Studies on the performance stability of mixed conducting BSCFO membranes in medium temperature oxygen permeation
Andre C. van
Veen
*,
Michael
Rebeilleau
,
David
Farrusseng
and
Claude
Mirodatos
CNRS-IRC, 2 avenue Albert Einstein, 69626, Villeurbanne Cedex, France. E-mail: vanveen@catalyse.univ-lyon1.fr; Fax: +33 (0)4 7244 5399; Tel: +33 (0)4 7244 5424
First published on 22nd November 2002
Abstract
A permeation study using bare Ba0.5Sr0.5Co0.8Fe0.2Ox membranes shows that stable oxygen fluxes are only achieved when operating the membrane at temperatures higher than 1023 K and indicates therefore that short contact time membrane reactors will be most suitable for future upgrading of light hydrocarbons.
Catalytic membrane reactors and related devices are attractive configurations to carry out large scale chemical processes as their multifunctional action leads to comparably small plant dimensions and lowered operational costs.1 An important scenario is the use of non-porous oxygen permeable membranes in the selective oxidation of light hydrocarbons allowing a process design without the cost intensive (cryogenic) oxygen separation unit. The use of highly perm-selective (i.e. dense) catalytic membranes might also lead to ‘green processes’ with enhanced selectivity towards value-added products as reactions with permeated nucleophilic lattice oxygen ions proceed more selectively than those with adsorbed electrophilic atoms formed from gas-phase oxygen.2,3 However, a profitable use of investment intensive membrane reactors depends on devices (i.e. membranes) allowing reasonably high production rates (i.e. oxygen fluxes) and a long term functionality. In turn, the used oxygen supplying inorganic membrane must exhibit a high kinetic stability (i.e. preserving an oxidised state of the bulk) and the working conditions have to match those of catalytic systems eventually necessary for the chemical reaction. One mixed conducting membrane material meeting the requirement of high permeation fluxes at high temperatures is the recently described Ba0.5Sr0.5Co0.8Fe0.2Ox perovskite (BSCFO).3–6 Unfortunately, information on the long time performance in oxygen permeation is limited,4 especially at comparably low temperatures (e.g. 923 K) recently suggested for the oxidative dehydrogenation of ethane (ODHE).3 We report therefore in this communication a in-depth study on the performance stability in oxygen permeation at comparably low temperature, i.e. in the range 973–1073 K. Besides observing formerly unreported high oxygen fluxes, we obtained detailed information on the causes of a membrane deactivation. Furthermore, some indications have been found that the sealing and/or the reactor geometry have an influence on the attainable oxygen permeation rate.
The synthesis of the BSCFO perovskite was carried out by an adapted variant of the so-called citrates method7,8 employing EDTA and citric acid in parallel as complex formation agents.4,5 Briefly, the required amounts of cation precursors were introduced as Ba(NO3)2, Sr(NO3)2, Co(NO3)2·6H2O and Fe(NO3)3·6H2O (purity >99.5%) and completely dissolved in a small portion of water using in respect to Ba and Sr an equimolar amount of EDTA and a 4 fold larger quantity of citric acid. By evaporation of water, a gel and subsequently a foam was formed. Finally all organic compounds were combusted by treating the foam at 1173 K for 8 h in air yielding a homogenous perovskite powder. Raw membrane discs were pressed by applying a pressure of 140 MPa for 1 min. A densification of the raw discs was accomplished by sintering them for 8 h at 1423 K in dense alumina boats.
The formation of a pure perovskite structure was checked by X-ray diffraction using a Bruker D5005 system in the 2θ range 3–80°, a step width of 0.02°, a counting time of 1 s and Cu Kα1+α2 radiation (1.54184 Å). The bulk elemental composition was determined by ICP-OES (Spectroflame) analysing a sample dissolved by heating at 523 to 573 K in a mixture of H2SO4 and HNO3. SEM micrographs were taken with a JEOL JSM-840A electron microscope, whereby a calibrated PGT spectrometer upgrade allowed to determine the element composition by EDX analysis (as mean value of several points analysed).
The oxygen permeation properties were studied using a reactor schematically depicted in Fig. 1. Membrane discs (1 mm thick) were sealed in between two dense alumina tubes (od: 12 mm, id: 8 mm) using gold rings as chemically inert sealants. Furthermore, the side wall of the discs was extensively covered with gold paste suppressing radial contributions to the oxygen flux permeating through the active cross-section of 0.5 cm2. The sealing was carried out in the beginning of experimental runs by heating to 1073 K for one night. After that the oxygen-rich side was fed at a constant total pressure adjusted to 120 kPa and a constant total flow rate of 50 ml min−1 using a mixed stream of 10 ml min−1 O2 (Air liquide) and N2 as balance (evaporated liquid N2, Air liquide) controlled individually by mass flow controllers (Brooks, type 5850TR). A flow of 20 ml min−1 He was passed to the oxygen-lean permeate side assuring a proper rinsing of permeated oxygen. Introduced gases were preheated on both membrane sides while passing the void space between the alumina tube and an inserted quartz tube. The analysis of the gas leaving via the tube centre was carried out with a calibrated computer controlled GC (HP 5890 series II, TCD, 5 Å sieve column). Hence, a separate monitoring of the oxygen and nitrogen partial pressures allowed to rule out major sealing problems or an incomplete densification of the membrane. Permeated oxygen fluxes were calculated from the oxygen partial pressure at the permeate side, the helium flow rate and the active membrane surface assuming a behaviour according to the law for ideal gases. Minor corrections (well below 4%) accounting for contributions from slight imperfections in the high temperature sealing were introduced on basis of the small detected nitrogen partial pressures, assuming a penetration of air.
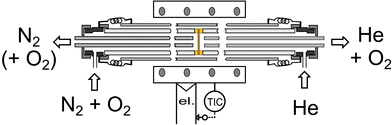 | ||
| Fig. 1 Scheme of the high temperature permeation reactor with gold seal. | ||
The stability of the membrane in terms of O2 permeability was studied by recording continuously the permeation flux under a partial pressure gradient of air/He for 34 days. As depicted in Fig. 2 the membrane was allowed to relax for approximately 4 days before cyclically changing the sample temperature within the range 973–1073 K.
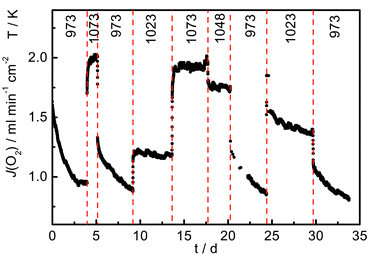 | ||
| Fig. 2 Oxygen permeation flux as a function of time at temperatures in the range 973–1073 K. | ||
Data collected at 973 K during the first days on stream indicated a steep decline of the initially high oxygen permeation flux. As no steady-state was reached after 4 days, the temperature was increased to 1073 K. In addition to the obvious instantaneous change related to a higher temperature, an additional much slower increase with time was observed. Upon decreasing the temperature back to 973 K the oxygen permeation was once more found to be non-stationary, resembling moreover the decline observed before. However, in this case the initial permeation fluxes were significantly lower than those measured during the first day on stream. Further temperature cycles suggested that the membrane performed in a stable manner at temperatures higher than 1023 K. At lower temperatures the permeability declined and a reactivation at high temperature was necessary to restore the initial status.
In order to investigate the reason of the observed decline in oxygen permeability, a spatial resolved analysis of the element composition after use was performed by means of SEM-EXD microscopy. Fig. 3 shows the element maps of Fe, Co, Sr and Ba along with a micrograph view in an interfacial sample region (showing the permeate side and the sample bulk after cracking the disc). The element maps suggest an enrichment of Ba at the surface of the permeate side, whereas the Fe and Co content strongly decreased. Quantitative measurements of the cation composition were performed at the air flushed feed side, the He swept permeate side and the sample bulk accessed by cracking. Table 1 reports the results along with those of a conventional ICP-OES analysis of two different samples before use. Within the analytical precision the results of the chemical analysis coincided well with those of the EDX analysis at the air exposed feed side and the bulk. However, in agreement with expectations drawn from the element maps, distinct changes become obvious when the EDX analysis of the He flushed permeate side is taken into account. The surface of the permeate side consists of almost 70% Ba, replacing almost completely Fe and Co, detected as traces below 4%, whereas Sr shows only a minor change in concentration.
 | ||
| Fig. 3 SEM-EDX element maps for Fe, Co, Sr, Ba and the respective image view showing both the bulk (top part) and the permeate side surface (bottom part) in the interfacial area obtained by cracking. | ||
In turn, the EDX results indicate a segregation of Ba to the surface of the permeate side during membrane operation at high temperature. A comparable behaviour was already observed in the case of La1−xSrxFeO3−δ membranes, where Sr segregated when the membrane was operated under large oxygen activity gradients.9
The results of the long term permeation tests indicate two distinctly different characteristics which might be best explained by changes in the sample constitution. First, an induction period was observed at the begin of the experiment where the oxygen permeation declined irreversibly from a high level (see results during day one at 973 K). This non-reversible induction period can be explained by the observed high temperature segregation of Ba to the surface near region at the permeate side. It is reasonable to assume a partial loss of mixed conductivity in this region leads in turn to an enlarged transport resistance and finally to decreased permeation fluxes. However, the observed decline in the permeation flux might be further reinforced by Ba specific properties (e.g. a formation stable adsorbate layers further hindering the permeation).
Second, at temperatures lower than 1023 K the oxygen permeation was non-stationary and declined with time. Contrary to the finding in the induction period the same decrease in oxygen permeability was periodically observed throughout the whole time on stream (see e.g. the cycles at 973 K between days 5 and 9, 20 and 25 and 30 and 34). Obviously, this deactivation was reversible and the former permeability could be restored by allowing the sample to relax to its former state, e.g. by performing measurements at temperatures above 1023 K. Moreover, the rate at which the deactivation occurred was found to increase with decreasing temperature leading at 973 K already to an important decline in oxygen permeation during a few hours. Furthermore, a performance decline could also be observed at a constant temperature of 1073 K when the membrane was operated with a slightly higher oxygen partial pressure enforced e.g. by a higher air side gas flow. It is therefore reasonable to explain the reversible deactivation phenomena by a changed ionic conductivity either resulting from a change in the oxygen vacancy concentration or by a change in the local oxidation state of the BSCFO material.
A. C. vV. and M. R. wish to thank Gerard Wicker for performing the SEM microscopy. Financial support was granted by the EC programme ‘CERMOX’ (G55RD-CT-2000-0035).
Notes and references
- J. A. Dalmon, Handbook of heterogeneous catalysis, Vol. 3, Wiley-VCH, Weinheim, 1997, 1387. Search PubMed.
- A. Pantazidis, S. A. Bucholz, H. W. Zanthoff, Y. Schuurman and C. Mirodatos, Catal. Today, 1998, 40, 207 CrossRef CAS.
- H. Wang, Y. Cong and W. Yang, Chem. Commun., 2002, 1468 RSC.
- Z. Shao, H. Dong, G. Xiong, Y. Cong and W. Yang, J. Membr. Sci., 2001, 183, 181 CrossRef CAS.
- Z. Shao, W. Yang, Y. Cong, H. Dong, J. Tong and G. Xiong, J. Membr. Sci., 2000, 172, 177 CrossRef CAS.
- H. Dong, Z. Shao, G. Xiong, J. Tong, S. Sheng and W. Yang, Catal. Today, 2001, 67, 3 CrossRef CAS.
- M. P. Pechini, US Pat., 3,330,697, 1967.
- T. Taguchi, S. Matsuura, M. Nagao, T. Choso and K. Tabata, J. Solid State Chem., 1997, 129, 60 CrossRef.
- J. E. ten Elshof, H. J. M. Bouwmeester and H. Verweij, Solid State Ionics, 1996, 89, 81 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |