 for carbon and fluorine, designated C,F(9s5p1d/4s2p1d), was augmented with diffuse s and p functions, specified in ref. 3 (Table 2 therein). Thus the final basis set may be described as C,F(10s6p1d/5s3p1d). For perfluoroanthracene (C14F10) there are 19 × 24 = 456 contracted gaussian functions in the basis set thus constructed.11,12
The stationary point geometries predicted here are reported in Figs. 1–3. The equilibrium geometry of the perfluorobenzene radical anion is displaced from D6h (the point group of neutral C6F6) to C2v, with an energy lowering of 0.40 eV. The magnitude of this splitting is greater than expected for a Jahn–Teller distortion, and indeed the singly-occupied molecular orbital (SOMO) of C6F6− is totally symmetric. The C–C bond length alternation for the radical anion is 1.410![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −1.379 = 0.31 Å. The perfluoronaphthalene radical anion distorts ever so slightly from D2h to C2h, but with an energy lowering of only 0.01 eV. The perfluoroanthracene radical anion retains the D2h symmetry of the neutral molecule.
−1.379 = 0.31 Å. The perfluoronaphthalene radical anion distorts ever so slightly from D2h to C2h, but with an energy lowering of only 0.01 eV. The perfluoroanthracene radical anion retains the D2h symmetry of the neutral molecule.
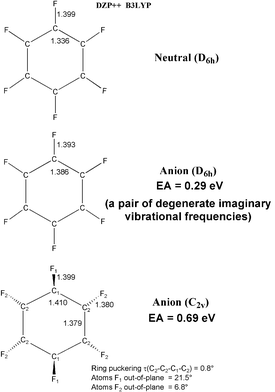 | ||
| Fig. 1 Geometrical structures of perfluorobenzene and its radical anion. Bond distances are in Å. | ||
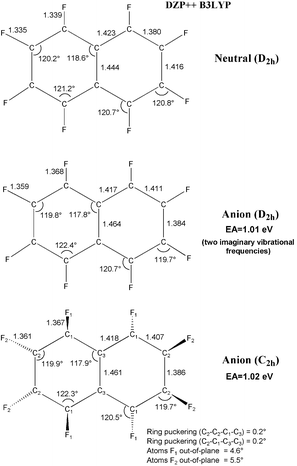 | ||
| Fig. 2 Geometrical structures of perfluoronaphthalene and its radical anion. Bond distances are in Å. | ||
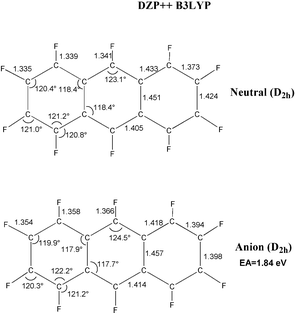 | ||
| Fig. 3 Geometrical structures of perfluoroanthracene and its radical anion. Bond distances are in Å. | ||
The predicted electron affinities are presented in Figs. 1–3 (numerical values printed) and in Fig. 4. Also shown in Fig. 4 are the analogously predicted electron affinities of unsubstituted benzene, naphthalene and anthracene.13 The experimental electron affinities of the latter are −1.12 ± 0.03, −0.19 ± 0.03, and 0.530 ± 0.005 eV, respectively.14–16 The theoretical EAs for these PAHs are −0.88, −0.20, and 0.58 eV, respectively. The good agreement with experiment encourages us to repose confidence in the present theoretical predictions for the perfluorinated species.
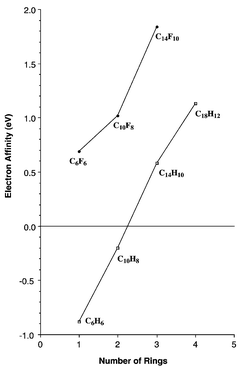 | ||
| Fig. 4 Theoretical electron affinities for simple polycyclic aromatic hydrocarbons (PAHs) and their perfluorinated counterparts. | ||
Fig. 1 gives the numerical value of our prediction for the EA (C6F6), namely 0.69 eV. Thus it is clear that perfluorination removes the strong tendency of benzene to repel an electron. Not surprisingly, then, perfluoronaphthalene will also bind an electron, with the predicted EA being 1.02 eV. The theoretical EA for perfluoroanthracene is even larger, 1.84 eV. Although we were not able to complete computations on perfluorotetracene, the EA for the unsubstituted tetracene is 1.13−0.58 = 0.55 eV, greater than that for anthracene. Thus we expect EA (C18F12) to be something like 1.84+0.55 = 2.39 eV. The trends in EAs in Fig. 4 are regular for both the PAHs and perfluorinated PAHs. The EAs are not yet falling off as a function of the number of benzene rings, although there is a diminution of the slope between anthracene and tetracene. It is abundantly clear that perfluorinated PAHs will be very effective electron acceptors and may thus be useful in the invention of new materials and new reactions.
This research was supported by current NSF grants to both the University of Georgia and the Texas A & M University. We thank Dr. Z Wang for helpful discussions.
Notes and references
- C. A. Reed and R. D. Bolskar, Chem. Rev., 2000, 100, 1075 CrossRef CAS.
- (a) M. R. Bryce, Chem. Soc. Rev., 1991, 20, 355 RSC; (b) A. E. Underhill, J. Mater. Chem., 1992, 2, 1 RSC.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, N. Sreela and G. B. Ellison, Chem. Rev., 2002, 102, 231 CrossRef CAS.
- C. M. Beck, J. Burdeniuc, R. H. Crabtree, A. L. Rheingold and G. P. Yap, Inorg. Chem. Acta, 1998, 270, 559 CrossRef CAS.
- I. Alkorta, I. Rogas and J. Elguero, J. Am. Chem. Soc, 2002, 124, 8593 CrossRef CAS.
- B3LYP combines Becke′s three parameter exchange functional with the correlation functional of Lee, Yang, and Parr: A. D. Becke, J. Chem. Phys., 1993, 98, 5648 Search PubMed; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1993, 37, 785 CrossRef CAS.
- J. M. Galbraith and H. F. Schaefer, J. Chem. Phys., 1996, 105, 862 CrossRef CAS.
- F. Jensen, J. Chem. Phys., 2002, 117, 9234 CrossRef CAS.
- S. Huzinaga, J. Chem. Phys., 1965, 42, 1293 CrossRef.
- T. H. Dunning, J. Chem. Phys., 1970, 53, 2823 CrossRef CAS.
- All computations were carried out with the Gaussian 94 program: M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Peterson, J. A. Montgomery, K. Raghavichari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzalez and J. A. Pople, Gaussian 94, (revision c.3); Gaussian, Inc., Pittsburgh, PA, 1995 Search PubMed.
- We note that for perfluorinated PAHs larger than C14F10, problems associated with the linear dependence of the DZP++ basis set appear. Therefore we also examined C14F10 and C14F10− with the smaller DZP basis set. However, the predicted EA is 1.61 eV, or 0.23 eV less than the reliably calibrated3 DZP++ result. Thus, it appears that diffuse basis functions will be necessary for the reliable theoretical predictions of the EA of perfluorinated tetracene..
- J. C. Rienstra-Kiracofe, C. J. Barden, S. T. Brown and H. F. Schaefer, J. Phys. Chem. A, 2001, 105, 524 CrossRef CAS.
- P. D. Burrow, J. A. Michejda and K. D. Kordan, J. Chem. Phys., 1987, 86, 9 CrossRef CAS.
- S. A. Lyapustina, S. Xu, J. M. Nilles and K. H. Bowen, J. Chem. Phys., 2000, 112, 6643 CrossRef CAS.
- J. Schiedt and R. Weinkauf, Chem. Phys. Lett., 1997, 266, 201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: calculated energies and electron affinities for perfluorinated benzene, naphthalene and anthracene and their anions. Calculated structures for perfluorinated naphthalene and anthracene and their anions. See http://www.rsc.org/suppdata/cc/b2/b208831m/ |
| This journal is © The Royal Society of Chemistry 2003 |