
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface barriers to mass transfer in nanoporous materials for catalysis and separations
Shuman
Xu
 ab,
Ke
Zheng
be,
Cristian-Renato
Boruntea
b,
Dang-guo
Cheng
*ac,
Fengqiu
Chen
ac,
Guanghua
Ye
d,
Xinggui
Zhou
d and
Marc-Olivier
Coppens
*b
ab,
Ke
Zheng
be,
Cristian-Renato
Boruntea
b,
Dang-guo
Cheng
*ac,
Fengqiu
Chen
ac,
Guanghua
Ye
d,
Xinggui
Zhou
d and
Marc-Olivier
Coppens
*b
aCollege of Chemical and Biological Engineering, Zhejiang Provincial Key Laboratory of Advanced Chemical Engineering Manufacture Technology, Zhejiang University, Hangzhou 310027, China. E-mail: dgcheng@zju.edu.cn
bEPSRC ‘‘Frontier Engineering’’ Centre for Nature Inspired Engineering & Department of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: m.coppens@ucl.ac.uk
cInstitute of Zhejiang University-Quzhou, Quzhou 324000, China
dState Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China
eBeijing Key Laboratory of Coal to Cleaning Liquid Fuels, National Energy R & D Center for Coal to Liquid Fuels, Synfuels China Co., Ltd, Beijing 101400, China
First published on 6th June 2023
Abstract
Surface barriers to mass transfer in various nanoporous materials have been increasingly identified. These past few years especially, a significant impact on catalysis and separations has come to light. Broadly speaking, there are two types of barriers: internal barriers, which affect intraparticle diffusion, and external barriers, which determine the uptake and release rates of molecules into and out of the material. Here, we review the literature on surface barriers to mass transfer in nanoporous materials and describe how the existence and influence of surface barriers has been characterized, aided by molecular simulations and experimental measurements. As this is a complex, evolving research topic, without consensus from the scientific community at the time of writing, we present various current viewpoints, not always in agreement, on the origin, nature, and function of such barriers in catalysis and separation. We also emphasize the need for considering all the elementary steps of the mass transfer process in optimally designing new nanoporous and hierarchically structured adsorbents and catalysts.
 Shuman Xu | Shuman Xu is a PhD candidate in the College of Chemical and Biological Engineering at Zhejiang University (ZJU). She was a visiting student at University College London (UCL) in 2019–2021. She was supervised by Professor Fengqiu Chen and Professor Dang-guo Cheng at ZJU and co-supervised by Professor Marc-Olivier Coppens at UCL. Her current research interest is the mass transfer process and its influence on catalytic performance in hierarchical zeolites. |
 Ke Zheng | Ke Zheng received his PhD (2019) degree in Chemical Engineering from the University of Chinese Academy of Sciences (UCAS), China. After postdoctoral training in Centre for Nature Inspired Engineering & Department of Chemical Engineering at the University College London (UCL), he joined Synfuels China Technology Co. Ltd. as a senior engineer in 2021. His major research interest is devoted to the development of modeling thermodynamic properties of fluids with equations of state and molecular simulation for various applications. |
 Cristian-Renato Boruntea | Cristian-Renato Boruntea is a postdoctoral researcher at the University of Antwerp. He received his PhD in 2020 from the Polytechnic University of Valencia (ITQ). After working nearly 3 years in the industry contributing to the low-carbon energy transition he was a postdoctoral researcher at the Centre for Nature Inspired Engineering at the University College London (UCL). His interests are focused on novel catalysts and engineering of porous materials for targeted applications, heterogeneous catalysis and renewable energy and chemicals production. |
 Dang-guo Cheng | Dang-guo Cheng is a professor in the College of Chemical and Biological Engineering at Zhejiang University. He is a recipient of the National Natural Science Foundation of China (NSFC) Outstanding Youth Fund and Zhejiang Province Excellent Youth Fund. He received his PhD from Tianjin University and joined Zhejiang University in 2006. He worked as a visiting scholar at the University of Illinois at Urbana-Champaign (UIUC) in 2014 and 2015. His research projects mainly focus on designing and preparing new catalytic materials for fossil energy conversion and catalytic reaction mechanisms. |
 Fengqiu Chen | Fengqiu Chen is a professor at the College of Chemical and Biological Engineering at Zhejiang University since 2001. He received his PhD in 1992 and has been teaching at ZJU since then. He worked as a visiting researcher at the University of Tokyo in 1998 and Karlsruhe University in 2003. As a leading researcher engaged in multiphase flow catalytic reaction engineering, he has published over 200 papers and 60 patents on catalyst engineering, catalytic reaction mechanism and kinetics, supercritical reaction engineering, gas–solid fluidization, etc. He was awarded the third prize for national technical invention in China. |
 Guanghua Ye | Guanghua Ye received his PhD (2016) degree in chemical engineering from the East China University of Science and Technology (ECUST), China. From 2014 to 2015, he was a visiting student at the University College London. During 2016–2018, he was a post-doctoral researcher at the ECUST and spent a short period as a visiting scholar at the Technical University of Hamburg-Harburg. Since 2018, he is an Associate Professor in Chemical Engineering at the ECUST. His research focuses on mass transfer in porous materials, especially heterogeneous catalysts. |
 Xinggui Zhou | Xing-Gui Zhou earned his bachelor's degree in Chemical Engineering in 1987 and his PhD degree in 1996 from East China University of Science and Technology (ECUST). Since joining the faculty of Chemical Engineering in 1996, he has been an active researcher at the State-Key Laboratory of Chemical Engineering (SKLoChE) at ECUST, where he currently serves as the director. As a full professor since 2002, he leads the catalysis and reaction engineering research group at SKLoCh. His research interests include controlled synthesis of solid catalysts, kinetics and mechanism of heterogeneous catalysis, transport in porous catalysts and fixed bed reactors, and process intensification with microfluids. |
 Marc-Olivier Coppens | Marc-Olivier Coppens is Ramsay Memorial Professor in Chemical Engineering at UCL, since 2012, after professorships at Rensselaer and TUDelft. He is also Vice-Dean for Engineering (Interdisciplinarity, Innovation). He directs the Centre for Nature-Inspired Engineering, which was granted EPSRC “Frontier Engineering” (2013) and “Progression”’ (2019) Awards. He is most recognized for pioneering nature-inspired chemical engineering (NICE): learning from fundamental mechanisms underpinning desirable traits in nature to develop innovative solutions to engineering challenges. He is Fellow of RSC, IChemE, AIChE, Corresponding Member of the Saxon Academy of Sciences (Germany), Qiushi Professor at Zhejiang University, and has delivered >50 named lectures, plenaries and keynotes. |
Key learning points1. Origins and nature of external and internal surface barriers.2. Experimental characterization of surface barriers. 3. Molecular simulation methods to investigate surface barriers. 4. Effects of synthesis and surface modification techniques on surface barriers. 5. New perspective on the overall mass transfer process in nanoporous materials. |
1. Introduction
Nanoporous materials are crucial in modern chemical, energy, and environmental technologies. Typical nanoporous materials span zeotype crystals, metal–organic frameworks (MOFs), mesoporous solids, and hierarchically structured materials that combine intrinsic micropores with additional mesopores or macropores.1 Due to their high specific surface area, the unique confinement-induced selectivity and controlled mass transfer within the nanopores, nanoporous materials have been widely applied as heterogeneous catalysts, membranes and adsorbents in critical industrial processes.The optimal design and tuning of processes using nanoporous materials requires fundamental understanding, including the identification of the rate-limiting step in the overall mass transfer. Traditionally, the mass transfer process is divided into external transport from the bulk phase onto the external surface of the solid particles and internal diffusion through the pore channels. For many catalytic and separation processes, the dominance of external transport limitations can be avoided by increasing the flow rate. Consequently, intraparticle diffusion has received major attention as the principal step. A widely applied way to enhance the mass transport efficiency of these materials is to enlarge the nanopore diameter or shorten the nanoporous diffusion pathlength, which has inspired the design of hierarchical nanoporous materials.2–5 However, some researchers found that when the dimensions of nanoporous materials, like zeolites, were decreased to tens of nanometers, the effective diffusivities were not improved as predicted, indicating the existence of some additional mass transport limitations.6–9
Direct experimental measurements and simulation work have further confirmed the effects of surface barriers and, thus, form the foundation for a new picture of the mass transfer process where the role of interfaces cannot be ignored. Some key techniques are summarized in Fig. 1. Measurements of uptake or release rates with the aid of macro- and microscopic methods can be used to identify mass transfer mechanisms in nanoporous materials. The apparent diffusivity represents the effective, overall diffusion behavior of adsorbates in a collection of samples. It is often determined using macroscopic techniques, such as zero-length column chromatography (ZLC),10,11 frequency response (FR),12,13 fast time-resolved infrared spectroscopy (IR),14,15 and intelligence gravimetric analyzer (IGA).16,17 Microscopic measurements, like pulsed field gradient nuclear magnetic resonance (PFG-NMR) and quasi-elastic neutron scattering (QENS), are particularly suitable for measuring intracrystalline transport resistance.18,19 To provide direct evidence of surface barriers, concentration profiles of guest molecules have been recorded using interference microscopy (IFM) and infrared microscopy (IRM) by Kärger and co-workers.20–25 Super-resolution fluorescence microscopy (FM)26,27 and structured illumination microscopy (SIM)28–30 have also been successfully extended to the characterization of mass transfer and heterogeneous catalysis. The adjustable confocal plane enables the observation of local density distributions of guest molecules, which enter and move through a specific zeolite crystal. The influence of surface barriers in nanoporous materials is also increasingly explored in the context of practical applications. Measuring catalytic reaction kinetics and membrane permeability analysis, combined with a precise structural characterization, could indirectly reflect the role of internal or external surface barriers.31,32 Molecular simulation methods to distinguish the surface barriers from intracrystalline diffusion are generally kinetic Monte Carlo (KMC)33 and molecular dynamics (MD) simulations, including dual control volume grand canonical molecular dynamics (DCV-GCMD),34,35 equilibrium molecular dynamics (EMD)36,37 and non-equilibrium molecular dynamics (NEMD).38
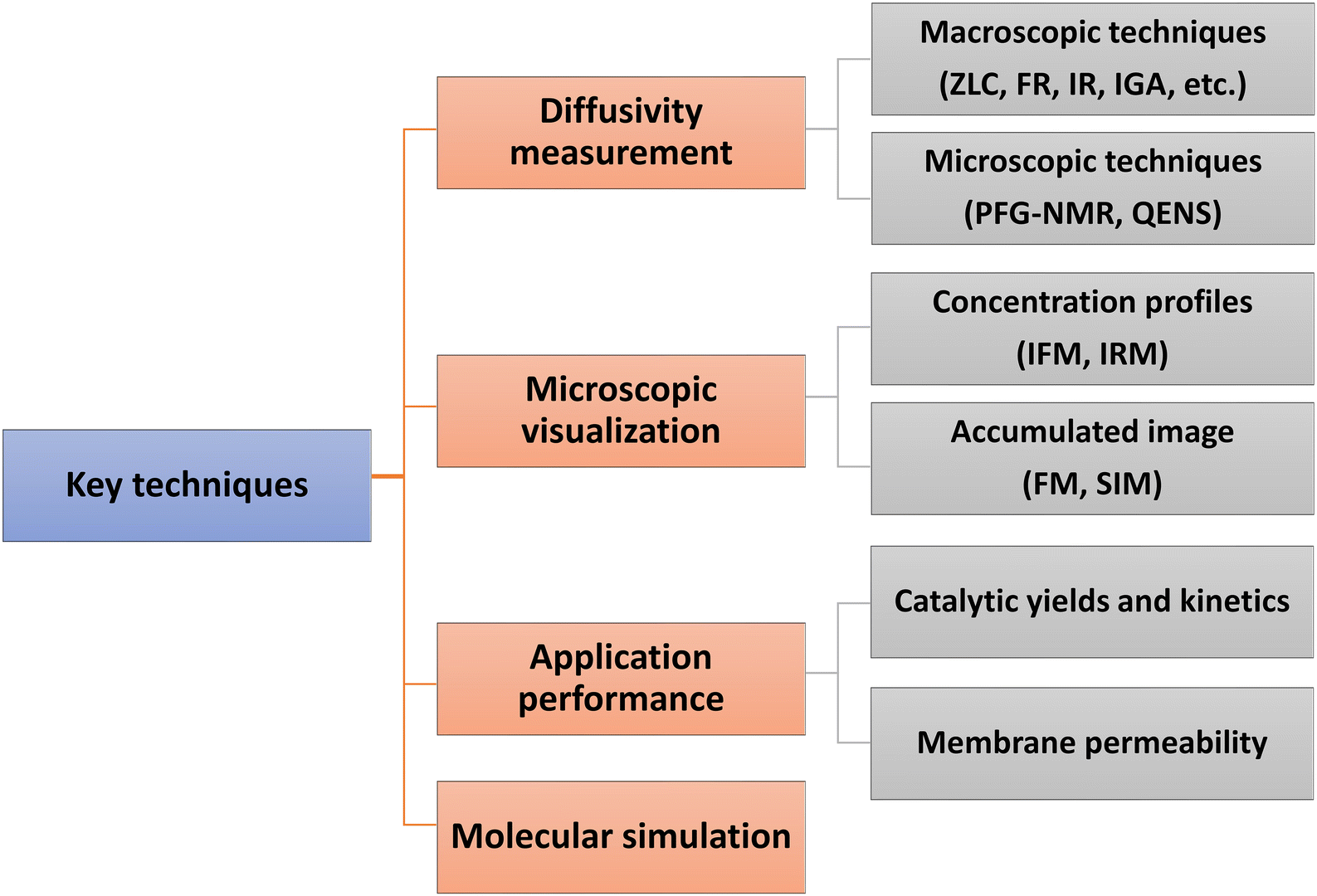 | ||
| Fig. 1 Key techniques used to study surface barriers. | ||
Noting the new insights on the importance of surface barriers, we give a schematic representation (Fig. 2) of the complete transport process in nanoporous materials, including a series of elementary steps: external transport via convection and diffusion in the bulk phase (i), surface diffusion (ii), pore entering (iii), intracrystalline diffusion in pore channels (iv) and across internal structural defects (v), intracrystalline intergrowth interfaces (vi) or intercrystalline grain boundaries (vii).6,12,15 In this overall picture, nanoporous diffusion is no longer the one and only determining step, and one could distinguish two classes of mass transport barriers. The first one consists of internal barriers, which disturb intraparticle mass transport. The second one is external barriers at the external surface and pore mouths.
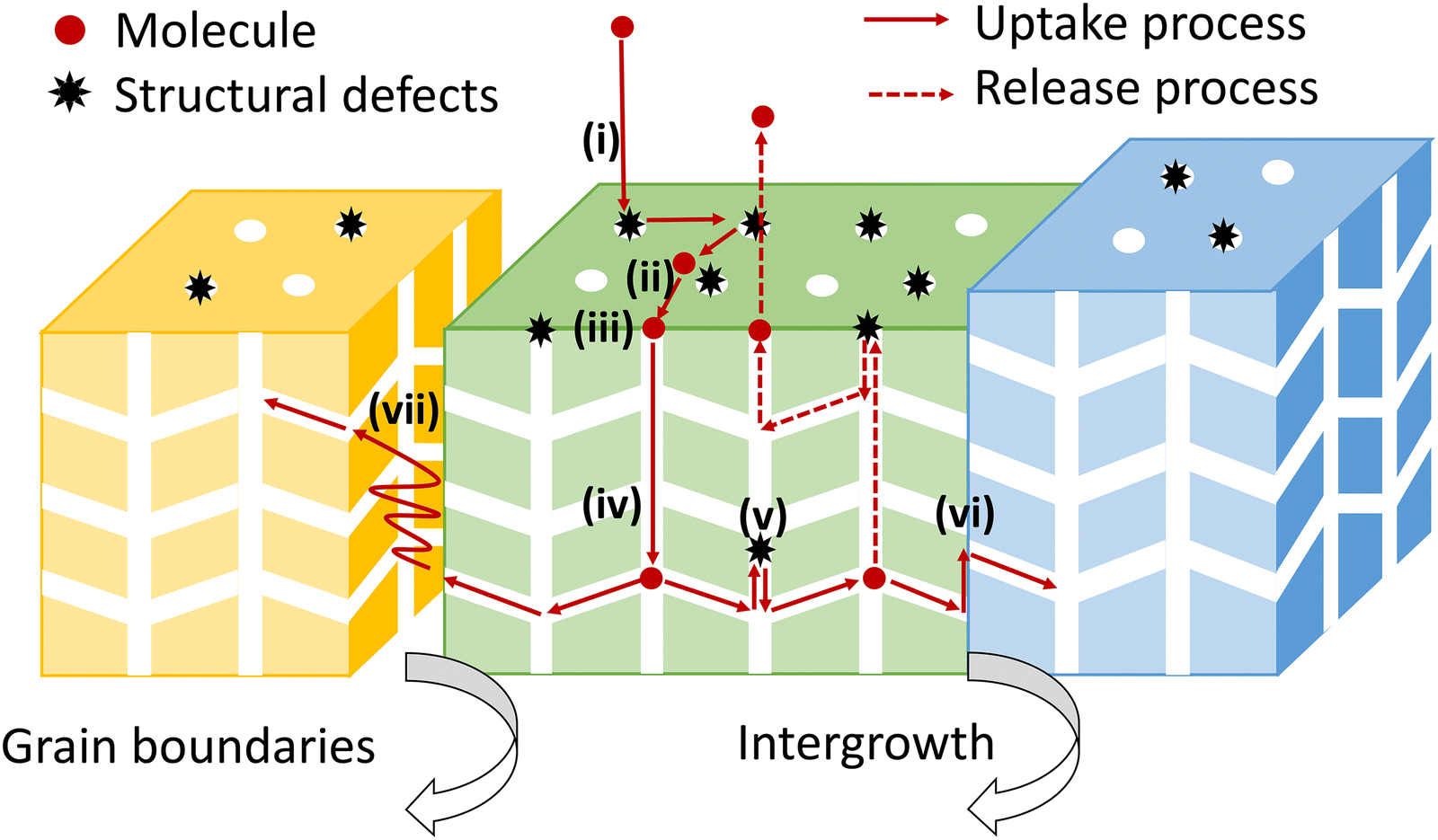 | ||
| Fig. 2 The complete transport process from the bulk phase into the interior of nanoporous materials. | ||
This review is organized in three sections, one for each specific type of surface barrier (Fig. 3). First, we summarize the experimental evidence of surface barriers. Second, we review the molecular simulation work on surface barriers. Finally, various viewpoints on the origins, nature, and functions of the surface barriers are discussed in detail. With the current knowledge of the surface barriers to mass transport, we provide a new perspective for the optimal design strategy of nanoporous materials. We also point out the research gap to unveil the nature of surface barriers at the atomistic level.
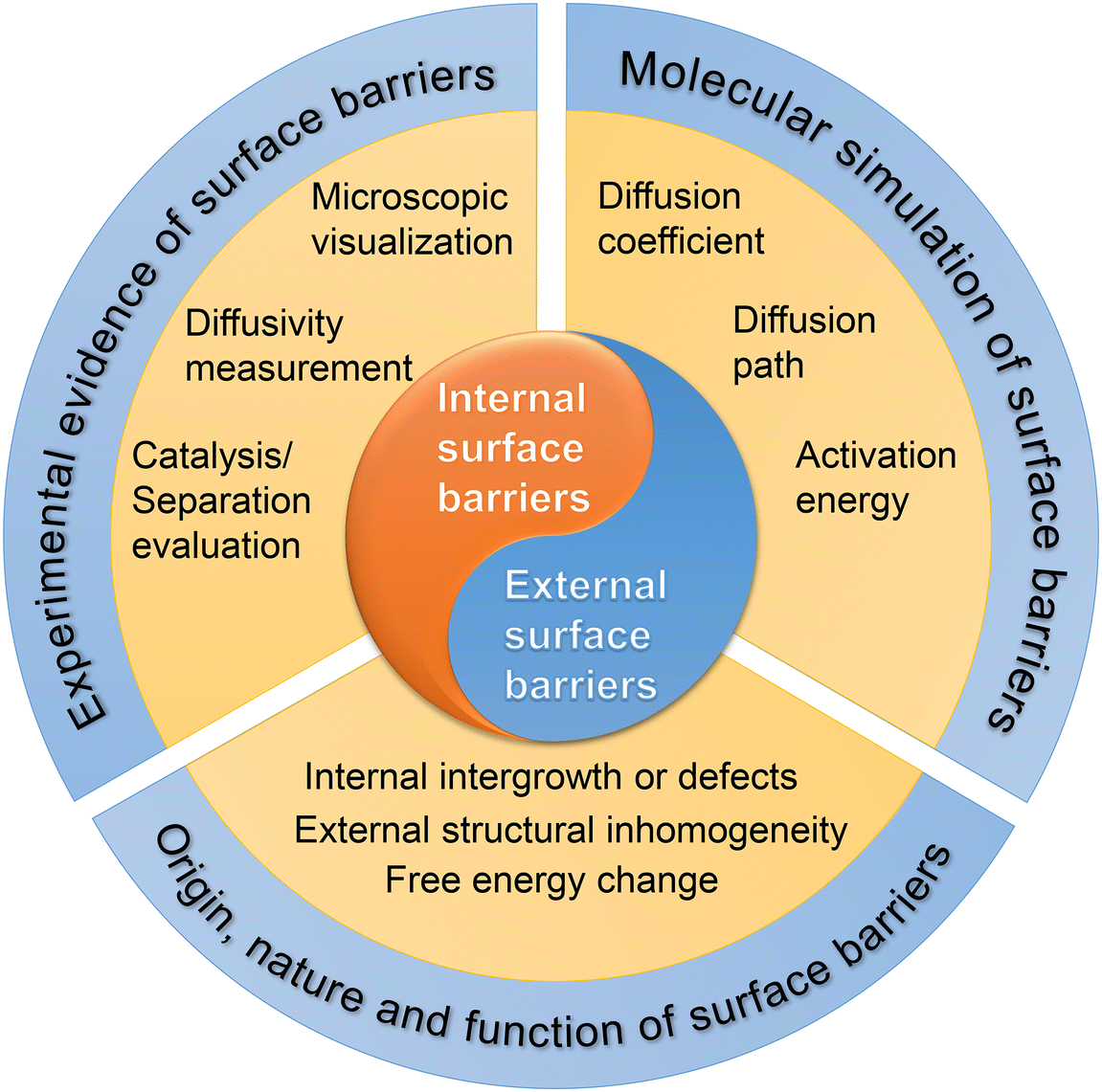 | ||
| Fig. 3 The scope of this review, including classification, research methods, and theoretical analysis of surface barriers. | ||
2. Internal surface barriers
As depicted in Fig. 4(A), internal surface barriers could be intra- or intercrystalline. Intergrowth interfaces or defects within single crystals contribute to internal surface barriers, while grain boundaries between aggregated nanocrystals lead to intercrystalline surface barriers in polycrystalline materials.39–41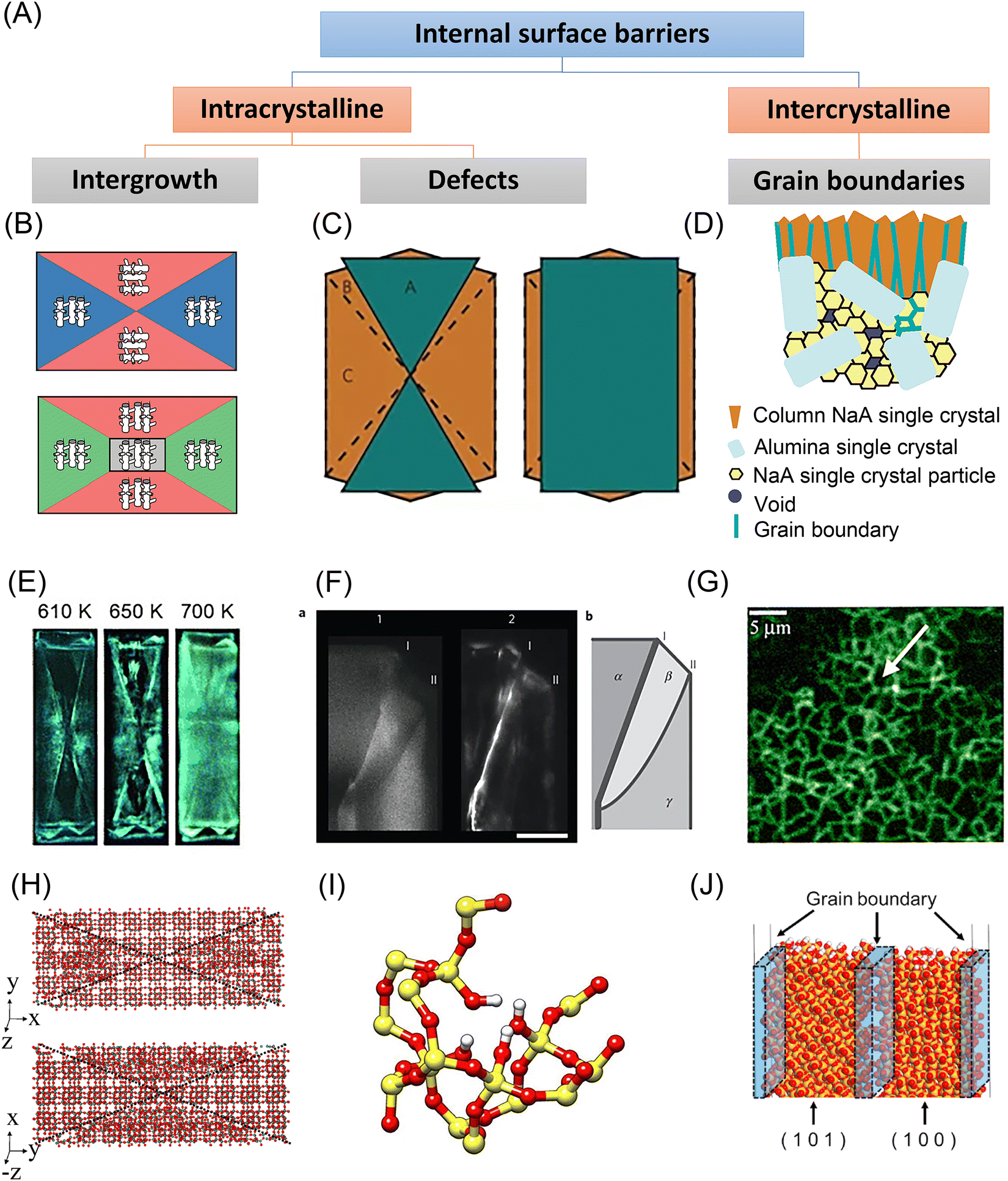 | ||
| Fig. 4 Experimental and simulation studies on the internal surface barriers in nanoporous materials. (A) Classification of internal surface barriers based on their origin and nature. (B) Scheme of MFI zeolite crystallography. Reprinted with permission from ref. 64. Copyright 2008 American Chemical Society. (C) Scheme of an MFI zeolite crystal with two intergrown components. Reprinted with permission from ref. 40. Copyright 2009 Springer Nature. (D) Scheme of NaA zeolite membrane on alumina substrate. Reprinted with permission from ref. 72. Copyright 2006 American Chemical Society. (E) Fluorescence microphotographs of ZSM-5 crystals taken during template removal. Reprinted with permission from ref. 39. Copyright 2007 John Wiley & Sons. (F) Left: Confocal fluorescence images of an MFI crystal corner. Right: Schematic showing the different subunits (α, β, and γ), along with the locations of the distinct diffusion barriers (I and II). Reprinted with permission from ref. 40. Copyright 2009 Springer Nature. (G) Fluorescence confocal optical microscopy (FCOM) images of grain boundaries in an MFI zeolite membrane. Reprinted with permission from ref. 73. Copyright 2001 Elsevier. (H) Simulation model used for twinned silicalite crystals. Reprinted with permission from ref. 53. Copyright 2006 American Chemical Society. (I) Silanol nests model of the hydrophilic defects induced by Al insertion in the silicalite-1 framework (silicon atoms are yellow; oxygen red; hydrogen white). Reprinted with permission from ref. 57. Copyright 2016 Springer Nature. (J) Atomistic models containing a grain boundary region in all-silica chabazite (CHA) zeolite membranes. Reprinted with permission from ref. 56. Copyright 2021 Elsevier. | ||
2.1 Experimental characterization
Since the 1990s, researchers have focused on developing new microscopic techniques that allow in situ scanning over one selected zeolite crystal and monitoring the concentration profiles of sorbate molecules during the mass transfer process. These experiments noted unusual mass transfer behavior, which could only be explained when taking internal surface barriers into account. Geus et al.42 used light microscopy and infrared microscopy to study the removal of microporous template tetrapropylammonium (TPA) from micro-sized single crystals of silicalite-1 zeolite. The hourglass pattern within the crystal was attributed to the hindered diffusion of water from the internal crystal sections during calcination. A similar internal structure was observed through fluorescence microscopy, see Fig. 4(E).39 Another category of evidence of internal surface barriers appeared when measuring the diffusivities of guest molecules. Vasenkov et al.43 applied PFG-NMR to measure the apparent intracrystalline diffusivity of alkanes in MFI-type zeolite crystals. As shown in Fig. 5(A), the measured, apparent diffusivities of methane at low temperatures gradually declined as a function of the root mean square displacement (RMSD). This change was ascribed to the role of internal surface barriers. Other possible explanations, like a broad distribution of diffusivities or restricted diffusion at external surfaces, were carefully ruled out. The dependence of the apparent diffusivity on diffusional length scales was further verified in various hydrocarbon-zeolite systems.19,44,45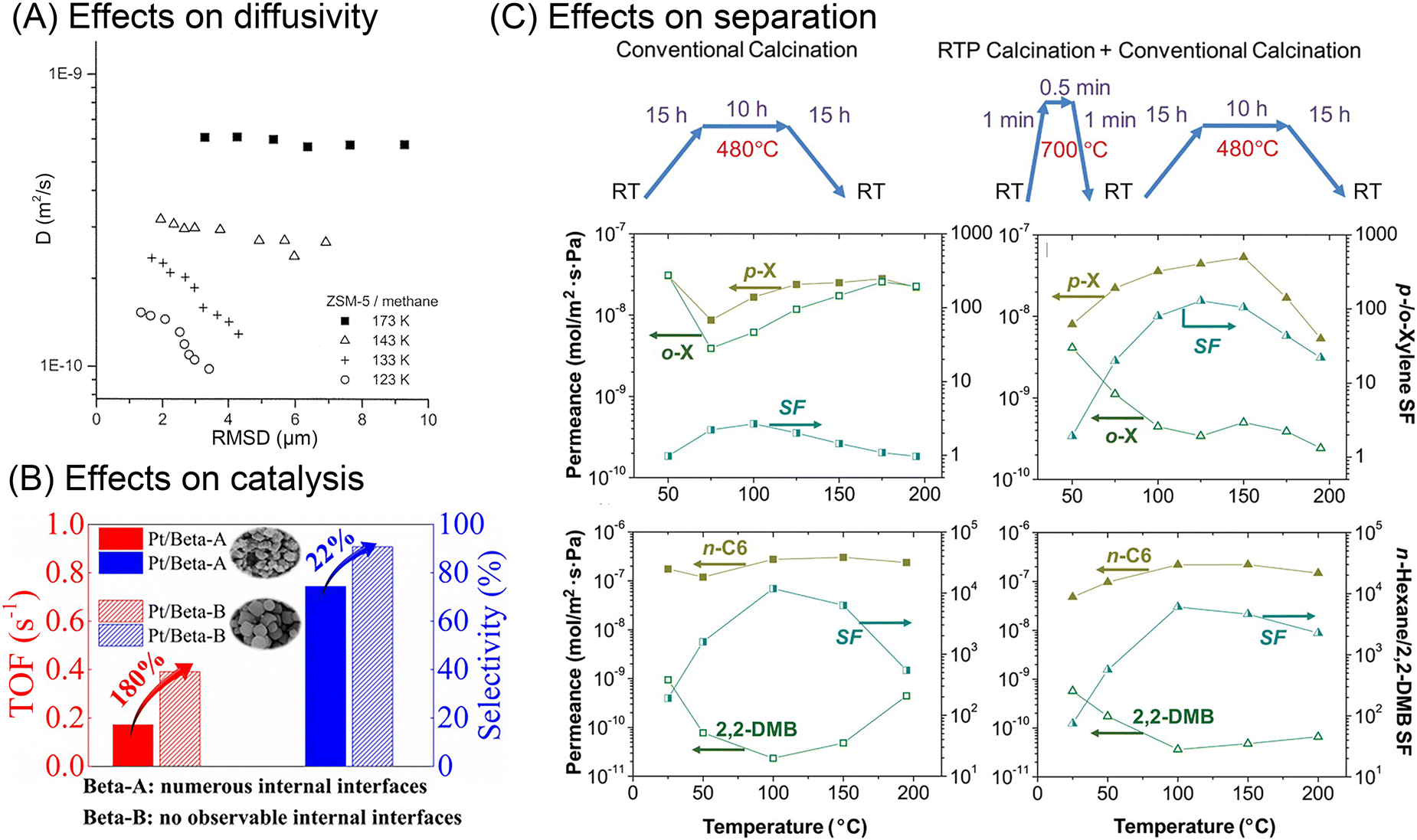 | ||
| Fig. 5 Effects of internal surface barriers on apparent diffusivity, catalysis, and separation performance. (A) Relationship between apparent diffusivities and root mean square displacements (RMSD) of methane in ZSM-5 zeolite at various temperatures. Reprinted with permission from ref. 43. Copyright 2001 American Chemical Society. (B) The effects of internal diffusion barriers in n-heptane isomerization catalyzed by Pt/Beta zeolite. Reprinted with permission from ref. 47. Copyright 2020 John Wiley & Sons. (C) Xylene and hexane isomers separation performance of MFI zeolite membranes treated through different thermal processes. Reprinted with permission from ref. 32. Copyright 2009 American Association for the Advancement of Science. | ||
All these experiments show the influence of internal surface barriers in large zeolites, micrometers in size. However, applying smaller particles with submicron and nanoscale dimensions is more common in actual industrial processes. Ye et al.46 and Guo et al.47 synthesized two types of Pt/Beta catalysts, based on either polycrystalline or monocrystalline submicron zeolite crystals. As shown in Fig. 5(B), the existence of internal grain boundaries has a negative effect on the mass transport and catalyzed isomerization of n-heptane. Despite limited reports on catalytic performance, lots of studies have focused on how grain boundaries affect membrane performance. The selectivity of nanoporous membranes relies typically on molecular sieving. Nonetheless, it is frequently discovered that the presence of grain boundaries reduces or at least modifies the selectivity by generating nonselective transport paths for guest molecules. For example, MFI zeolite membranes with intercrystalline grain boundaries showed extremely small separation factors (<5) for binary xylene isomers. In comparison, another type of MFI membrane with sealed cracks showed higher separation factors, even up to 300.48 Several researchers report that grain boundaries can be selectively repaired or plugged in order to improve membrane performance.32,49,50 Choi et al.32 thus developed a rapid thermal processing (RTP) method to heal the defects. As shown in Fig. 5(C), compared with the conventional calcined membranes, RTP treatment led to a significantly improved separation factor for xylene isomers. However, the separation performance showed no obvious change for hexane isomer mixtures. Therefore, the influence of internal grain boundaries on the membrane performance depends heavily on the properties of the guest molecules.
2.2 Molecular simulations
From the dependence of the experimental diffusivity on the temperature,43 the mass transfer process across the internal surface barrier is observed to be energetically activated. Vasenkov and Kärger51 thus applied Monte Carlo simulations based on transition state theory (TST) to study alkane diffusion in cubic lattices with intracrystalline transport barriers. The simulated results, including RMSD dependence and activation energies, were consistent with their previous experimental findings.43In recent decades, simulations have progressed to more precise atomistic models thanks to growing computational resources, methodological advancements, and more accurate parameters to characterize atomic interactions.1,52 Newsome and Sholl53 constructed an atomistic model to mimic the internal grain boundaries in twinned silicalite zeolites, see Fig. 4(H). Based on this model, they applied DCV-GCMD to simulate the mass transfer of CH4 and CF4 molecules. The results revealed that the interfacial resistances become more dominant for molecules of bulky size and strong adsorption strength, and at low temperatures.
Thomas et al.54 also used MD simulations to explore the role of intracrystalline space for xenon diffusion in MFI zeolites. The significant energy minima in the interfacial region function as a bottleneck at low temperatures, as evidenced by the calculated density distribution and diffusivities. With increasing temperature, molecules can more easily overcome the energy barrier, increasing transport across the intercrystalline region. Similarly, Takaba et al.55 showed that the intercrystalline area reduces the methane diffusivity in MFI-type zeolite membranes by an order of magnitude.
According to NEMD simulations by Hirosawa et al.,56 the grain boundary inside an all-silica chabazite membrane (see Fig. 4(J)) could improve the selectivity and permeability for a CO2/CH4 gas mixture. It is also noteworthy that the grain boundary orientation affected the separation performance. The molecular sieving action was enhanced when the grain boundary was parallel to the bulk phase. In comparison, the membrane showed higher CO2 permeability but moderate selectivity when the grain boundary extended to the membrane surface. These results again show that the specific influence of internal surface barriers is complex and should be applied with particular caution when transplanting between different systems. The internal structural defects in the membrane were also simulated for aluminum site-induced barriers. Fasano et al.57 examined how defect concentration and silanol hydrophilicity affected effective diffusivities. As shown in Fig. 4(I), The increase in pore hydrophilicity caused by Al atom replacement is mimicked by the introduction of silanol nests in the MFI structure.57,58 The simulated self-diffusivities and experimentally measured corrected diffusivity show similar trends in that the diffusivity for low amounts of water decreases with higher defect density.57,59
Although these simulations help consider the physical origin of internal surface barriers, the discrepancies between the simulation model and a genuine zeolite crystal are still noticeable. Due to computational limitations, the simulated crystal sizes are restricted to a few nanometers in length. However, experimentally observed grain boundaries in actual zeolite crystals usually appear over micrometer scales. The current efforts have also been limited by the lack of experimental data on the atomic-scale structure of the interfaces and structure defects. Future analyses of the internal surface barriers could be improved by more precise atomic-scale depictions.
2.3 Origin, nature and function
There are various kinds of inhomogeneities in nanoporous materials that lead to internal surface barriers. One example is intergrowth structures, wherein guest molecules may encounter diffusion barriers between the basic crystal building blocks. Early electron microscopic studies of MFI-type zeolite crystals indicated that adjacent crystals showed a 90° rotation in crystallographic orientation around the c-axis.60,61 Subsequent research studied the interior region of large single crystals by optical microscopy with polarized light and always revealed an hourglass pattern inside MFI-type zeolites.42,62 Based on such observations, the interior structure of MFI crystals was traditionally simplified to a two-component model with a 90° rotation inside. Notably, this model suggests that the straight pathways are hardly accessible from the external surface. However, this speculation did not apply to the observation of a silicalite-1 crystal by atomic force microscopy (AFM) that elevated terraces appeared on both the (010) and the (100) faces but with different step heights.63 In rationalizing these observations, a three-component model containing three sets of components with varying defect concentrations was proposed, as shown in Fig. 4(B).64The literature also extensively discusses irregular stacking faults as an origin for internal surface barriers. Zeolite beta was found to be an intergrown hybrid of two different but closely similar structures, showing considerable stacking disorder.65 Stacking faults were also noted as a potential cause of internal diffusion barriers in zeolite X. Mirror twins were discovered on the (111) plane of FAU-type zeolite by high-resolution transmission electron microscope (HRTEM), which was later validated by diffraction pattern analysis.19 For specific MFI-type zeolite crystal morphologies, Karwacki et al.40 used a powerful combination of various microscopic and spectroscopic techniques to show that internal surface barriers originate from small-angle differences of 0.5–2° in addition to a 90° mismatch in pore alignment, as shown in Fig. 4(C) and (F).
In recent years, various new zeolites have been obtained as intergrowths that share a similar periodic building unit but are connected in different ways.66,67 New microscopic techniques with an increased resolution, like annular dark-field scanning transmission electron microscopy (ADF-STEM), assisted by selected area electron diffraction (SAED), allow for the differentiation of the boundaries of each domain.68 In view of these new findings, the international zeolite association (IZA) has recently updated its database for zeolite frameworks with intergrowth structures, moving them to a separate classification, while discontinuing the previous distinguishing method with an asterisk before the three-letter code of the zeolite material. The assembly of nanosheets with intergrowth domains, such as MFI/MEL and FAU/EMT families, results in intercrystalline porosity, thus serving as a new route to synthesize hierarchically structured zeolites.69–71 However, the relevant reports barely examined the potential existence of internal surface barriers.
In addition to the intergrowth interfaces in single crystals, grain boundaries and cracks in polycrystalline nanoporous materials may also act as internal transport barriers, especially in zeolite membranes. When synthesizing polycrystalline zeolite membranes, zeolite seed crystals jointly grow on or within a porous support, inevitably resulting in the formation of cracks, as seen in Fig. 4(D).72 The microscopy image shows that, when the structure-directing agent is removed, sudden zeolite unit cell contraction and an imbalance in the thermal expansion coefficients of the substrate and the zeolite film result in tensile tensions that cause these defects to develop. Bonilla et al.73 applied fluorescence confocal optical microscopy (FCOM) to observe the grain boundary network in MFI membranes, see Fig. 4(G). Hong et al.74 further applied FCOM to quantitatively assess the impact of grain boundaries in MFI zeolite membranes. They discovered that the grain boundary defects had diameters of around 1–2 nm, and the crack defects had sizes of 7–8 nm. Even though the presence of cracks was minimal (less than 1%), they were responsible for 52–58% of the total molar flux, which deteriorated the selectivity of xylene isomers. On this basis, the healing of internal defects has been widely explored. The RTP process described earlier could strengthen grain bonding by condensing Si–OH groups, thus reducing grain boundaries in zeolite membranes.32
3. External surface barriers
Apart from the internal intergrowths or defects, external surface barriers could contribute significantly to the overall mass transfer process. Such external surface barriers have been asserted in the literature through diffusional measurements, catalytic investigations, and molecular simulations. They have also been invoked as the original cause for discrepancies in diffusivity values between measurement techniques or different crystals. Still, there is no definite description of the atomistic nature of external surface barriers. Fig. 6(A) summarizes several hypotheses, which are compared hereafter.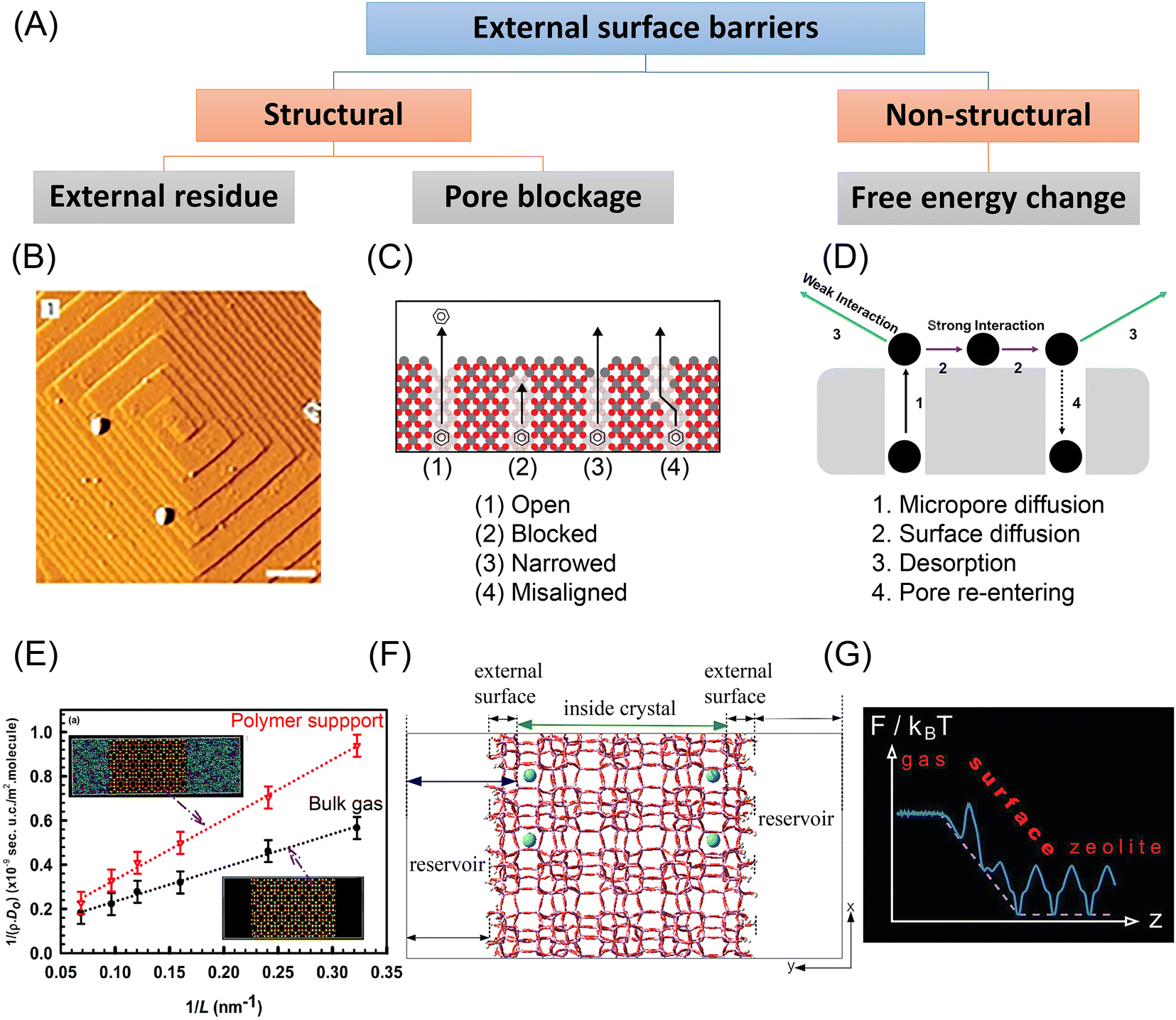 | ||
Fig. 6 Experimental and simulation studies on the external surface barriers in nanoporous materials. (A) Classification of external surface barriers based on their origin and nature. (B) AFM images (scale bar: 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μm) obtained on one facet of an MFI-type crystal. Reprinted with permission from ref. 40. Copyright 2009 Springer Nature. (C) Surface pore blockages including bridging, narrowing, and pore misalignment. Reprinted with permission from ref. 11. Copyright 2015 American Chemical Society. (D) Pore re-entering caused by strong sorbate–sorbent interaction. Reprinted with permission from ref. 104. Copyright 2019 American Chemical Society. (E) Length dependence of CH4 diffusivity in the interface region of SAS zeolite in the presence of the bulk gas and a polymer support at T = 300 K. Reprinted with permission from ref. 95. Copyright 2018 Royal Society of Chemistry. (F) Atomistic model of a silicalite crystal with blocking atoms. Reprinted with permission from ref. 91. Copyright 2018 American Chemical Society. (G) Free energy changes in ideal zeolite structures. Reprinted with permission from ref. 99. Copyright 2012 American Chemical Society. μm) obtained on one facet of an MFI-type crystal. Reprinted with permission from ref. 40. Copyright 2009 Springer Nature. (C) Surface pore blockages including bridging, narrowing, and pore misalignment. Reprinted with permission from ref. 11. Copyright 2015 American Chemical Society. (D) Pore re-entering caused by strong sorbate–sorbent interaction. Reprinted with permission from ref. 104. Copyright 2019 American Chemical Society. (E) Length dependence of CH4 diffusivity in the interface region of SAS zeolite in the presence of the bulk gas and a polymer support at T = 300 K. Reprinted with permission from ref. 95. Copyright 2018 Royal Society of Chemistry. (F) Atomistic model of a silicalite crystal with blocking atoms. Reprinted with permission from ref. 91. Copyright 2018 American Chemical Society. (G) Free energy changes in ideal zeolite structures. Reprinted with permission from ref. 99. Copyright 2012 American Chemical Society. | ||
3.1 Experimental characterization
Since the 1970s, surface barriers have been proposed to interpret the dependencies of measured apparent diffusivities on zeolite crystal sizes.75,76 PFG-NMR experiments revealed apparent molecular exchange rates that were lower than those predicted by intracrystalline diffusivities.77 Recent ZLC experiments measured apparent diffusivities that spanned several orders of magnitude from nano-sized to micro-sized zeolites, as shown in Fig. 7(A).10 Similar conclusions have been extended to hierarchically structured zeolites. Despite being faster than in micro-sized silicalite-1, the diffusion of cyclohexane in hierarchically structured MFI-type zeolites did not reach the high rate that was theoretically predicted by assuming that the characteristic diffusion length equals the nanocrystal radius.7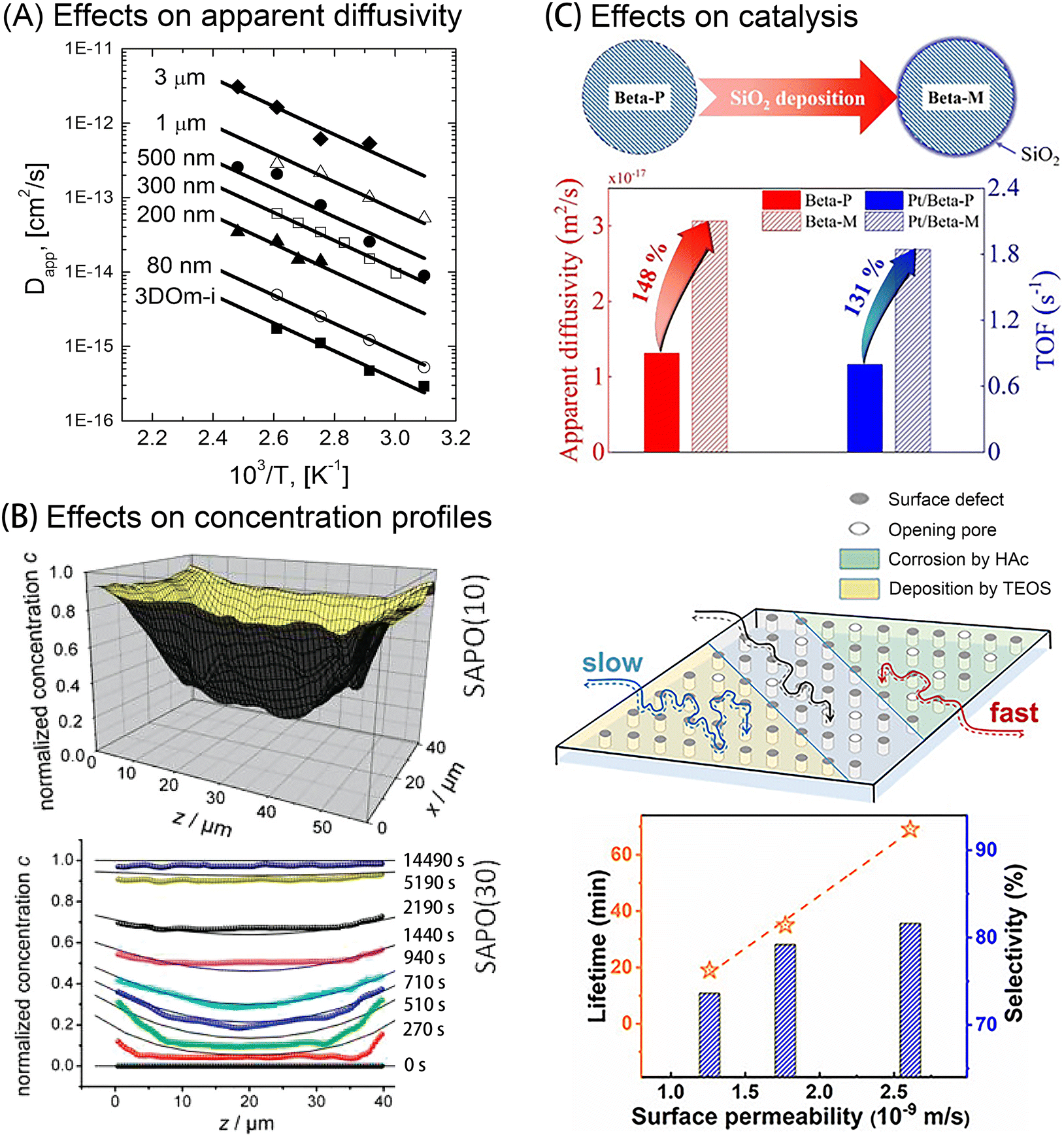 | ||
| Fig. 7 Effects of external surface barriers on apparent diffusivity, concentration profiles, and catalysis performance. (A) Cyclohexane/silicalite-1 Arrhenius plots of apparent diffusivity measured in zeolites with varying crystal sizes. Reprinted with permission from ref. 10. Copyright 2013 American Chemical Society. (B) Concentration gradients of methanol uptake in two SAPO samples. Reprinted with permission from ref. 79. Copyright 2010 American Chemical Society. (C) Top: Improved apparent diffusivity (red) and catalytic isomerization activity (blue) after SiO2 deposition on zeolite Beta crystals Reprinted with permission from ref. 31. Copyright 2021, John Wiley & Sons. Bottom: Regulated surface permeability and methanol-to-olefins (MTO) catalytic performance in SAPO-34 zeolites favored by acid etching (green), but deteriorated by SiO2 deposition (yellow). Reprinted with permission from ref. 83. Copyright 2020, John Wiley & Sons. | ||
Kärger and colleagues used novel micro-imaging techniques (e.g., IRM and IFM) to precisely observe molecular transport behavior at the single crystal level, including across the external surface.22,23 Based on the concentration gradients of permeating molecules, these methods enabled to quantify the effects of surface barriers in nanoporous materials. They discovered a noticeable jump in boundary concentration compared with the equilibrium value, thus indicating the influence of external surface barriers.20,29 In comparison, the equilibrium value is quickly achieved after a quite short time interval in the absence of notable external surface barriers, as shown in Fig. 7(B).24,78,79 Aided by these techniques, the existence of surface barriers has been widely confirmed in nanoporous materials, including zeolites, zeotype materials, and MOFs.20,79,80 Notably, due to the limited spatial resolution, these microimaging approaches can at present only be used to study nanoporous materials with well-defined morphologies, large crystal dimensions (>20 μm), and excellent optical transparency. However, synthesizing such large nanoporous crystals often takes a long time and necessitates harsh experimental conditions. In addition, the most widely applied nanoporous materials are made of nano-sized or micrometer-sized crystals to maximize utilization efficiency. Therefore, new methods to characterize surface barriers with higher resolution and applicable to smaller crystals must be found. Recently, single-molecule fluorescence imaging has been developed as a technique to reveal spatiotemporal gradients of product molecules in zeolite crystals.26,29
The quantification of surface barriers used to be complicated due to the requirement of both apparent diffusivities acquired from macroscopic measurements, as well as intracrystalline diffusivity data derived from microscopic measurements or molecular simulation. Recently, Gao et al.17 put forward a dual-resistance model (DRM) to decouple the surface barriers from intracrystalline diffusivity in nanoporous materials without the need for any preliminary data. By applying this method, several conclusions could be drawn. The derived intracrystalline diffusivities among varying-sized zeolite samples or obtained by different techniques showed surprising consistency at small adsorbate loading, suggesting that intracrystalline diffusivity solely depends on the topological structure of the zeolite in this case. Moreover, the surface permeability was shown to be sensitive to both the physical and chemical non-idealities of the external crystal surface and their interactions with the guest molecules.
In some circumstances, the external diffusion barriers could be the deciding factor in the total mass transfer rate, and thus notably affect the observed catalytic activity. For instance, only when external surface barriers were taken into account, could multiscale modelling results agree with experiments for benzene ethylation catalyzed by ZSM-5/silica composites.81 Later experimental work confirmed that by improving the surface permeabilities of zeolites through post-treatment, the catalytic activity, selectivity, and resistance to deactivation were greatly enhanced in methanol-to-olefins (MTO), isomerization, and cracking reactions.31,82,83 In recent work, Peng et al.28 examined the effects of external surface barriers in ZSM-5 zeolites catalyzed furfuryl alcohol oligomerization. The spatiotemporal evolution of product molecules observed at the single-crystal level clearly demonstrated that increased surface permeability by acid etching promoted the catalytic reaction rate.
Although there is abundant evidence to support the existence of external surface barriers, it is still unclear how to precisely control them. Numerous efforts have been reported to enhance surface permeabilities by optimizing external surficial components or pore mouth shapes. Although similar post-treatment strategies were applied, various studies noted contradictory effects. For example, chemical liquid deposition (CLD) of organosilanes like tetraethyl orthosilicate (TEOS) can create an amorphous silica (SiO2) layer on the external surface. This amorphous layer may obstruct or constrict surface pores, thus hampering mass transfer due to steric hindrance. From an energetic perspective, however, this layer may increase the sticking coefficients of molecules, leading to improved surface permeabilities. Lercher et al.15,84,85 showed that the surface deposition could either increase or decrease the mass transfer rate, depending on the host–guest system. Hu et al.31 showed that the apparent diffusivity and catalytic activity of n-pentane molecules was dramatically increased after SiO2 deposition onto the external surface of zeolite Beta crystals, as illustrated in Fig. 7(C). According to Liu et al.,82 the SiO2-modified zeolites showed a 90% increase in surface permeability compared with the parent zeolite, which reduces deactivation in the olefin catalytic cracking (OCC) reaction. However, further structural characterization revealed different mechanisms. For zeolites with low Si/Al ratios (∼30), TEOS molecules would preferentially react with the bridging hydroxyl groups (Si–OH–Al), thus significantly reducing the amount of external acidic sites, by forming Si–O–Al bonds.31 In comparison, there are much fewer available external acid sites in zeolites with a high Si/Al ratio (∼300). Therefore, the reactions mainly happen at external neutral silanols by forming Si–O–Si bonds.82 Both groups noted that excessive SiO2 deposition could clog many pore entrances. This could help to interpret the results published by Peng et al.83 that SiO2 deposition leads to pronounced lower surface permeability and shortened catalyst lifetime in the MTO reaction over SAPO-34 zeolites, as shown in Fig. 7(C).
Other conflicting results were reported when acid etching was applied to modify the external surfaces of zeolite crystals. Wloch86 reported that the uptake rate of hexane molecules increased in HF-etched ZSM-5 zeolites, and similar results were found for other alkane–zeolite systems.78,87 In comparison, some experimental measurements show no obvious change in uptake rates in silicalite-1 zeolite crystals before and after HF etching.16,88 These contradicting results indicate that the role of external surface barriers also depends on the properties of guest molecules and host materials. Inter-crystal diversity appears because seemingly identical crystals (i.e., with comparable size and morphology) gathered from the same batch can demonstrate drastically variable mass transfer rates.21 Besides, the intra-crystal diversity of external surface barriers has also been tracked by recording the local surface permeabilities in single crystals.29 Therefore, one may draw incorrect assumptions about the origin and nature of surface barriers by merely comparing the results of surface treatment for different collections of zeolite samples.
3.2 Molecular simulations
During experimental studies of external surface barriers, it can be difficult to decouple the effects of interfacial material imperfections from those arising from a resistance in the fluid adjoining the surface. Molecular simulations, on the other hand, can be carried out on atomistic models of the interfacial and crystal structures, to distinguish between both types of external surface resistances.MD simulations demonstrated the presence of non-structural external surface barriers in nano-sized zeolite crystals, even without any surface deposition or distortion. Bai et al.89 conducted MD simulations to compare hexane diffusion in pure microporous MFI-type zeolites and hierarchical nanosheets. Because of tortuous diffusional paths, the self-diffusivity in nanosheets was relatively low at low loadings. Knio et al.90 also applied EMD simulations to study the diffusion of light gases in 2 nm MFI zeolite nanosheets and found a correlation with the adsorption heat of those gases. To be more specific, the diffusivity of the more strongly adsorbing CO2 was decreased in nanosheets, while the diffusivity of H2 in nanosheets was comparable to that in bulk microporous zeolite.
The effects of structural changes at the external surface were also explored by molecular simulation. Sastre et al.91–93 constructed an atomistic model for an MFI-type zeolite with varying degrees of pore mouth blocking, see Fig. 6(F). MD simulations of alkane diffusion in the model confirmed the existence of pore-blockage-induced external surface barriers. Other simulation work showed the relationship between fluxes and hydrophilicity of the membrane, and the estimated permeabilities agreed well with the experimental measurements.94 Dutta et al.95 simulated light gas transport in nanosized zeolite membranes. The effects of external surface barriers to mass transfer became more evident in the presence of a dense polymer support, as shown in Fig. 6(E).
3.3 Origin, nature and function
External surface barriers could originate from energetic or mechanical changes. From a structural perspective, the imperfections around the external surface of nanoporous materials may be a source of external surface barriers. A crust layer was observed on the surface region of MFI-type zeolite by Karwacki et al., as shown in Fig. 6(B).40 The guest molecules could not directly enter a pore mouth because of such sediment hindrance, which causes detours along the external surface and slows down surface permeation.86,96 Another important source of surface barriers is ascribed to pore blockage, including pore misalignment, pore bridging, and pore narrowing, as shown in Fig. 6(C).11 However, current experimental tools remain incapable of clearly recognizing these structural changes at the pore entrance. By comparing ZLC and FR tests with molecular simulations, Dauenhauer et al.11 estimated that over 99.9% of the surface pores in silicalite-1 zeolites appeared to be blocked. Kärger et al.20 drew a similar conclusion for Zn(tbip), a nanoporous MOF material, that the external surface barriers result from the nearly total occlusion of surface pores. This counterintuitive conclusion suggests that the pore blockage mechanism alone might not be sufficient justification for external surface barriers.97Free energy changes, either from enthalpic or entropic origins, could be another important cause of external surface barriers. This means that even a “perfect” zeolite crystal with no surface defects could also trigger surface barriers – the surface is the defect. Keil et al.98–101 attributed the external surface barriers in perfect zeolite crystals to the significant free energy changes in the interfacial region, as shown in Fig. 6(G). Additionally, temperature and the thermophysical properties of the bulk fluid play an essential role in the barrier height. As the authors have shown, the more the critical pressure is approached, the more the impact of surface barriers decreases because the transport resistances in the boundary layer and the zeolite become equal. Varanasi et al.102 discovered that water molecules experience significant entropy gains when moving across the external surface into the interior region of carbon nanotubes. Bai et al.89 claimed that the prolonged diffusion paths were caused by an enthalpic cost when escaping from the microporous area. Dauenhauer et al.97,103,104 found that the strong host–guest interaction leads to pore re-entering of gas molecules, as shown in Fig. 6(D). Additional experimental investigation revealed that the importance of non-structural surface barriers differs dramatically with the adsorption heat of guest molecules.104,105 These findings point out that for host–guest systems with weak interactions, pore mouth opening aided by acid etching is more efficient to regulate external surface barriers. On the other hand, for strong host–guest interactions, adsorption at the surface could be reduced by depositing a mesoporous silica layer106 or the opportunity for micropore re-entering could be diminished by introducing an inert surface.97
New synthesis methods for the controlled growth of zeolite crystals with enhanced mass transfer properties have been increasingly reported.107,108 Rimer and colleagues showed markedly enhanced molecular uptake rates in finned and egg-shell structured zeolite catalysts.109,110 They also applied cooperative surface passivation and hierarchical structuring to improve the catalytic activity of zeolite beta catalysts.111 Now over a decade ago, Ryoo and colleagues first synthesized single-unit-cell nanosheets, demonstrating higher catalytic activity and better stability than conventional microporous zeolites.112,113 Their remarkable materials aimed to improve the overall mass transfer performance but, at the time, no analysis of the external surface permeability was performed. With the introduction of hierarchical porosity into the zeolite framework and other nanoporous materials, the external surface area tends to increase sharply. Therefore, it is necessary to examine if there exists a critical point that balances the shortening microporous diffusional length and the possible increase of external surface barriers.
In the context of the mechanisms underpinning external surface barriers, the symmetry of the adsorption and desorption processes has been extensively debated. Teixeira et al.13 argued that the external surface barrier is asymmetric, more important for desorption than adsorption, based on the postulated disparities in mass transfer path lengths. However, Sastre et al.92,93 pointed out that this statement appears to violate the microscopic reversibility principle. The similar path length distributions for adsorption and desorption in a zeolite crystal with varying degrees of pore blockage, as well as mathematical thermodynamic analysis, were used to further illustrate the symmetry of the external surface barriers. The adsorption and desorption paths based on these two theories are shown in Fig. 8.93
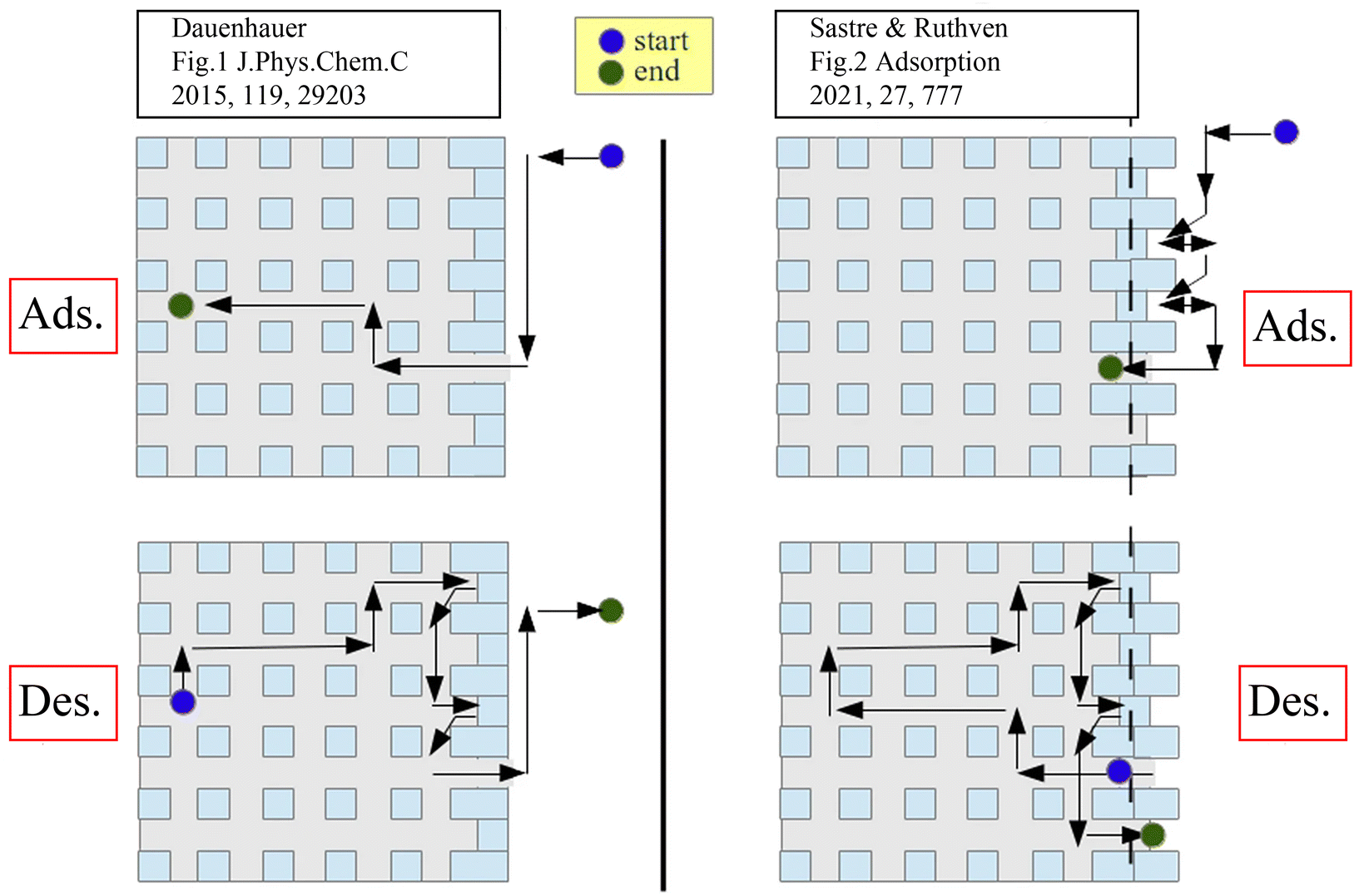 | ||
| Fig. 8 Schematic representation of adsorption/desorption models with asymmetric (left) and symmetric (right) barriers. Reprinted with permission from ref. 93. Copyright 2021 Springer Nature. | ||
4. Conclusions and outlook
Nobel Laureate Wolfgang Pauli once stated: “God made the bulk; the surface was invented by the devil”. This review has demonstrated the significant role of the surface barriers on the overall mass transport processes in nanoporous materials, the complexity of identifying their (likely multiple) origins, and the tremendous opportunities for catalysis and separation processes when we are able to control them.Table 1 lists the characteristics and challenges based on the current knowledge of surface barriers in nanoporous materials. Surface barriers act as a critical parameter prompting new opportunities to fine-tune them for applications. Controlling these effects may optimize energy usage and impact product selectivity when running specific reactions, thus leading to more sustainable chemical production.
| Types | Internal surface barriers | External surface barriers | ||
|---|---|---|---|---|
| Classification | Intracrystalline | Intercrystalline | Structural | Non-structural |
| Origin and nature | Intergrowth; defects | Grain boundaries | Disordered residue; pore blockage (bridging, narrowing, or misalignment) | Strong sorbate–sorbent interaction (free energy change) |
| Modulation strategy | Precisely controlled synthesis of perfect crystal; post-treatment for defect healing | Controlled etching to remove residue and open pore mouth | Post-treatment for weakening the interaction (deposition, inert mixture, epitaxial growth, etc.) | |
| Main challenges | Precise identification of the crystal growth mechanism; precise identification of the structural nature of surface barriers at the atomistic level; utilization of the surface barriers to control the catalysis and separation performance in nanoporous materials. | |||
Despite extensive experimental and simulation studies from numerous groups, there is still no real consensus on the mass transfer mechanisms in nanoporous materials, especially when surface barriers are accounted for. During the design and application of nanoporous materials, it is crucial to take the impacts of internal and external surface barriers into consideration. On the one hand, minimizing or eliminating the effects of these barriers makes sense because they always slow down the mass transfer of guest molecules. On the other hand, however, these barriers might also be utilized to regulate selectivity among complex components. Additional research on this subject should be carried out to investigate the nature of these effects in-depth and see how they can be either avoided or exploited. Aided by molecular simulations, the atomistic origins of surface barriers could be further explored. Future developments in microimaging technology that have single-molecule or single-atom resolution could offer a powerful tool for quantifying the effects down to increasingly smaller scales. We emphasize the need to address both internal and external surfaces for the rational design of efficient catalysts and membrane separations. Accounting for the impact of surface composition on the surface barriers could encourage the exploration of new nanoporous materials as well, where both the bulk and the interfaces are properly considered in materials and process design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support from the EPSRC of the UK (EP/S03305X/1), the National Natural Science Foundation of China (22278353) and the Zhejiang Provincial Natural Science Foundation of China (LZ23B060001) are greatly appreciated. S. X. acknowledges the support from the China Scholarship Council (201906320255).Notes and references
- B. C. Bukowski, F. J. Keil, P. I. Ravikovitch, G. Sastre, R. Q. Snurr and M.-O. Coppens, Adsorption, 2021, 27, 683–760 CrossRef CAS.
- M.-O. Coppens, T. Weissenberger, Q. Zhang and G. Ye, Adv. Mater. Interfaces, 2021, 8, 2001409 CrossRef CAS.
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam, M. Klumpp and A. Inayat, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC.
- X. Liu, C. Wang, J. Zhou, C. Liu, Z. Liu, J. Shi, Y. Wang, J. Teng and Z. Xie, Chem. Soc. Rev., 2022, 51, 8174–8200 RSC.
- B. Coasne, New J. Chem., 2016, 40, 4078–4094 RSC.
- O. C. Gobin, S. J. Reitmeier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2009, 113, 20435–20444 CrossRef CAS.
- C.-C. Chang, A. R. Teixeira, C. Li, P. J. Dauenhauer and W. Fan, Langmuir, 2013, 29, 13943–13950 CrossRef CAS PubMed.
- W.-Y. Gao, A. D. Cardenal, C.-H. Wang and D. C. Powers, Chem. – Eur. J., 2019, 25, 3465–3476 CrossRef CAS PubMed.
- L. Heinke and C. Wöll, Adv. Mater., 2019, 31, 1806324 CrossRef PubMed.
- A. R. Teixeira, C.-C. Chang, T. Coogan, R. Kendall, W. Fan and P. J. Dauenhauer, J. Phys. Chem. C, 2013, 117, 25545–25555 CrossRef CAS.
- A. R. Teixeira, X. Qi, W. C. Conner, T. J. Mountziaris, W. Fan and P. J. Dauenhauer, Chem. Mater., 2015, 27, 4650–4660 CrossRef CAS.
- O. C. Gobin, S. J. Reitmeier, A. Jentys and J. A. Lercher, Microporous Mesoporous Mater., 2009, 125, 3–10 CrossRef CAS.
- A. R. Teixeira, X. Qi, C.-C. Chang, W. Fan, W. C. Conner and P. J. Dauenhauer, J. Phys. Chem. C, 2014, 118, 22166–22180 CrossRef CAS.
- S. J. Reitmeier, R. R. Mukti, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2008, 112, 2538–2544 CrossRef CAS.
- O. C. Gobin, S. J. Reitmeier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 1171–1179 CrossRef CAS.
- L. Gueudré, E. Jolimaîte, N. Bats and W. Dong, Adsorption, 2010, 16, 17–27 CrossRef.
- M. Gao, H. Li, M. Yang, S. Gao, P. Wu, P. Tian, S. Xu, M. Ye and Z. Liu, Commun. Chem., 2019, 2, 43 CrossRef.
- J. Kärger, W. Heink, H. Pfeifer, M. Rauscher and J. Hoffmann, Zeolites, 1982, 2, 275–278 CrossRef.
- A. Feldhoff, J. Caro, H. Jobic, J. Ollivier, C. B. Krause, P. Galvosas and J. Kärger, ChemPhysChem, 2009, 10, 2429–2433 CrossRef CAS PubMed.
- F. Hibbe, C. Chmelik, L. Heinke, S. Pramanik, J. Li, D. M. Ruthven, D. Tzoulaki and J. Kärger, J. Am. Chem. Soc., 2011, 133, 2804–2807 CrossRef CAS PubMed.
- J. C. S. Remi, A. Lauerer, C. Chmelik, I. Vandendael, H. Terryn, G. V. Baron, J. F. M. Denayer and J. Kärger, Nat. Mater., 2016, 15, 401–406 CrossRef CAS PubMed.
- J. Kärger, Microporous Mesoporous Mater., 2014, 189, 126–135 CrossRef.
- J. Kärger, T. Binder, C. Chmelik, F. Hibbe, H. Krautscheid, R. Krishna and J. Weitkamp, Nat. Mater., 2014, 13, 333–343 CrossRef PubMed.
- P. Kortunov, C. Chmelik, J. Kärger, R. A. Rakoczy, D. M. Ruthven, Y. Traa, S. Vasenkov and J. Weitkamp, Adsorption, 2005, 11, 235–244 CrossRef CAS.
- P. Kortunov, L. Heinke, S. Vasenkov, C. Chmelik, D. B. Shah, J. Kärger, R. A. Rakoczy, Y. Traa and J. Weitkamp, J. Phys. Chem. B, 2006, 110, 23821–23828 CrossRef CAS PubMed.
- Z. Ristanović, J. P. Hofmann, G. De Cremer, A. V. Kubarev, M. Rohnke, F. Meirer, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 6559–6568 CrossRef PubMed.
- Z. Ristanović, M. M. Kerssens, A. V. Kubarev, F. C. Hendriks, P. Dedecker, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2015, 54, 1836–1840 CrossRef PubMed.
- S. Peng, H. Li, W. Liu, J. Yu, Z. Xu, M. Ye and Z. Liu, Chem. Eng. J., 2022, 430, 132760 CrossRef CAS.
- S. Peng, Y. Xie, L. Wang, W. Liu, H. Li, Z. Xu, M. Ye and Z. Liu, Angew. Chem., Int. Ed., 2022, 61, e202203903 CAS.
- J. J. E. Maris, D. Fu, F. Meirer and B. M. Weckhuysen, Adsorption, 2021, 27, 423–452 CrossRef CAS.
- S. Hu, J. Liu, G. Ye, X. Zhou, M.-O. Coppens and W. Yuan, Angew. Chem., Int. Ed., 2021, 60, 14394–14398 CrossRef CAS PubMed.
- J. Choi, H.-K. Jeong, M. A. Snyder, J. A. Stoeger, R. I. Masel and M. Tsapatsis, Science, 2009, 325, 590–593 CrossRef CAS PubMed.
- S. Vasenkov and J. Kärger, Microporous Mesoporous Mater., 2002, 55, 139–145 CrossRef CAS.
- G. Arya, E. J. Maginn and H.-C. Chang, J. Phys. Chem. B, 2001, 105, 2725–2735 CrossRef CAS.
- M. G. Ahunbay, J. R. Elliott and O. Talu, J. Phys. Chem. B, 2002, 106, 5163–5168 CrossRef CAS.
- D. A. Newsome and D. S. Sholl, J. Phys. Chem. B, 2005, 109, 7237–7244 CrossRef CAS PubMed.
- D. A. Newsome and D. S. Sholl, Nano Lett., 2006, 6, 2150–2153 CrossRef CAS PubMed.
- L. Liu, D. Nicholson and S. K. Bhatia, J. Phys. Chem. C, 2016, 120, 26363–26373 CrossRef CAS.
- L. Karwacki, E. Stavitski, M. H. F. Kox, J. Kornatowski and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2007, 46, 7228–7231 CrossRef CAS PubMed.
- L. Karwacki, M. H. F. Kox, D. A. Matthijs de Winter, M. R. Drury, J. D. Meeldijk, E. Stavitski, W. Schmidt, M. Mertens, P. Cubillas, N. John, A. Chan, N. Kahn, S. R. Bare, M. Anderson, J. Kornatowski and B. M. Weckhuysen, Nat. Mater., 2009, 8, 959–965 CrossRef CAS PubMed.
- L. Karwacki, H. E. van der Bij, J. Kornatowski, P. Cubillas, M. R. Drury, D. A. M. de Winter, M. W. Anderson and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2010, 49, 6790–6794 CrossRef CAS PubMed.
- E. R. Geus, J. C. Jansen and H. van Bekkum, Zeolites, 1994, 14, 82–88 CrossRef CAS.
- S. Vasenkov, W. Bohlmann, P. Galvosas, O. Geier, H. Liu and J. Karger, J. Phys. Chem. B, 2001, 105, 5922–5927 CrossRef CAS.
- H. Takaba, A. Yamamoto, K. Hayamizu and S.-I. Nakao, J. Phys. Chem. B, 2005, 109, 13871–13876 CrossRef CAS PubMed.
- K. Ulrich, D. Freude, P. Galvosas, C. Krause, J. Kärger, J. Caro, P. Poladli and H. Papp, Microporous Mesoporous Mater., 2009, 120, 98–103 CrossRef CAS.
- G. H. Ye, Y. Y. Sun, Z. Y. Guo, K. K. Zhu, H. L. Liu, X. G. Zhou and M. O. Coppens, J. Catal., 2018, 360, 152–159 CrossRef CAS.
- Z. Guo, X. Li, S. Hu, G. Ye, X. Zhou and M.-O. Coppens, Angew. Chem., Int. Ed., 2020, 59, 1548–1551 CrossRef CAS PubMed.
- G. Xomeritakis, Z. Lai and M. Tsapatsis, Ind. Eng. Chem. Res., 2001, 40, 544–552 CrossRef CAS.
- J. Kim, E. Jang, S. Hong, D. Kim, E. Kim, H. Ricther, A. Simon, N. Choi, D. Korelskiy, S. Fouladvand, J. Nam and J. Choi, J. Membr. Sci., 2019, 591, 117342 CrossRef CAS.
- D. Korelskiy, P. Ye, M. S. Nabavi and J. Hedlund, J. Mater. Chem. A, 2017, 5, 7295–7299 RSC.
- S. Vasenkov and J. Karger, Microporous Mesoporous Mater., 2002, 55, 139–145 CrossRef CAS.
- F. J. Keil, R. Krishna and M.-O. Coppens, Rev. Chem. Eng., 2000, 16, 71–197 CAS.
- D. A. Newsome and D. S. Sholl, J. Phys. Chem. B, 2006, 110, 22681–22689 CrossRef CAS PubMed.
- A. M. Thomas and Y. Subramanian, J. Chem. Phys., 2018, 149, 064702 CrossRef PubMed.
- H. Takaba, A. Yamamoto and S.-I. Nakao, Desalination, 2006, 192, 82–90 CrossRef CAS.
- F. Hirosawa, M. Miyagawa and H. Takaba, J. Membr. Sci., 2021, 632, 119348 CrossRef CAS.
- M. Fasano, T. Humplik, A. Bevilacqua, M. Tsapatsis, E. Chiavazzo, E. N. Wang and P. Asinari, Nat. Commun., 2016, 7, 12762 CrossRef CAS PubMed.
- F. Cailliez, G. Stirnemann, A. Boutin, I. Demachy and A. H. Fuchs, J. Phys. Chem. C, 2008, 112, 10435–10445 CrossRef CAS.
- T. Humplik, R. Raj, S. C. Maroo, T. Laoui and E. N. Wang, Langmuir, 2014, 30, 6446–6453 CrossRef CAS PubMed.
- G. D. Price, J. J. Pluth, J. V. Smith, J. M. Bennett and R. L. Patton, J. Am. Chem. Soc., 1982, 104, 5971–5977 CrossRef CAS.
- D. G. Hay, H. Jaeger and K. G. Wilshier, Zeolites, 1990, 10, 571–576 CrossRef CAS.
- O. Geier, S. Vasenkov, E. Lehmann, J. Kärger, U. Schemmert, R. A. Rakoczy and J. Weitkamp, J. Phys. Chem. B, 2001, 105, 10217–10222 CrossRef CAS.
- J. R. Agger, N. Hanif, C. S. Cundy, A. P. Wade, S. Dennison, P. A. Rawlinson and M. W. Anderson, J. Am. Chem. Soc., 2003, 125, 830–839 CrossRef CAS PubMed.
- M. B. J. Roeffaers, R. Ameloot, M. Baruah, H. Uji-i, M. Bulut, G. De Cremer, U. Müller, P. A. Jacobs, J. Hofkens, B. F. Sels and D. E. De Vos, J. Am. Chem. Soc., 2008, 130, 5763–5772 CrossRef CAS PubMed.
- M. M. J. Treacy and J. M. Newsam, Nature, 1988, 332, 249–251 CrossRef CAS.
- H. Lee, J. Shin, K. Lee, H. J. Choi, A. Mayoral, N. Y. Kang and S. B. Hong, Science, 2021, 373, 104–107 CrossRef CAS PubMed.
- D. Schwalbe-Koda, S. Kwon, C. Paris, E. Bello-Jurado, Z. Jensen, E. Olivetti, T. Willhammar, A. Corma, Y. Román-Leshkov, M. Moliner and R. Gómez-Bombarelli, Science, 2021, 374, 308–315 CrossRef CAS PubMed.
- P. Kumar, D. W. Kim, N. Rangnekar, H. Xu, E. O. Fetisov, S. Ghosh, H. Zhang, Q. Xiao, M. Shete, J. I. Siepmann, T. Dumitrica, B. McCool, M. Tsapatsis and K. A. Mkhoyan, Nat. Mater., 2020, 19, 443–449 CrossRef CAS PubMed.
- M. Khaleel, A. J. Wagner, K. A. Mkhoyan and M. Tsapatsis, Angew. Chem., Int. Ed., 2014, 53, 9456–9461 CrossRef CAS PubMed.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed.
- D. Xu, G. R. Swindlehurst, H. Wu, D. H. Olson, X. Zhang and M. Tsapatsis, Adv. Funct. Mater., 2014, 24, 201–208 CrossRef CAS.
- Z. Liu, T. Ohsuna, K. Sato, T. Mizuno, T. Kyotani, T. Nakane and O. Terasaki, Chem. Mater., 2006, 18, 922–927 CrossRef CAS.
- G. Bonilla, M. Tsapatsis, D. G. Vlachos and G. Xomeritakis, J. Membr. Sci., 2001, 182, 103–109 CrossRef CAS.
- S. Hong, D. Kim, H. Richter, J.-H. Moon, N. Choi, J. Nam and J. Choi, J. Membr. Sci., 2019, 569, 91–103 CrossRef CAS.
- J. Kärger and J. Caro, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1363–1376 RSC.
- M. Bülow, P. Struve, G. Finger, C. Redszus, K. Ehrhardt, W. Schirmer and J. Kärger, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 597–615 RSC.
- J. Kärger, M. Bülow, G. R. Millward and J. M. Thomas, Zeolites, 1986, 6, 146–150 CrossRef.
- P. Kortunov, S. Vasenkov, C. Chmelik, J. Kärger, D. M. Ruthven and J. Wloch, Chem. Mater., 2004, 16, 3552–3558 CrossRef CAS.
- D. Tzoulaki, L. Heinke, M. Castro, P. Cubillas, M. W. Anderson, W. Zhou, P. A. Wright and J. Kärger, J. Am. Chem. Soc., 2010, 132, 11665–11670 CrossRef CAS PubMed.
- J. Kärger, P. Kortunov, S. Vasenkov, L. Heinke, D. B. Shah, R. A. Rakoczy, Y. Traa and J. Weitkamp, Angew. Chem., Int. Ed., 2006, 45, 7846–7849 CrossRef PubMed.
- S. M. Rao, E. Saraçi, R. Gläser and M.-O. Coppens, Chem. Eng. J., 2017, 329, 45–55 CrossRef CAS.
- X. Liu, Y. Wang, J. Zhou, C. Wang, J. Shi, Y. Ye, Y. Wang, J. Teng and Z. Xie, Chem. Commun., 2023, 59, 470–473 RSC.
- S. Peng, M. Gao, H. Li, M. Yang, M. Ye and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 21945–21948 CrossRef CAS PubMed.
- S. J. Reitmeier, O. C. Gobin, A. Jentys and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 533–538 CrossRef CAS PubMed.
- S. J. Reitmeier, O. C. Gobin, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2009, 113, 15355–15363 CrossRef CAS.
- J. Wloch, Microporous Mesoporous Mater., 2003, 62, 81–86 CrossRef CAS.
- L. Gueudré, N. Bats and E. Jolimaître, Microporous Mesoporous Mater., 2012, 147, 310–317 CrossRef.
- C. Chmelik, A. Varma, L. Heinke, D. B. Shah, J. Kärger, F. Kremer, U. Wilczok and W. Schmidt, Chem. Mater., 2007, 19, 6012–6019 CrossRef CAS.
- P. Bai, E. Haldoupis, P. J. Dauenhauer, M. Tsapatsis and J. I. Siepmann, ACS Nano, 2016, 10, 7612–7618 CrossRef CAS PubMed.
- O. Knio, H. Fang, S. E. Boulfelfel, S. Nair and D. S. Sholl, J. Phys. Chem. C, 2020, 124, 15241–15252 CrossRef CAS.
- G. Sastre, J. Kärger and D. M. Ruthven, J. Phys. Chem. C, 2018, 122, 7217–7225 CrossRef CAS.
- G. Sastre, J. Kärger and D. M. Ruthven, J. Phys. Chem. C, 2019, 123, 19596–19601 CrossRef CAS.
- G. Sastre, J. Kärger and D. M. Ruthven, Adsorption, 2021, 27, 777–785 CrossRef CAS.
- S. Turgman-Cohen, J. C. Araque, E. M. V. Hoek and F. A. Escobedo, Langmuir, 2013, 29, 12389–12399 CrossRef CAS PubMed.
- R. C. Dutta and S. K. Bhatia, Phys. Chem. Chem. Phys., 2018, 20, 26386–26395 RSC.
- W. L. Duncan and K. P. Möller, Adsorption, 2005, 11, 259–273 CrossRef CAS.
- X. Qi, V. Vattipalli, P. J. Dauenhauer and W. Fan, Chem. Mater., 2018, 30, 2353–2361 CrossRef CAS.
- N. E. R. Zimmermann, B. Smit and F. J. Keil, J. Phys. Chem. C, 2010, 114, 300–310 CrossRef CAS.
- N. E. R. Zimmermann, S. P. Balaji and F. J. Keil, J. Phys. Chem. C, 2012, 116, 3677–3683 CrossRef CAS.
- N. E. R. Zimmermann, B. Smit and F. J. Keil, J. Phys. Chem. C, 2012, 116, 18878–18883 CrossRef CAS.
- N. E. R. Zimmermann, T. J. Zabel and F. J. Keil, J. Phys. Chem. C, 2013, 117, 7384–7390 CrossRef CAS.
- S. R. Varanasi, Y. Subramanian and S. K. Bhatia, Langmuir, 2018, 34, 8099–8111 CrossRef CAS PubMed.
- V. Vattipalli, X. Qi, P. J. Dauenhauer and W. Fan, Chem. Mater., 2016, 28, 7852–7863 CrossRef CAS.
- X. Qi, V. Vattipalli, K. Zhang, P. Bai, P. J. Dauenhauer and W. Fan, Langmuir, 2019, 35, 12407–12417 CrossRef CAS PubMed.
- J. Hedlund, M. S. Nobandegani and L. Yu, J. Membr. Sci., 2022, 641, 119893 CrossRef CAS.
- G. Ye, Z. Guo, Y. Sun, K. Zhu, H. Liu, X. Zhou and M.-O. Coppens, Chem. Ing. Tech., 2017, 89, 1333–1342 CrossRef CAS.
- X. Liu, J. Shi, G. Yang, J. Zhou, C. Wang, J. Teng, Y. Wang and Z. Xie, Commun. Chem., 2021, 4, 107 CrossRef CAS PubMed.
- Q. Yang, Y. Li, Z. Chen, L. Hu, Z. Li, Y. Wang, Z. Zhao, C. Xu and G. Jiang, AIChE J., 2021, 67, e17130 CAS.
- H. Dai, Y. Shen, T. Yang, C. Lee, D. Fu, A. Agarwal, T. T. Le, M. Tsapatsis, J. C. Palmer, B. M. Weckhuysen, P. J. Dauenhauer, X. Zou and J. D. Rimer, Nat. Mater., 2020, 19, 1074–1080 CrossRef CAS PubMed.
- T. T. Le, K. Shilpa, C. Lee, S. Han, C. Weiland, S. R. Bare, P. J. Dauenhauer and J. D. Rimer, J. Catal., 2022, 405, 664–675 CrossRef CAS.
- S. Han, N. Linares, T. Terlier, J. B. Hoke, J. García Martínez, Y. Li and J. D. Rimer, Angew. Chem., Int. Ed., 2022, 61, e202210434 CAS.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- K. Na, M. Choi, W. Park, Y. Sakamoto, O. Terasaki and R. Ryoo, J. Am. Chem. Soc., 2010, 132, 4169–4177 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |