Highly stable performance of catalytic methane dehydrocondensation towards benzene on Mo/HZSM-5 by a periodic switching treatment with H2 and CO2
Yuying
Shu
,
Hongtao
Ma
,
Ryuichiro
Ohnishi
and
Masaru
Ichikawa
*
Catalysis Research Center, Hokkaido University, Kita-Ku, N-11, W-10, Sapporo, 060-0811, Japan. E-mail: michi@cat.hokudai.ac.jp; Fax: +81 011 706 4957; Tel: +81 011 706 2912
First published on 26th November 2002
Abstract
Excellent stability and high catalytic activity of methane dehydrocondensation towards benzene and naphthalene on Mo/HZSM-5 were achieved at 1023–1073 K by a periodic H2 or CO2 switching operation, owing to the efficient removal of coke deposition.
The catalytic dehydroaromatization of methane towards benzene and naphthalene is of great scientific importance and industrial benefit for the effective utilization of natural gas for alternative petrochemical feed stocks and bulky hydrogen production for fuel cells. In the last decade, a lot of research work on methane dehydroaromatization has been conducted,1–8 but the problem of decrease in catalytic activity with time on stream is still unsolved. Therefore, it is of vital importance to improve the catalytic performance by stabilizing the catalytic activity, in particular, from the viewpoint of potential industrial application of this process. Effective methods to solve the problems by adding a few percent of CO or CO2 to the methane feed on HZSM-5 supported Mo and Re catalysts and the modification of the catalyst by dealumination treatment have been previously reported.9–14 Here we present a novel and effective method for the stabilization of catalytic activity at 1023–1073 K by a periodic switching operation of gas feeds between methane and hydrogen or carbon dioxide, due to the effective removal of coke deposited on Mo/HZSM-5 catalyst.
The Mo/HZSM-5 catalyst (6wt% Mo loading) was prepared by the wet impregnation technique with an NH4ZSM-5 zeolite (SiO2/Al2O3 = 40, gifted by Tosoh Co., Japan) and an aqueous solution of ammonium heptamolybdate (Kanto Chem. Co. of Japan).11 The methane dehydrocondensation reaction was carried out in a fixed-bed continuous-flow system at 1023–1073 K, a methane flow rate of 15–70 ml min−1 and a pressure of 0.3 MPa, and with alternate flows by switching with methane and H2 or 20% CO2/He. All the products were analyzed on-line by two GCs with FID and TCD, as described in previous reports.9–11,13 Mo K-edge XAFS (X-ray absorption fine structure) measurements were conducted on samples of Mo/HZSM-5 at the Photon Factory of the National Laboratory for High Energy Physics (KEK-PF, Tsukuba, Japan) to characterize the Mo/HZSM-5 catalyst before and after a periodic switching operation with H2 or 20% CO2/He flow at 1023–1073 K. The energy and current of the electrons (or positions) were 2.5 GeV and 250 mA, respectively, and a Si(311) channel-cut monochromator was used.15
The periodic switching operation of gas feed with methane and 100% H2 or 20% CO2/He on Mo/HZSM-5 resulted in the excellent improvement of the catalyst stability in methane dehydrocondensation to benzene and naphthalene at 1073 K, as shown in Figs. 1 and 2. Fig. 1 presents the formation rates of benzene and hydrogen on 6% Mo/HZSM-5 at 1073 K, 0.3 MPa and methane space velocities of 2700–12600 ml g−1 h−1, with switching operation between CH4 and H2 for 30 min each, in comparison with those without the switching treatment. For the switching treatment of gas feed with H2, a stable and high formation rate of benzene (10300 nmol g-cat−1 s−1) and H2 (20800 nmol g-cat−1 s−1; benzene/H2 molar ratio = 12) at 15% conversion of methane, close to thermodynamically limited conversion of methane at 1073 K, was obtained in a prolonged time on stream. In contrast, without the switching treatment, the benzene formation rate decreased drastically and reached zero after only 2 h on stream. Furthermore, a periodic switching treatment between methane and carbon dioxide was performed. Fig. 2 shows the benzene formation rates with time on stream for a switching operation between 20% CO2/He or 100% H2 and methane, and without the switching operation, at 1073 K, 0.3 MPa and 2700 ml g−1 h−1. Again, a serious decreasing trend for benzene formation without the switching treatment, i.e., in a conventional continuous mode of methane flow, and the distinct promotion effect on the catalyst stability with the switching operation between CH4 and 20% CO2/He or 100% H2 could be observed. However, comparing with the H2 switching treatment, the 20% CO2/He treatment was less efficient for the improvement of the catalyst stability.
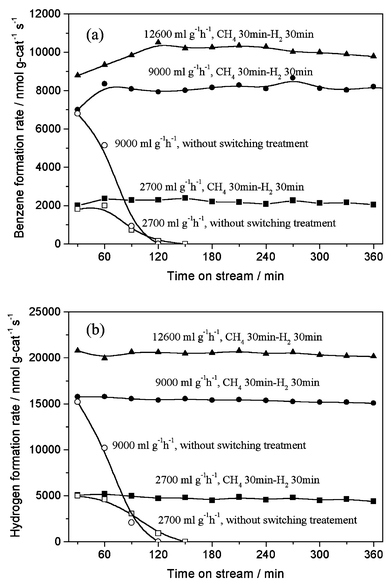 | ||
| Fig. 1 Formation rates of benzene (a) and hydrogen (b) in methane dehydrocondensation reaction on 6% Mo/HZSM-5 at 1073 K, 0.3 MPa and 2700–12600 ml g−1 h−1 with a periodic switching treatment between CH4 for 30 min and H2 for 30 min (solid symbols) and without the treatment (open symbols). | ||
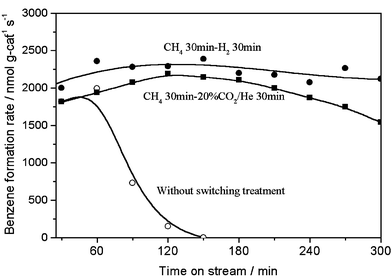 | ||
| Fig. 2 Formation rates of benzene in methane dehydrocondensation reaction on 6% Mo/HZSM-5 at 1073 K, 0.3 MPa and 2700 ml g−1 h−1 with a periodic switching treatment between CH4 and 20% CO2/He (■) or H2 (●) and without the treatment (○). | ||
Note that the coke formation was substantially suppressed by the periodic switching operation between methane and H2 or CO2, as compared with that using a methane flow alone. TPO profiles of used Mo/HZSM-5 catalysts under a continuous methane flow at 1073 K for 30 min (a), and then treated with 20% CO2/He for 30 min (b), or with 100% H2 for 30 min (c), are depicted in Fig. 3. Two peaks were observed at ca. 760 and ca. 850 K. The low-temperature peak was attributed to carbon associated with molybdenum, while the high-temperature peak was attributed to carbonaceous deposits on the Brønsted acid sites of the zeolite.13 Peak heights corresponding to both low- and high-temperature peaks decreased remarkably with CO2 or H2 treatment, demonstrating their effectiveness for coke removal from the catalyst surface. Moreover, the decrease in the amount of coke for the H2 treatment is more pronounced, indicating that H2 treatment is more efficient for coke elimination than CO2 treatment. Thus, the TPO results demonstrate that the notable improvement of the catalyst stability with the periodic switching treatment was due to the effective removal of coke.
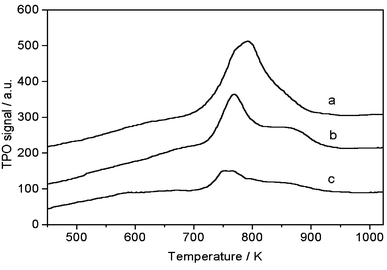 | ||
| Fig. 3 TPO profiles of used Mo/HZSM-5 samples under CH4 stream at 1073 K, 0.3 MPa and 2700 ml g−1 h−1 for 30 min (a), and then treated with 20% CO2/He for 30 min (b) or with 100% H2 for 30 min (c). (TPO experiments were carried out using 30 mg of catalyst at a 10% O2/He flow rate of 15 ml min−1 and temperature raising rate of 10 K min−1.) | ||
Mo K-edge EXAFS experiments were conducted to reveal the change in the active phase of the Mo/HZSM-5 catalyst by the periodic switching operation of methane and 20% CO2/He or 100% H2, the Fourier transform (FT) spectra of which are shown in Fig. 4. As has been described in ref. 15, the FT function of the sample that reacted with methane at 1073 K for 30 min showed a characteristic peak at 2.7 Å, the same as for the Mo2C standard. After the H2 treatment that followed the methane dehydrocondensation at 1073 K for 30 min, the features of the FT spectrum were unchanged, but its intensity increased greatly, i.e., about a two fold increase. These results suggest that the contribution of Mo2C as the active phase is unchanged, and is relatively enhanced by the H2 switching treatment at 1073 K, compared with the results for a continuous feed of methane. In contrast, after the 20% CO2/He treatment at 1073 K for 30 min, the FT spectrum of the Mo/HZSM-5 changed remarkably: the characteristic peak for Mo2C was lost and a peak for Mo–O at ca. 1.5 Å was increased.15 This suggests that the switching CO2 treatment results in the partial oxidation of active Mo carbide during its effective removal of the surface coke.
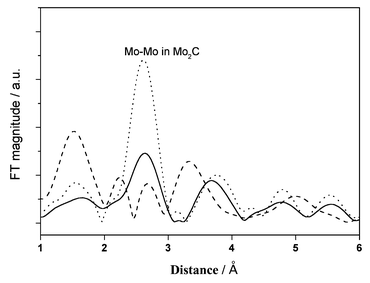 | ||
| Fig. 4 Mo K-edge Fourier transform spectra of used Mo/HZSM-5 samples under CH4 stream at 1073 K, 0.3 MPa and 2700 ml g−1 h−1 for 30 min (solid line), and then treated with 100% H2 for 30 min (dotted line) or with 20% CO2/He for 30 min (dashed line). | ||
In conclusion, the periodic switching treatment with hydrogen or carbon dioxide is substantially effective to regenerate the active Mo/HZSM-5 catalyst due to the efficient removal of coke deposited in the methane dehydrocondensation reaction to benzene and naphthalene at high temperatures of 1023–1073 K. As a result, a high and stable catalytic performance is achieved by a periodic switching treatment with methane and H2 or CO2 in contrast to that obtained for a continuous flow of methane alone.
This work was sponsored by the New Energy and Industrial Technology Development Organization (NEDO) of Japan through the 2000-2002FY Regional Consortium R&D Program for New Energy Processes of NEDO (12S1001).
Notes and references
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35 Search PubMed.
- F. Solymosi, A. Erdohelyi and A. Szoke, Catal. Lett., 1995, 32, 43 Search PubMed.
- L. Chen, L. Lin, Z. Xu, X. Li and T. Zhang, J. Catal., 1995, 157, 190 CrossRef CAS.
- D. Wang, J. H. Lunsford and M. P. Rosynek, Top. Catal., 1996, 3, 289 Search PubMed.
- S. Liu, Q. Dong, R. Ohnishi and M. Ichikawa, Chem. Commun., 1997, 1455 RSC.
- R. W. Borry, E. C. Lu, Y. H. Kim and E. Iglesia, Stud. Surf. Sci. Catal., 1998, 119, 403 Search PubMed.
- Y. Shu, D. Ma, L. Xu, Y. Xu and X. Bao, Catal. Lett., 2000, 70, 67 Search PubMed.
- Y. Shu and M. Ichikawa, Catal. Today, 2001, 71, 55 CrossRef CAS.
- S. Liu, L. Wang, Q. Dong, R. Ohnishi and M. Ichikawa, Chem. Commun., 1998, 1217 RSC.
- L. Wang, R. Ohnishi and M. Ichikawa, J. Catal., 2000, 190, 276 CrossRef CAS.
- Y. Shu, R. Ohnishi and M. Ichikawa, J. Catal., 2002, 206, 134 Search PubMed.
- Y. Lu, D. Ma, Z. Xu, Z. Tian, X. Bao and L. Lin, Chem. Commun., 2001, 2048 RSC.
- Y. Shu, R. Ohnishi and M. Ichikawa, Catal. Lett., 2002, 81, 9 Search PubMed.
- W. Ding, G. D. Meitzner and E. Iglesia, J. Catal., 2002, 206, 14 Search PubMed.
- S. Liu, L. Wang, R. Ohnishi and M. Ichikawa, J. Catal., 1999, 181, 175 CrossRef.
| This journal is © The Royal Society of Chemistry 2003 |