Optimisation of extraction procedures for metallothionein-isoforms and superoxide dismutase from liver samples using spiking experiments
Volker Nischwitz*, Bernhard Michalke and Antonius Kettrup
GSF-National Research Centre for Environment and Health, Institute of Ecological Chemistry, Ingolstädter Landstr. 1, 85764 Neuherberg, Germany
First published on 11th December 2002
Abstract
The speciation of trace element species in solid matrices like liver samples is still problematic due to two reasons. On the one hand direct methods with sufficient selectivity and sensitivity are currently not available. Therefore extraction procedures have to be applied which are often problematic in respect to species stability. On the other hand there are no reference materials with known amounts of metal proteins like metallothionein-isoforms (MT) and superoxide dismutase (SOD) for quality control. So the aim of this study was to develop and optimise procedures for the species-preserving extraction of the model compounds MT and SOD from liver samples. Spiking experiments were performed to overcome the lack of appropriate reference materials. In a first step the stability of the model species without liver matrix was investigated by the variation of several extraction parameters. The extractant and exposure to ultrasonic energy especially had a great influence on the recovery of the species while temperature, buffer concentration and atmospheric conditions were less critical. In a second step spiked liver samples were extracted with a selection of procedures taken from the literature. Most of these methods provided recoveries between 70% and 100%. Additionally the buffer concentration and the extractant-to-liver ratio were varied for optimisation. The metal balance of an extraction showed recoveries of 81% for Cd, 94% for Cu and 87% for Zn.
Introduction
The need of trace element speciation is generally accepted and there is a rising number of applications in this promising field. The establishment of coupling techniques like HPLC-ICP-MS enabled the characterization of trace element species in liquid samples.1,2 Even volatile or gaseous samples have been analysed by GC-ICP-MS.3 However there are no sufficiently selective and sensitive methods for the direct determination of trace element species in solid samples currently available.4In the biomedical field the speciation analysis of body fluids is not sufficient to understand the complex transport and function of metal binding proteins. Tissue samples are at least equally important. The liver especially contains a variety of enzymes and further trace element species which are essential for the health of the organism. A change in the distribution or concentration of these species is often an indication of pathological processes.
Currently, the only suitable possibility is the use of extraction procedures to transfer the target species from solid to solution. But this is a critical step due to many reasons. Firstly, the addition of an extractant and the extraction conditions (e.g. temperature, pH, buffer concentration, use of energy) change chemical equilibria in the sample and therefore might influence the species stability. Secondly, a homogenisation of the tissue is necessary to achieve a sufficient extraction efficiency for the cytosolic target proteins. Thirdly, there is a solid residue after extraction which is not accessible for speciation. Depending on the centrifugation speed and homogenisation technique it contains e.g. mitochondria, nuclei and cell membranes.
Another difficulty is the lack of appropriate reference materials for metal proteins in liver samples for quality control of analytical methods. So spiking experiments are necessary to produce samples with known amounts of the target species. Moreover the homogeneity of the liver tissue has to be considered because there are different cell types which differ in their metal protein content.
The major point of criticism of spiking experiments is the question whether the spiked species are present in the same binding form as the naturally occurring amounts.5,6 In the case of the liver samples this prerequisite is certainly not given before the homogenisation of the spiked liver sample. But in respect to species stability the critical step starts only with the disruption of the cell walls and the mixture of cell components with the extractant. Simultaneously the cytosolic i.e. dissolved native MT and SOD are mixed in the same way as the spiked i.e. also dissolved MT and SOD with the extractant and the liver matrix. So an equal binding form of native and spiked species is likely. But this approach is of course limited to procedures which are applied during or after homogenisation.
Considering this background the aim of this study was to develop and optimise mild species-preserving extraction procedures for trace element species from real (bio-)matrices, such as liver samples and to prove the species stability during extraction. Metallothionein (MT) and superoxide dismutase (SOD) were used as model compounds for the comparison of different extraction conditions. The optimisation was performed in two steps. 1. Investigations on the stability of the model compounds without liver matrix: Spike solutions of the model species were mixed with different extractants and the parameters temperature, pH, buffer concentration, ultrasonic exposure and atmospheric conditions were varied within a realistic range to investigate the stability of the species. 2. Extraction of liver samples spiked with MT and SOD: A selection of six extraction procedures taken from the literature was applied with minor or no modifications to spiked liver samples and compared by the recovery for the spiked species. Additionally the buffer concentration and the extractant-to-liver ratio were varied to optimise these parameters. Three procedures with the best recoveries were chosen for further experiments. The species stability during extraction was determined qualitatively by the identification of the model species with HPLC-ESI-MS and quantitatively by the recovery of the spiked species derived from HPLC-ICP-MS.
Results of both steps are presented in the following. Later unspiked liver samples will be extracted with the three optimised procedures to characterize the natural contents of the model compounds i.e. the porcine liver MT and the porcine liver SOD.
Background
Metallothioneins are rather well known and investigated by countless studies for more than 40 years. However there still remain many questions concerning the universal functions ranging from metal transport, and metal detoxification to radical scavenging, immune response and genotoxicity.7 Moreover there is a broad range of extraction procedures for MT from liver samples. A literature survey showed e.g. pH values of the extractant from 7 to 9, extractant-to-liver ratios (v/w) from 1 to 10 and buffer concentrations from 0 (without buffer) to 100 mM.8–13 So the necessity of a harmonization and selection of the methods for MT quantification is obvious.7 The preparatively isolated rabbit liver MT I mixture from Sigma was used for spiking in this study. The focus was laid on the two dominant Cd–MT-isoforms in this preparation with a molecular weight of 6988 Da (Cd7MT-2d) and 7014 Da (Cd7MT-2e) respectively.14Superoxide dismutases occur in relatively high concentrations in many tissues especially in liver, kidney and brain. Their important function lies in the protection of cells against the highly toxic superoxide radical-anion.15 Extraction procedures described in the literature use pH values of the extractant from 7 to 8.2, extractant-to-liver ratios (v/w) from 1 to 9 and buffer concentrations from 0 (no buffer) to 500 mM.16–19 A bovine liver CuZn–SOD with a molecular weight of 31443 Da (2 apo-subunits of 15593 Da with one atom of Cu and Zn each) from Sigma was applied for the spiking experiments in this study.14
Experimental
Liver, model compounds and chemicals
Fresh porcine liver from a local store was used for the experiments. It was obtained few hours after slaughtering and processed immediately after transport to the lab. So the risk of changes by autolytic processes was minimized.Rabbit liver metallothionein I (Lot 80K7012; Cd content 8%, Zn content 1.2%) and bovine liver superoxide dismutase (EC 1.15.1.1; Lot 129H7002) were bought from Sigma, Deisenhofen. Tris(hydroxymethyl)aminomethane (TRIS) was obtained from Serva, Heidelberg. [4-(2-Hydroxyethyl)-piperazino]-ethansulfonic acid (HEPES) and ammonium formate were bought from Sigma, St. Louis and acetonitrile Chromasolv from Riedel de Haen, Seelze. Methanol Lichrosolv, Titriplex III-solution (Na2EDTA) and hydrochloric acid (30%, suprapur) were purchased from Merck, Darmstadt. Ammonium acetate, puriss. and Triton X-100 were obtained from Fluka, Buchs. Deionised water (18 MΩ cm) was prepared with a Millipore system. All reagents could be used without further purification.
Preparation of the extracts from the first step
The extraction was performed in a glove bag (Atmos Bag, Sigma-Aldrich) under an argon atmosphere, extractants were purged with argon before use and samples were cooled in an ice bath (approximately 4 °C). All experiments were carried out in duplicate; the conditions are listed in Table 1. Aliquots of a MT-I/SOD-spike solution were mixed with the respective extractant in a closed plastic tube. After 20 min in an ice or water bath at the respective temperature the extracts were transferred into plastic containers, frozen and lyophilised (freezedryer Beta from Christ, Osterode; about 0.03 mbar, sample temperature −10 °C). Sonication of extracts was performed with a Branson sonifier either at 30 W or 50 W. Samples were cooled in an ice bath during sonication and exposure times were 30 s or 60 s (30 s exposure, 30 s break to avoid heating by sonication, 30 s exposure). The influence of oxygen on the species stability was investigated by comparing samples prepared in the glove bag, samples prepared at the air and samples purged with oxygen gas.| Number | Extractant | Temperature/°C | Sonication |
|---|---|---|---|
| 1.1 | 10 mM Tris–HCl (pH 7.4) | 4 | — |
| 1.2 | 10 mM (NH4)2HPO4 (pH 8.1) | 4 | — |
| 1.3 | 10 mM Tris–HCl, 250 mM sucrose (pH 7.4) | 4 | — |
| 1.4 | 50% Acetonitrile in water (pH 6.6) | 4 | — |
| 1.5 | 10 mM Tris–HCl, 20% methanol (pH 7.4) | 4 | — |
| 1.6 | Water (pH 8.7) | 4 | — |
| 1.7 | 10 mM HEPES (pH 7.4) | 4 | — |
| 1.8 | 10 mM Tris–HCl (pH 7.4) | 22 | — |
| 1.9 | 10 mM Tris–HCl (pH 7.4) | 35 | — |
| 1.10 | 10 mM Tris–HCl (pH 7.4) | 50 | — |
| 1.11 | 10 mM Tris–HCl (pH 7.4) | 65 | — |
| 1.12 | 10 mM Tris–HCl (pH 6.4) | 4 | — |
| 1.13 | 10 mM Tris–HCl (pH 8.4) | 4 | — |
| 1.14 | 20 mM Tris–HCl (pH 7.4) | 4 | — |
| 1.15 | 40 mM Tris–HCl (pH 7.4) | 4 | — |
| 1.16 | 80 mM Tris–HCl (pH 7.4) | 4 | — |
| 1.17 | 10 mM Tris–HCl (pH 7.4) | 4 | 30 W; 30 s |
| 1.18 | 10 mM Tris–HCl (pH 7.4) | 4 | 30 W; 60 s |
| 1.19 | 10 mM Tris–HCl (pH 7.4) | 4 | 50 W; 30 s |
| 1.20 | 10 mM Tris–HCl (pH 7.4) | 4 | 50 W; 60 s |
| 1.21 | 10 mM Tris–HCl (pH 7.4) (at the air) | 4 | — |
| 1.22 | 10 mM Tris–HCl (pH 7.4) (purged with oxygen) | 4 | — |
Preparation of extracts from the second step
In analogy to the first step the extracts were prepared in a glove bag and ice cooling was applied if possible. Two replicates of each experiment were prepared for quality control. Experimental conditions are given in Table 2. Pieces of approximately 1 g were cut from the same lobe of a fresh porcine liver. Only homogenous inner tissue was used; the surface skin was removed. Additionally, ten small pieces of liver tissue covering the whole used region of the liver were collected, later digested and analysed for their total metal contents. Relative standard deviations below 10% (2.1% for Mn, 2.3% for Zn, 3.3% for Fe, 3.6% for Cu and 9.9% for Cd) prove the homogeneity of the trace element content. The other samples were weighed exactly and 50 μL of a spike solution containing each 100 μg MT-I and SOD were added to each sample. Afterwards they were homogenized manually with half of the extractant volume in a glass–Teflon-homogeniser. The homogenate was transferred into a centrifugation tube and residues were washed with the second half of the extractant volume from the homogeniser into the tube. After centrifugation (Biofuge 17 RS, Heraeus Sepatech Osterode) the supernatant was collected with a pipette, frozen and lyophilised. The extracts were redissolved in deionised water and filtered before analysis (0.45 μm, membrex PES).| Number | Extractant | Ratioa | Centrifugationb | Reference |
|---|---|---|---|---|
| a Extractant-to-liver ratio (v/w).b Temperature 4 °C. | ||||
| 2.1 | 100 mM Ammonium formate (pH 7.4) | 4 | 19118 g; 30 min | 13 |
| 2.2 | 50 mM NaH2 PO4 (pH 7.0) | 3 | 12000 g; 30 min | 31 |
| 2.3 | 10 mM Tris–HCl, 250 mM sucrose (pH 7.4) | 1 | 10700 g; 20 min | 10 |
| 2.4 | 10 mM Tris–HCl, 250 mM sucrose, 1 mM EDTA (pH 7.4) | 3 | 10000 g; 15 min | 33 |
| 2.5 | 20 mM Tris–acetic acid, 0.2% Triton X 100 (pH 7.8) | 2 | 13700 g; 30 min | 32 |
| 2.6 | 10 mM Tris–HCl (pH 7.4) | 3 | 10000 g; 15 min | 30 |
| 2.7 | 20 mM Tris–HCl (pH 7.4) | 3 | 10000 g; 15 min | — |
| 2.8 | 50 mM Tris–HCl (pH 7.4) | 3 | 10000 g; 15 min | — |
| 2.9 | 100 mM Tris–HCl (pH 7.4) | 3 | 10000 g; 15 min | — |
| 2.10 | 10 mM Tris–HCl (pH 7.4) | 1 | 10000 g; 15 min | — |
| 2.11 | 10 mM Tris–HCl (pH 7.4) | 2 | 10000 g; 15 min | — |
| 2.12 | 10 mM Tris–HCl (pH 7.4) | 4 | 10000 g; 15 min | — |
| 2.13 | 10 mM Tris–HCl (pH 7.4) | 5 | 10000 g; 15 min | — |
Analysis of MT and SOD in the extracts
Unfortunately, a quantification of the whole metal proteins by HPLC-ESI-MS was not successful because of the low ionisation of the target species in ESI at neutral conditions. So ICP-MS detection of the bound metals was used which is less specific. However no significant interferences by coeluting species were noticed. SOD is routinely quantified by activity assays, but these cannot distinguish between the spiked bovine CuZnSOD and the porcine CuZnSOD.Two Cd7MT-isoforms and the CuZn-SOD were quantified in the extracts of the first step by reversed phase HPLC (HyPurity C4 column, 30 mm × 4.6 mm, Thermo Hypersil, Kleinostheim) coupled to ICP-MS. A HP 1050 Titan system with variable wavelength UV-detector was used and an ELAN 5000 quadrupole ICP-MS equipped with a Meinhard nebulizer and a cyclon spray chamber. Separation was achieved with a 10 mM ammonium acetate buffer and a methanol gradient at pH 7.5. The 114Cd signal was used for the MT-isoforms and the 63Cu signal for the SOD. Both copper isotopes are interfered in quadrupole ICP-MS so the more frequent 63Cu was chosen. Due to the chromatographic separation the risk of interferences is reduced to coeluting species. In blank runs or runs with unspiked extracts no interfering peak was detected at the retention time of the bovine CuZnSOD. Calibration was performed with MT/SOD solutions in 10 mM ammonium acetate (pH 7.5) which were prepared from the spike solution. So it was not necessary to determine the exact concentration of each isoform in the mixture. Concentrations of MT-isoforms and SOD refer to the used preparations i.e. 10 mg L−1 Cd7MT-2d means the concentration of Cd7MT-2d in a solution of 10 mg L−1 Sigma MT-I preparation. Peak areas were used for quantification.
For the liver extracts of the second step a preseparation with size exclusion chromatography (TSK column HW 55 (S), 50 cm × 8 mm, TosoHaas, Stuttgart) was necessary to separate the high contents of high molecular weight proteins (>200 kDa) from the comparatively low molecular weight MT/SOD fraction. In contrast to the first step calibration was performed with spiked liver extracts to compensate matrix effects. The Cu peak of the spiked SOD appears relatively small in Fig. 1 because the scale is guided by the large native Cu peaks. In fact the signal to noise ratio is 27.
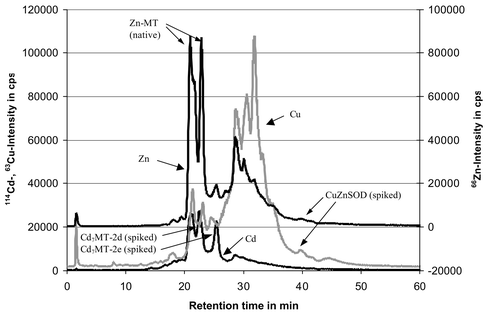 | ||
| Fig. 1 HPLC-ICP-MS chromatogram of the SEC-fraction (molecular weight range of MT and SOD) of the TRIS extract from a spiked liver sample (C4 reversed phase column; flow rate 0.45 mL min−1; 10 mM ammonium acetate buffer with methanol gradient; isotopes 114Cd, 63Cu, 66Zn are displayed). For further details refer to Nischwitz et al.14 | ||
For both steps ESI-MS spectra of RP-HPLC fractions of the MT isoforms and the SOD were recorded for further identification via the molecular weight. These measurements were performed with a TSQ 700 ESI-MS applying 5.5 kV electrospray voltage, 1400 V multiplier voltage, 200 °C capillary temperature and a sample flow rate of 5 to 10 μL min−1 using a Harvard syringe pump. MT containing fractions were pre-concentrated and partially desalted by ultrafiltration (centrifugal filters FUGISEP MIDI with 4000 Da molecular weight cut off).
Total metal contents were determined by ICP-AES (JY-70, Jobin Yvon Instruments SA, France) because the Cu- and Zn-isotopes are interfered by cluster ions in quadrupole-ICP-MS. Detection limits of ICP-AES were sufficient for the interesting metals. Previously the liver samples as well as the extracts and residues were pressure digested with concentrated HNO3 at 170 °C for 8 h.20
Further details of the analytical methods, instruments and the quantitative data evaluation were described earlier.14
Results and discussion
Recovery
Concentrations with confidence intervals were determined for two Cd7MT-isoforms and CuZn-SOD by HPLC-ICP-MS. A typical chromatogram of a sample from the second step is shown in Fig. 1. Mean values were calculated from the two replicates. The confidence interval of the mean was derived from the confidence intervals of the replicates by propagation of error, if the confidence intervals of the replicates overlapped. Otherwise the difference between the mean and the individual replicates was used. The recovery was calculated as percentile ratio of the measured concentration in the extracts and the spiked concentration.Proof of species stability
The retention times and signal patterns of the HPLC-ICP-MS chromatograms were the same for calibration solutions and extracts. For samples with an acceptable recovery (>60%) ESI-MS spectra of the RPC-fractions of Cd7MT-2d, Cd7MT-2e and CuZnSOD were recorded. Signals were detected at the same m/z values (within the quadrupole resolution of approximately 0.5–1 Da) as previously for the model compounds before extraction.14 Examples of ESI-MS spectra of Cd7MT-2d and SOD in an extract from the second step are shown in Fig. 2. The MT spectrum shows the +4 and +5 charged molecular ions of the Cd7MT-2d in an unacidified sample. The calculated molecular weight of (6996 ± 1) Da is in good agreement with the molecular weight of (7000 ± 2) Da for this species determined previously by ESI-MS in a calibration solution without liver matrix.14 After acidification the +4 and +5 charged molecular ions of the apo-MT were detected (spectrum is not shown). From the difference of the resulting molecular weight in both cases the presence of 7 Cd-ions can be derived. ESI-MS spectra of SOD could only be recorded after acidification of the samples so only the identification of the protein was possible. The molecular weight of (15595 ± 6) Da matches very good with the respective result of (15593 ± 4) Da derived from a calibration solution in the absence of liver matrix.14 Calibration of the RPC-ICP-MS with spiked liver extracts provided reproducible and linear calibration curves. | ||
| Fig. 2 ESI-MS spectra of RPC fractions of a TRIS extract from a spiked liver sample (see Fig. 1). ESI-MS conditions: 5.5 kV electrospray voltage, 1400 V multiplier voltage, 200 °C capillary temperature. Intensities are relative to the highest peak whose absolute intensity is given at the top of the right y axis. (a) Cd7MT-2d (not acidified). The MT spectrum shows the +4 and +5 charged molecular ions. The calculated molecular weight is (6996 ± 1) Da. (b) CuZnSOD (acidified). The spectrum of the apo-SOD shows the +11, +10 and +9 charged molecular ions. The resulting molecular weight is (15595 ± 6) Da. | ||
Additionally, ESI-MS spectra of the intensive Zn-species detected near the retention time of the spiked Cd-MT were recorded. The resulting molecular weights of about 6000 Da (acidified fractions) and about 6430 Da (not acidified fractions) correspond with the respective values of 5970 Da to 6017 Da for seven porcine liver apo-MT isoforms21 and a mass difference of 443 Da for seven Zn atom (minus 14 H+). This supports the obvious suggestion that these species are porcine Zn-MT.
Step 1: Investigations on the stability of the model species without liver matrix
At first the stability of the Cd7MT isoforms and the CuZnSOD was investigated without the liver matrix under variation of several extraction parameters. These experiments (Table 1) provide direct information about the stability ranges of the model species without stabilizing or destabilizing effects of the liver matrix. The most important parameter is the extractant. Most of the published extraction procedures for MT or SOD use buffers especially TRIS or phosphate. Sometimes sucrose is added10 to match the osmotic conditions in the liver or organic solvents like acetonitrile, ethanol and chloroform12,22,23 are used after the homogenisation to remove matrix proteins by precipitation. The reducing agents 2-mercaptoethanol or dithiotreitol are applied in several procedures to prevent oxidation of MT.24,25 Seven extractants were compared in this study. The recoveries are given in Fig. 3. The TRIS-buffer provided the best result with recoveries around 100% for MT and SOD. Slightly higher recoveries were found for TRIS with 20% methanol while the phosphate buffer yielded only recoveries between 80 and 100%. The SOD recovery in TRIS + sucrose is very good, but only 80% of the MT isoforms were found. The HEPES-buffer is acceptable for MT while SOD recovery is far beyond 100%. The worst results were obtained with water and water + acetonitrile. In both cases only between 60 and 80% of the MT-bound Cd is recovered and the SOD recovery is again rather high.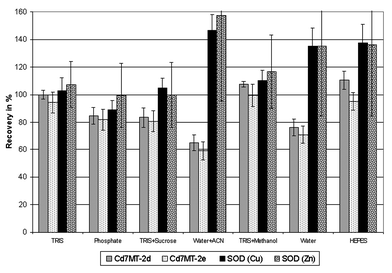 | ||
| Fig. 3 Recovery of MT-isoforms and SOD after extraction with different solvents (experimental conditions are given in Table 1). The MT-isoforms were quantified via the 114Cd-signal and the CuZnSOD via the 63Cu-signal and additionally via the 66Zn-signal according to Nischwitz et al.14 (ACN = acetonitrile.) | ||
In summary the buffered extractants provided better results than the unbuffered. Especially the TRIS containing extractants stabilized the Cd-MT isoforms as well as the CuZnSOD, so this buffer was used for all further experiments of the first step. Reduced recoveries for the Cd7MT are probably due to a release of Cd from the MT. Increased recoveries for the SOD-bound Cu are an indication of unstable conditions, too, but the explanation is more difficult. As further information the SOD-quantification via the 66Zn-signal is also presented in Fig. 3. The results match well with the quantification via the 63Cu-signal; however the standard deviation is considerably higher due to the lower peak height. Moreover, the replicates were analysed on different days so an analytical error is quite improbable. Maybe a transfer of Cu and Zn from destabilized MT to unsaturated SOD occurs. Due to the low metal content of the SOD (<0.5% of Cu and Zn) even small amounts of these metals would be sufficient to produce this effect.
In the following single extraction parameters were varied while the others were held constant on standard conditions i.e. 10 mM TRIS buffer, pH 7.4, 4 °C and argon atmosphere. The effect of the temperature was investigated between 4 °C and 65 °C. The recoveries are within a very good range of 100 ± 15% and no significant trend was found. So the model species are stable under these conditions. This is consistent with the well known heat stability of MT which is frequently used for the quick removal of matrix proteins by heat denaturation.26
The TRIS buffer concentration was varied from 10 mM to 80 mM. Again no significant trend was found and good recoveries between 83% and 104% were obtained for all species and buffer concentrations.
A more critical parameter is the pH value because acidification leads to a release of the complexed metal ions from MT and SOD.27 In respect to the native liver pH of about 6.9 to 7.428 the pH was changed only between 6.4 and 8.4. Recoveries of the MT-bound Cd from 98% to 108% prove the stability of the Cd7MT isoforms. SOD bound Cu was recovered to 91.2 ± 6.7% and 95 ± 13% at pH 7.4 and 8.4 respectively, but only to 74 ± 13% at pH 6.4. So a release of Cu from the SOD already starts between pH 7.4 and 6.4.
Sonication is an alternative method to the mechanical homogenisation for small amounts of tissue.12 But as far as we know stability tests for MT and SOD under sonication have not been published yet. So TRIS extracts of MT/SOD-spikes were exposed to ultrasonic power of 30 W or 50 W for 30 s or 60 s (Fig. 4). This treatment had quite a negative influence on Cd–MT stability. The recovered amount of MT-bound Cd decreased rapidly with increasing ultrasonic power. A doubled exposure time led to no significant further reduction. Probably the MT protein was denatured and ionic Cd was released. In the RP-chromatogram of the sample 1.20 (50 W; 60 s) no relevant Cd peak was detected. An injection of a Cd standard solution showed that ionic Cd is not eluted from the column at neutral conditions. This supports the hypothesis that the Cd–MT was decomposed and the Cd was present in ionic form. In contrast to this the SOD was not affected by the sonication. Recoveries throughout are near 100%.
 | ||
| Fig. 4 Ultrasonic exposure of extracts of MT and SOD with 30 W or 50 W ultrasonic power and 30 s or 60 s exposure time (experimental conditions are given in Table 1). | ||
Finally the influence of the atmospheric conditions on the stability of MT and SOD were investigated in this first step. An oxidation of MT during isolation in air in the time scale of days was described by Minkel et al.29 but this purification procedure involved other problematic steps like heat treatment and dialysis over several hours. In the present study the samples were handled in a glove bag under an argon atmosphere during extraction as a precaution against oxidation. For an estimation of the effect of oxygen on the stability of the model species two extracts were prepared at the air and another two were additionally purged with oxygen for 1 min. In the air no relevant losses of the species occurred (recoveries: 89 ± 11% for Cd7MT-2d, 94 ± 19% for Cd7MT-2e and 93 ± 16% for CuZnSOD). Purging with oxygen reduced the recovery below 80%, but even in this worst case experiment more than 70% of the metal ions remained bound to the protein (79 ± 11% for Cd7MT-2d, 74 ± 18% for Cd7MT-2e and 71 ± 16% for CuZnSOD).
Step 2: Extraction of spiked liver samples
In the first step the species stability was investigated in a simplified system containing only the model species and the extractants, but in real samples the complex liver matrix is present and a lot of stabilizing or destabilizing interactions between the model species and matrix compounds are possible. So liver samples were spiked with a MT/SOD preparation to produce samples with a defined amount of the model species as a basis for the comparison of different extraction procedures. An additional Cd-MT peak was detected in the extracts of these spiked samples compared to the extracts without liver matrix although there were no relevant contents of Cd–MT in the unspiked liver. Probably a redistribution of Cd between the spiked rabbit liver Cd–MT and the native porcine liver Zn–MT took place. The use of spiked extracts from unspiked liver samples as calibration standards compensated this effect. The stability of metal–MT complexes increases in the order Zn < Cd < Cu. Therefore, two interactions between the spiked Cd–MT and the native CuZn–MT are possible: First the exchange of Zn from the porcine MT by Cd from the spiked MT as discussed above and second an exchange of Cd from the spiked MT by Cu from porcine species. But this requires easily available Cu in the extract which is not present; otherwise it would have exchanged the less firmly bound Zn from the porcine MT.A selection of six extraction procedures for MT or SOD was taken from the literature and applied to the spiked samples (Fig. 5) to confirm the results from the first step regarding the TRIS buffer. The ammonium formate buffer13 showed the best recoveries and the confidence intervals for the three species included 100%. The MT recovery in the TRIS extract30 is a bit lower; the SOD recovery is lower, too, but this is only due to one of the replicates. SOD recovery with TRIS + sucrose10 is the second best, but Cd7MT-2d recovery is below 80%. The phosphate buffer31 is slightly better for the MT and slightly worse for the SOD. The addition of the surface active substance Triton X-100 to the TRIS buffer32 reduces the mean recoveries below 80% for MT and SOD. Completely different is the result of TRIS + EDTA33 which was described for the purification of a SOD. According to this the SOD–Cu recovery is acceptable with nearly 80%. However most of the Cd from the MT was bound by EDTA and eluted near the void volume of the reversed phase column. Zn was totally removed from the native MT while Cu was released only to a small degree. Obviously Zn and Cd are complexed more firmly by EDTA than by MT so this extractant is not suitable for MT extraction under preservation of the native metal content.
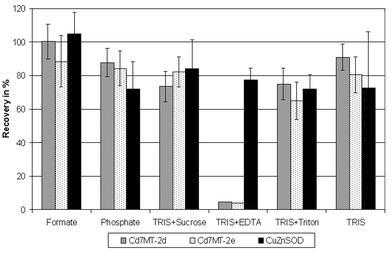 | ||
| Fig. 5 Comparison of six methods for the extraction of liver samples spiked with MT and SOD (experimental conditions are given in Table 2). | ||
In analogy to the first step the TRIS buffer concentration was varied to confirm the results in the presence of the liver matrix. The mean recovery of the MT isoforms is nearly constant for all buffer concentrations at 83% to 91% for Cd7MT-2d and 77% to 87% for Cd7MT-2e. The losses of about 15% in comparison to the extraction without liver matrix (step 1) are probably mainly due to incomplete recovery from the liver matrix and from the homogenizing equipment. SOD recoveries are fluctuating between 70% and 109% without significant differences.
Another interesting parameter is the extractant-to-liver ratio i.e. the extractant volume in mL per g of fresh liver. The recovery of the Cd7MT-2d is presented in Fig. 6 (upper curve). A maximum is reached for an extractant-to-liver-ratio of 3 although not significantly. Similar results were obtained for Cd7MT-2e and CuZnSOD. Another study came to the same conclusion for MT.26 The lower curve in Fig. 6 represents the contents of one native Zn-MT. It was quantified via the Zn content of the peak in the reversed phase HPLC. Both curves are quite similar. This shows that the spiked Cd-MT and the native Zn-MT behave similarly in the homogenized sample. So the use of spiked liver samples is suitable for the optimisation and comparison of different extraction procedures.
 | ||
| Fig. 6 Optimisation of the extractant-to-liver ratio for the extraction of spiked liver samples on the basis of the recovery of spiked Cd-MT as well as the contents of native Zn-MT (experimental conditions are given in Table 2; Zn-MT-contents refer to the liver fresh weight). | ||
Metal balance for the extraction of spiked liver samples
An important aspect for the validation of extraction procedures for trace element species is the metal balance. The total contents of Cd, Cu and Zn in unspiked liver samples, spike solution, liver extracts and extraction residues were determined by ICP-AES. The results of the formate buffer are listed as an example in Table 3. Good balances were found for Cu and Zn with less than 15% loss of the reconstituted initial contents of the spiked liver sample. The native contents of both metals were far higher than the spiked contents and relevant amounts remained in the extraction residue. The gap in the Cd balance is a bit bigger but still below 20%. In contrast to Cu and Zn the native Cd content was very low and nearly the total initially present Cd was introduced with the spike solution. Accordingly the Cd content of the extraction residue was comparatively low, but after all 10% of spiked Cd remained in the solid residue as a result of the interaction between the spike solution and the liver matrix. Extracted metal contents are more easily lost than residually bound amounts through adsorption to tube walls or to the homogeniser. Moreover the extracts were filtered with a membrane filter (0.45 μm) before SEC separation and total metal analysis which might account for some metal losses in the balance, too.| Cd | Cu | Zn | |
|---|---|---|---|
| a Mean of ten samples from the same part of the liver as the samples for the extraction.b 100 μg Sigma MT-I and 100 μg SOD per extraction experiment.c After filtration with a membrane filter (0.45 μm).d Sum of unspiked liver and spiked amount.e Percentile ratio of [extract + residue] to spiked liver sample. | |||
| Unspiked livera | (0.045 ± 0.004) μg g−1 | (25.28 ± 0.90) μg g−1 | (73.66 ± 1.70) μg g−1 |
| Spiked amountb | (5.423 ± 0.314) μg g−1 | (0.16 ± 0.01) μg g−1 | (1.06 ± 0.06) μg g−1 |
| Extractc | (3.906 ± 0.332) μg g−1 | (8.98 ± 0.51) μg g−1 | (36.26 ± 1.39) μg g−1 |
| Extraction residue | (0.538 ± 0.006) μg g−1 | (14.90 ± 0.39) μg g−1 | (28.49 ± 0.59) μg g−1 |
| Spiked liverd | (5.468 ± 0.314) μg g−1 | (25.43 ± 0.90) μg g−1 | (74.71 ± 1.70) μg g−1 |
| Extract + residue | (4.444 ± 0.332) μg g−1 | (23.87 ± 0.64) μg g−1 | (64.75 ± 1.51) μg g−1 |
| Recoverye | 81.3 ± 0.3% | 93.9 ± 0.6% | 86.7 ± 1.5% |
Conclusion
The effect of the relevant extraction parameters on the stability of the model species MT and SOD was investigated in a simplified system without the liver matrix as well as with spiked liver samples. The extraction of the species alone (step 1) showed that in particular the choice of the extractant, the pH and the exposure towards ultrasonic energy have a major influence on the stability of the model species. Temperature, buffer concentration and exposure to air were less critical.The optimised conditions from this first step were 10 mM TRIS–HCl, pH 7.5, 4 °C, argon atmosphere and no sonication. The buffer concentration was set to the lowest value to avoid unnecessary amounts of buffer salts in the extracts. Cooling and inert atmosphere were chosen as precautions especially in respect to a later application where further may be less stable species shall be characterized. The pH was kept near the native liver pH.
The extraction of spiked liver samples confirmed the insignificant effect of the buffer concentration. A new aspect was the extractant-to-liver ratio whose optimum was around 3. The comparison of several methods from the literature resulted in an overall good performance except for the EDTA method in respect to MT. The ammonium formate procedure, the TRIS–HCl-method and the TRIS–sucrose combination were selected for further experiments in order to use two different buffers as well as the osmotic effect of the sucrose.
Although the samples had passed a complex preparation process the losses in the metal balance are acceptable for Cd and even low for Cu and Zn. The extraction yields of Cu and Zn from the residues will probably rise if the extraction process is repeated with the same method applied to the residue of the first extraction.
The presented experiments contribute to the demand of Nordberg7 towards a harmonisation of extraction methods for MT. Additionally SOD was investigated in the same way which is a first approach into the direction of a universal extraction method for trace element species from liver samples.
Acknowledgement
The author thanks Prof. Dr P. Schramel for support and Mr P. Grill for performing the ICP-AES-measurements.References
- B. Michalke, Trends Anal. Chem., 2002, 21, 154–165 CrossRef CAS.
- J. Szpunar, Trends Anal. Chem., 2000, 19, 127–137 CrossRef CAS.
- F. Vanhaecke and L. Moens, Fresenius’ J. Anal. Chem., 1999, 364, 440–451 CrossRef CAS.
- F. Adams and S. Slaets, Trends Anal. Chem., 2000, 19, 80–85 CrossRef CAS.
- P. Quevauviller, J. Chromatogr., A, 1996, 750, 25–33 CrossRef CAS.
- R. Morabito, Fresenius’ J. Anal. Chem., 1995, 351, 378–385 CrossRef CAS.
- M. Nordberg, Talanta, 1998, 46, 243–254 CrossRef CAS.
- C. B. Knudsen, I. Bjornsdottir, O. Jons and S. H. Hansen, Anal. Biochem., 1998, 265, 167–175 CrossRef CAS.
- R. W. Briggs and I. M. Armitage, J. Biol. Chem., 1982, 257, 1259–1262 CAS.
- D. Klein, J. Lichtmannegger, U. Heinzmann and K. H. Summer, J. Hepatol., 2000, 32, 193–201 CrossRef CAS.
- A. K. Vogiatzis and N. S. Loumbourdis, Arch. Environ. Contam. Toxicol., 1998, 34, 64–68 CrossRef CAS.
- J. H. Beattie and M. P. Richards, J. Chromatogr., A, 1994, 664, 129–134 CrossRef CAS.
- J. S. Garvey, R. J. Vander Mallie and C. C. Chang, Methods Enzymol., 1982, 84, 121–138 Search PubMed.
- V. Nischwitz, B. Michalke and A. Kettrup, Anal. Bioanal. Chem., Search PubMed in the press.
- W. P. Michalski, J. Chromatogr., B: Biomed. Appl., 1996, 684, 59–75 Search PubMed.
- C. C. Conrad, D. T. Grabowski, C. A. Walter, M. Sabia and A. Richardson, Free Radical Biol. Med., 2000, 28, 447–462 CrossRef CAS.
- H. Van Beek and A. J. Baars, At. Spectrosc., 1990, 11, 70–74 Search PubMed.
- M. Nakano, Methods Enzymol., 1990, 186, 227–232 Search PubMed.
- M. L. Salin and W. W. Wilson, Mol. Cell. Biochem., 1981, 36, 157–161 CAS.
- P. Schramel, A. Wolf, R. Seif and B. J. Klose, Fresenius’ Z. Anal. Chem., 1980, 302, 62–64 CAS.
- SWISS PROT protein sequence database (http://us.expasy.org/sprot)..
- C. B. Knudsen and J. H. Beattie, J. Chromatogr. A, 1997, 792, , 463–473 CrossRef CAS.
- S. Klausner, J. H. R. Kägi and K. J. Wilson, Biochem. J., 1983, 209, 71–80.
- P. E. Hunziker and J. H. R. Kägi, Biochem. J., 1985, 231, 375–382 CAS.
- C. Wolf, U. Rösick and P. Brätter, Fresenius’ J. Anal. Chem., 2000, 368, 839–843 CrossRef CAS.
- K. Nöstelbacher, Untersuchungen zur Bestimmung von Metallothionein-Isoformen in Rattenlebern, sowie deren Induzierbarkeit bei nutritivem oxidativem Streß, Herbert Utz Verlag, München, 1999, p. 8 and pp. 51–52 Search PubMed.
- J. H. R. Kägi and Y. Kojima, Experientia Supplementum 52, Metallothionein II, Birkhäuser, Basel, 1987, p. 39 Search PubMed.
- J. L. Boyer, J. Graf and P. J. Meier, Annu. Rev. Physiol., 1992, 54, 415–438 CrossRef CAS.
- D. T. Minkel, K. Poulsen, S. Wielgus, C. F. Shaw and D. H. Petering, Biochem. J., 1980, 191, 475–485 CAS.
- D. L. Eaton and B. F. Toal, Toxicol. Appl. Pharmacol., 1982, 66, 134–142 CrossRef CAS.
- J. R. Riordan and V. Richards, J. Biol. Chem., 1980, 255, 5380–5383 CAS.
- J. V. Bannister and W. H. Bannister, Methods Enzymol., 1984, 105, 88–93 Search PubMed.
- B. L. Geller and D. R. Winge, Methods Enzymol., 1984, 105, 105–114 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |