
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Divalent metallocenes of the lanthanides – a guideline to properties and reactivity
Sebastian
Schäfer
a,
Sebastian
Kaufmann
a,
Esther S.
Rösch
 b and
Peter W.
Roesky
*a
b and
Peter W.
Roesky
*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
bBaden-Württemberg Cooperative State University Karlsruhe, Erzbergerstr. 121, 76133 Karlsruhe, Germany
First published on 15th May 2023
Abstract
Since the discovery in the early 1980s, the soluble divalent metallocenes of lanthanides have become a steadily growing field in organometallic chemistry. The predominant part of the investigation has been performed with samarium, europium, and ytterbium, whereas only a few reports dealing with other rare earth elements were disclosed. Reactions of these metallocenes can be divided into two major categories: (1) formation of Lewis acid–base complexes, in which the oxidation state remains +II; and (2) single electron transfer (SET) reductions with the ultimate formation of Ln(III) complexes. Due to the increasing reducing character from Eu(II) over Yb(II) to Sm(II), the plethora of literature concerning redox reactions revolves around the metallocenes of Sm and Yb. In addition, a few reactivity studies on Nd(II), Dy(II) and mainly Tm(II) metallocenes were published. These compounds are even stronger reducing agents but significantly more difficult to handle. In most cases, the metals are ligated by the versatile pentamethylcyclopentadienyl ligand: (C5Me5). Other cyclopentadienyl ligands are fully covered but only discussed in detail, if the ligand causes differences in synthesis or reactivity. Thus, the focus lays on three compounds: [(C5Me5)2Sm], [(C5Me5)2Eu] and [(C5Me5)2Yb] and their solvates. We discuss the synthesis and physical properties of divalent lanthanide metallocenes first, followed by an overview of the reactivity rendering the full potential of these versatile reactants.
 Sebastian Schäfer | Sebastian Schäfer graduated from the Technical University of Kaiserslautern in 2012. He joined the group of Peter W. Roesky at KIT for his doctoral studies, focusing on low valent silicon and coinage metal chemistry. After receiving his degree in 2016 and a brief postdoctoral stay at KIT, he joined the chemical industry for a two-year post doc position working on the development of heterogeneous catalysts. Since 2019, he has been working as a head of laboratory and project lead in the field of inorganic materials in the chemical industry. |
Sebastian Kaufmann graduated in 2020, receiving his doctoral degree from the Karlsruhe Institute of Technology (KIT). After a short postdoctoral stay at the KIT as the head of a student's laboratory practice, he moved on to the Baden-Württemberg Cooperative State University Karlsruhe. Where he was responsible for the laboratories in the Department of Safety, Health and Environment and assisted in the development of the new study course “sustainable science and technology”. In 2022 he went on to the chemical industry, joining a GLP laboratory. |
 Esther S. Rösch | Esther S. Rösch graduated from the University of Karlsruhe in 2007 and received her doctoral degree from the Karlsruhe Institute of Technology (KIT) in 2009. After a short postdoctoral stay at the KIT, she moved to the pharmaceutical industry. In 2013, she was appointed professor for Bioanalytics at the Pforzheim University of Applied Sciences. In 2018, she joined the Baden-Württemberg Cooperative State University Karlsruhe, where she headed the Department of Safety, Health and Environment from 2020–2022. Her current research focuses on magnetic nanoparticles and their biomedical applications, as well as on the utilisation of organic and inorganic luminophores, especially lanthanide-based ones. |
 Peter W. Roesky | Peter W. Roesky obtained his diploma in chemistry in 1992 from the University of Würzburg and his doctoral degree from the Technical University of Munich in 1994. After postdoctoral work at Northwestern University, USA (1995–1996), he completed his habilitation at the University of Karlsruhe in 1999. He was appointed a full professor at the Freie Universität Berlin in 2001, during which he joined the faculty of chemistry and biochemistry. In 2008, he became a Full Professor of inorganic functional materials at the Karlsruhe Institute of Technology (KIT). From 2013 to 2015, he served as Dean of the Faculty of Chemistry and Biosciences at KIT. His current research interest revolves around the synthetic inorganic and organometallic chemistry of s-block metals, silicon, phosphorus, gold, and lanthanides. |
1. Introduction
The revolution of organometallic chemistry in the mid of the last century, starting from the landmark (but rather serendipitous) isolation of ferrocene1,2 and the groundbreaking recognition of its sandwich structure3,4 set off an avalanche of cyclopentadienyl chemistry that did not directly capture the lanthanides. In that time associated with a somewhat meek redox chemistry limited to the oxidation state +III, trivalent cyclopentadienyl compounds were isolated but displayed no noteworthy chemistry besides the formation of Lewis acid–base adducts. Also, cyclopentadienyl compounds of the rare earths are typically pretty insoluble in ethereal and hydrocarbon solvents as they tend to form polymeric structures.5 This proved especially true for divalent lanthanide compounds. Classically, three lanthanide elements have a well accessible divalent oxidation state: europium, samarium and ytterbium. Reactions of the corresponding Ln(II) halide precursors LnX2 with cyclopentadienyl salts or metal/ammonia systems with cyclopentadiene resulted usually in insoluble organometallic polymers that did allow only a very few further reactivity studies.6 This changed with the emergence of one of the most prominent ligands in organometallic chemistry, pentamethylcyclopentadienyl. With its enhanced sterical bulk and higher electron density compared to the parent cyclopentadienyl, it allowed the isolation of unusual and reactive species. In combination with its excellent crystallization properties, it played an immense role in the history of organometallics. It also allowed the isolation of the first well-soluble, divalent metallocenes of Sm, Eu and Yb, which resulted in the investigation of the fascinating chemistry of these reactive molecules. Today, we consider these compounds as the “classic” divalent metallocenes. In addition, a few divalent metallocenes of the other rare elements ligated by even more bulky cyclopentadienyl ligands are known. Although divalent metallocenes of Tm,7 Dy,8 and Nd9 were established some years ago, metallocenes of most of the other rare earth metallocenes were published only very recently.10,11These newly accessible divalent lanthanides are mostly considered as the “non-classical” divalent compounds. This review focuses mainly on the “classical” divalent lanthanide metallocenes of Sm, Eu and Yb ligated by the permethylated cyclopentadienyl ligand C5Me5. Their rich and colourful chemistry is still being actively researched all over the world even after 40 years of discovery and has not ceased to surprise to this day. Other substitution patterns of the cyclopentadienyl ligand are also known. However, they are only discussed herein if the influence of the substituents strongly deviates from the corresponding permethylated cyclopentadienyl ligand derivatives. The first part of this review will deal with the synthesis, structure and physical properties of the divalent metallocenes of the lanthanides. The second part covers the reactivity, divided into reactivity as a Lewis acid, yielding Ln(II) complexes, and reactivity in redox reactions resulting in Ln(III) complexes.
2. Synthesis
[(C5Me5)2Yb] was the first divalent decamethylmetallocene. Its synthesis was published independently by Watson and Andrews in 1980.12,13 By reacting YbBr2 with KC5Me5 or NaC5Me5 with YbCl2 in ethereal solvents like tetrahydrofuran (THF), diethylether (Et2O) or 1,2-dimethoxyethane (DME) the corresponding metallocene was obtained as its respective ether adduct. The THF solvate of [(C5Me5)2Sm] was first reported by Evans in 1981.14 By diffusing samarium vapour into a hexane solution of HC5Me5, the diene is reduced with concomitant hydrogen evolution. A dark mixture was obtained that was filtered. Extraction of the remaining dark solid with THF yields [(C5Me5)2Sm(THF)2] alongside hydride-containing side products. A few years later, a salt metathesis route to the metallocene starting from SmI2 and KC5Me5 was reported (Scheme 1).15 In contrast to [(C5Me5)2Sm(solvent)n] and [(C5Me5)2Yb(solvent)n], [(C5Me5)2Eu(solvent)n] could be prepared from EuCl3 and three equivalents of Na(C5Me5) in THF and crystallised as etherate.13 Here, one equivalent of pentamethylcyclopentadienyl acts as a reducing agent by transferring one electron to the Eu(III) centre with the release of a formal (C5Me5) radical that combines to a dimer. The observed reactivity is a result of the redox potential of the Eu(III)/Eu(II) couple.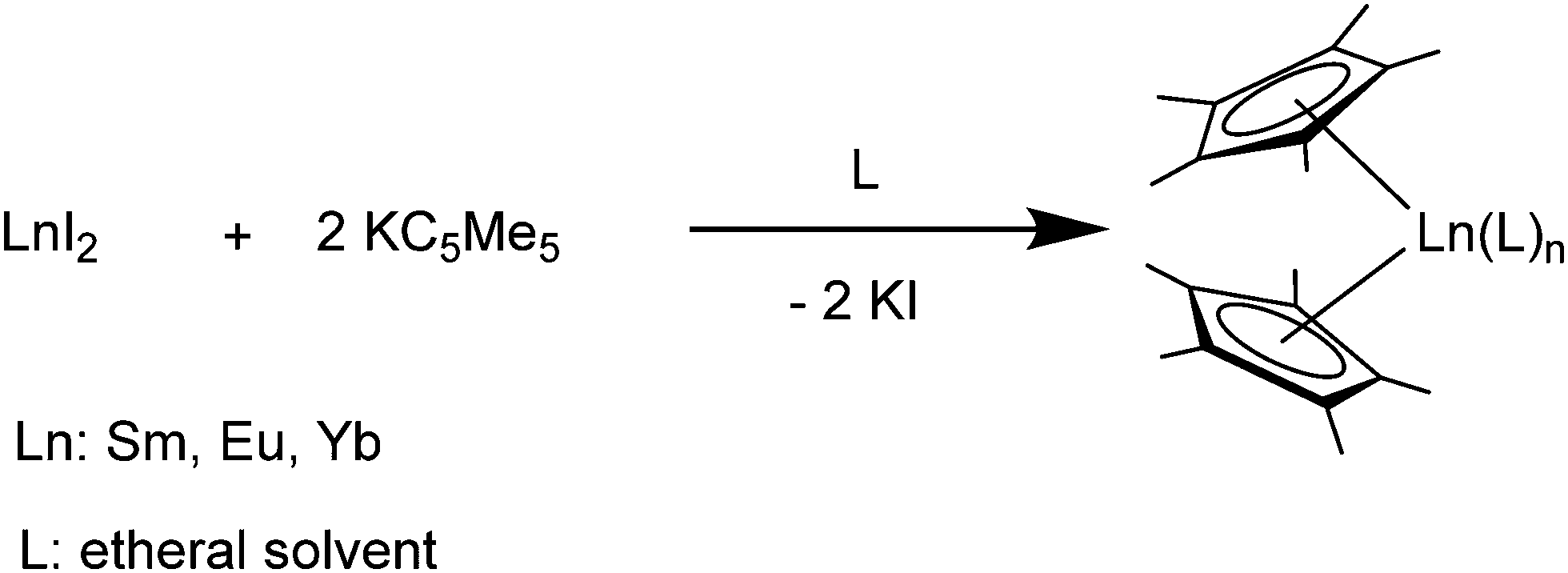 | ||
| Scheme 1 Synthesis of the title compounds via salt metathesis.15 | ||
Today, the most established and convenient route to THF-solvated [(C5Me5)2Ln(solvent)n] and similar metallocenes is the reaction of two equivalents of the cyclopentadienyl derivative with the corresponding divalent iodides in THF.16–19 The divalent iodides can be cleanly prepared using the respective elemental metals and 1,2-diiodoethane.20 Metallocenes with other substitution patterns on the five-membered ring including larger entities such as indenyl, fluorenyl and ansa-metallocenes are obtained similarly.11,21–35 With a few exceptions in which the substituents strongly influence the structure, these compounds are not discussed here in detail. Thus, salt metathesis of other cyclopentadienyl derivatives such as pentabenzylcyclopentadienyl (CpBn5) likewise led to the corresponding metallocenes [(CpBn5)2Ln] (Ln = Sm, Eu, Yb) (Scheme 2).36 Due to the sterically demanding ligand as well as the saturated coordination sphere of the lanthanides by π-interaction with one phenyl ring per ligand, these compounds are present without any coordinating solvent at the metal centre.
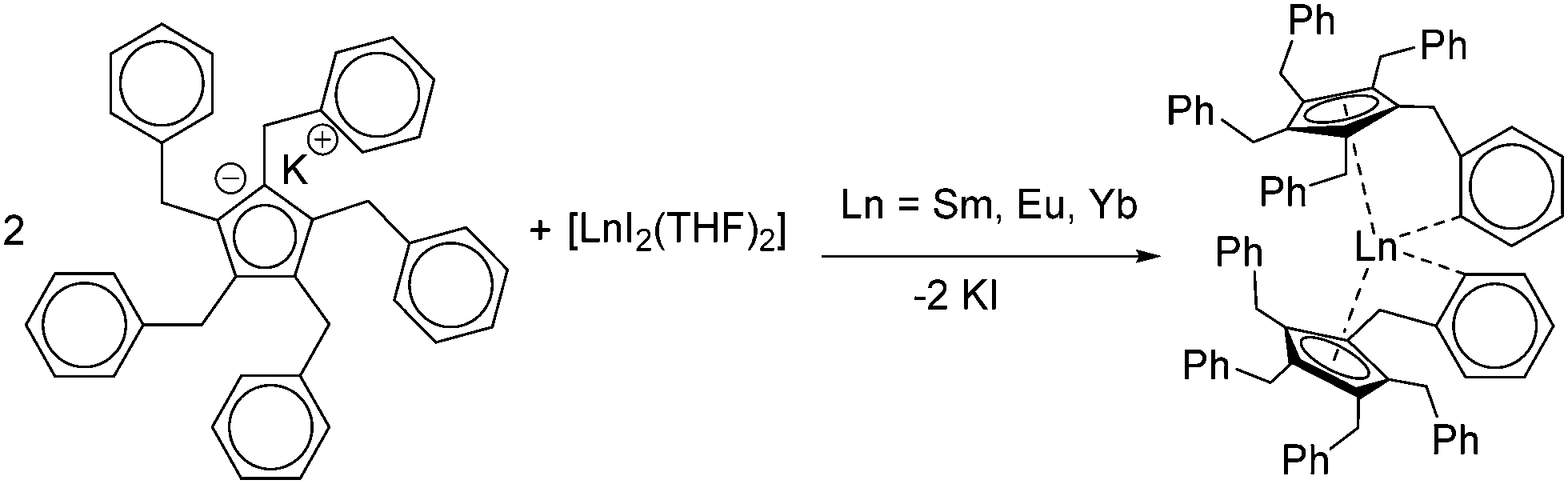 | ||
| Scheme 2 Synthesis [(CpBn5)2Ln] (Ln = Sm, Eu, Yb) via salt metathesis.36 | ||
The synthesis of [TmI2(DME)3] in 1997 paved the way for thulium in organometallic chemistry.37 However, metallocenes of thulium have not been synthesised with the pentamethylcyclopentadienyl ligand since bulkier ligands are needed to stabilise the highly reactive metal ions in the divalent oxidation state. In general, thulium metallocenes were synthesised similarly by salt metathesis from the corresponding alkali metal cyclopentadienyl derivatives and [TmI2(THF)3]. Thus, [(1,3-C5H3(SiMe3)2)2Tm(THF)],38 [(1,2,4-C5H2(SiMe3)2)3Tm(THF)],39 [(1,3-C5H3tBu2)2Tm],40 [(1,2,4-C5H2tBu3)2Tm],41 and [(1,2,4-C5H2tBu3)2Tm(THF)]7 were obtained by this route.
In contrast, all solution-based synthetic routes yield ethereal adducts of [(C5Me5)2Ln(solvent)n] (Ln = Sm, Eu, Yb). However, for some applications, it is necessary to start with the unsolvated, more reactive metallocenes. To obtain the solvent-free metallocenes of Sm and Eu, sublimation of the solvated compounds can be carried out. Evans and co-workers studied the synthesis of solvent-free samarocene.42 Upon heating [(C5Me5)2Sm(THF)2] to 85 °C at a pressure of 1 × 10−5 mbar, the compound desolvates readily and solvent-free samarocene was obtained in 75% yield as a sublimate. The desolvation is a two-stage process. The intermediate monosolvate [(C5Me5)2Sm(THF)] is formed first and was successfully isolated and confirmed by X-ray crystallography.43 In contrast, [(C5Me5)2Eu(THF)] does not release THF that easily and three consecutive sublimations at 85 °C and 1 × 10−5 mbar are necessary to obtain a THF-free product.44 In comparison, the solvent-free ytterbium compound cannot be obtained from the sublimation of either [(C5Me5)2Yb(THF)] or [(C5Me5)2Yb(OEt2)]. The base-free ytterbocene was obtained by vacuum removal of the solvent from a strong boiling toluene solution of [(C5Me5)2Yb(OEt2)].19 The desolvation of the THF-solvate is not possible with this method, indicating the strong bond between the oxygen and ytterbium that appears to be stronger than in the related Eu and Sm metallocenes. When bulkier substituted cyclopentadienyl ligands were used (e.g., [(1,3-C5H3(SiMe3)2)], the removal of the solvent is facilitated.45 In the case of very crowded cyclopentadienyl ligands (e.g. 1,2,4-C5H2b3, 1,2,4-C5H2(SiMe3)3, C5Ph5, CpBz5 (pentabenzylcyclopentadienyl) the metallocenes are obtained solvent free.29,36,46–49
It is also possible to synthesise the decamethylmetallocenes of Eu and Yb directly from the elements. Solutions of Yb or Eu metal in liquid ammonia reacted with HC5Me5 to first yield ammonia solvates of europocene and ytterbocene. Further extraction with THF or Et2O subsequently resulted in the formation of [(C5Me5)2Eu(THF)] or the mixed solvate, [(C5Me5)2Yb(NH3)(Et2O)] (Scheme 3).50 An analogous reaction with samarium metal has not been reported in the literature. [(C5Me5)2Eu(THF)2] was also synthesised from the metal and the plumbocene derivative [(C5Me5)2Pb].51
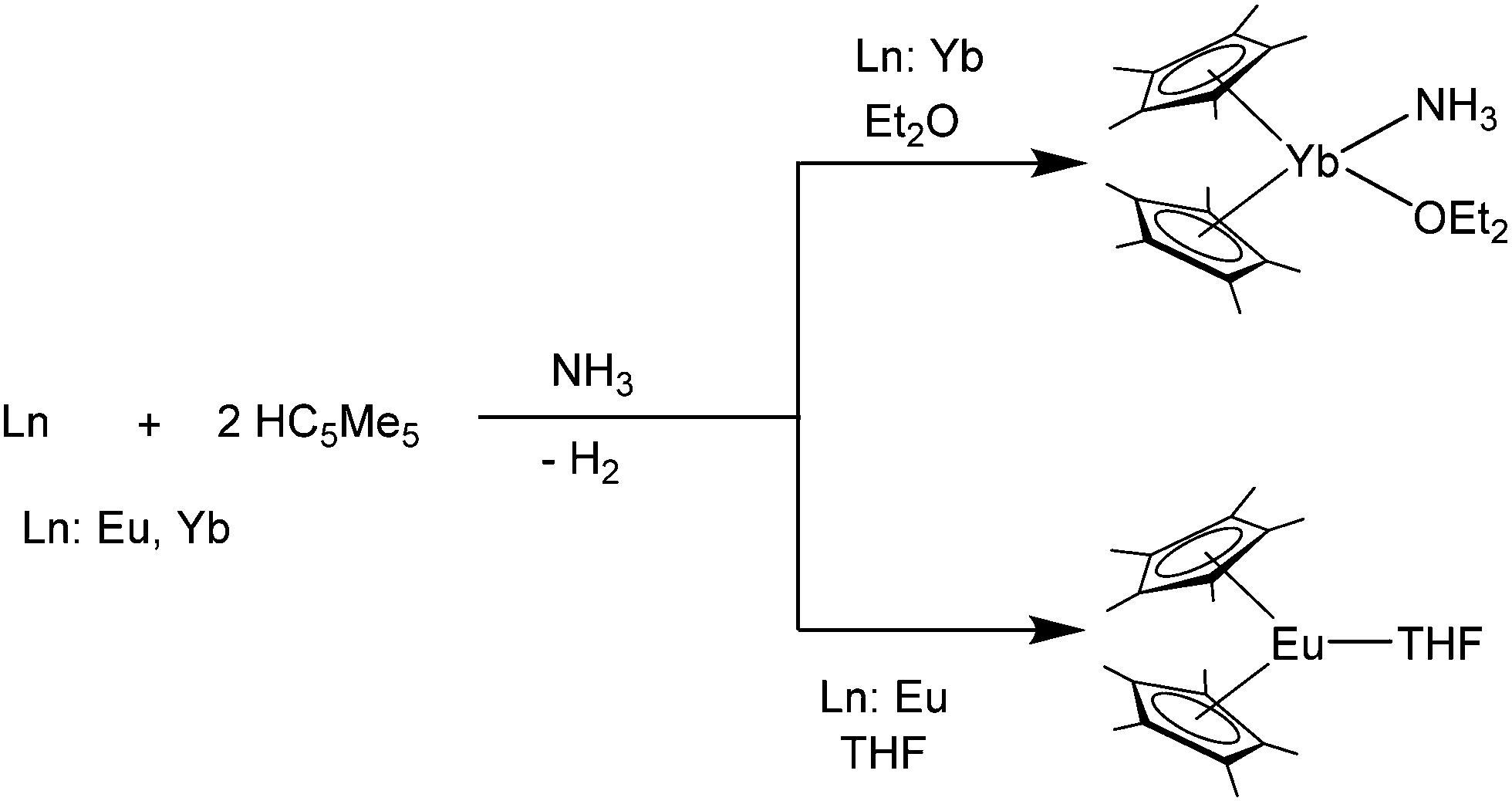 | ||
| Scheme 3 Syntheses of title compounds starting from metals Eu and Yb in liquid ammonia.50 | ||
The synthesis of metallocenes with very bulky cyclopentadienyl ligands is different in some cases due to sterical reasons. In some cases, the desired metallocenes were directly obtained from the metal. Thus, the reaction of Yb metal with two equivalents of the bulky pentaphenylcyclopentadiene ligand and one equivalent of diphenylmercury in THF at room temperature did not yield the sandwich complex [(C5Ph5)2Yb]. Instead, the ionic species [Yb(THF)6][C5Ph5]2 was obtained. However, the addition of non-coordinating solvents led to the desired sandwich complex [(C5Ph5)2Yb], which remained insoluble in any nonpolar solvent.47 This redox-transmetalation/protolysis (RTP) reaction was later extended to Sm and Eu.52 Thus, these metals react with one equivalent of HgPh2, and two equivalents of C5Ph5H to give at 40 °C for several days the decaphenyllanthanocenes [Ln(C5Ph5)2] (Ln = Sm, Eu). When using Hg(C6F5)2 in place of HgPh2 the reaction was performed at room temperature. The octaphenyllanthanocenes [Sm(C5Ph4H)2(THF)] and [Eu(C5Ph4H)2(DME)] were obtained likewise from HgPh2 and two equivalents of C5Ph4H2. A similar oxidative approach starting from the metal was used for the synthesis of the phosphine functionalised metallocenes [(η5-C5H4PPh2)2Eu(DIME)] and [(η5-C5H4PPh2)2Yb(DIME)] (DIME = diethylene glycol dimethyl ether). Here, [Tl(C5H4PPh2)] was reacted with metallic europium or ytterbium powder in THF in the presence of mercury, followed by crystallization from a solvent mixture of DME and DIME.53 In a similar way bis(tris(trimethylsilyl)cyclopentadienyl)europium was synthesised from europium powder and Tl(1,2,4-C5H2(SiMe3)3).49
A surprising alternative to this route to form tetra- and penta-phenylcyclopentadienyldiphenylphosphines was disclosed by Junk, Deacon, and co-workers. They underwent selective C–P bond cleavage with Eu, Sm, or Yb metal in the presence of catalytic amounts of I2 to give [(C5Ph4H)2Ln(solvent)] or [(C5Ph5)2Ln] (Scheme 4).54
 | ||
| Scheme 4 Syntheses of [(C5Ph4H)2Ln] or [(C5Ph5)2Ln].54 | ||
Also ansa-metallocenes were obtained directly from the metals, e.g., acenaphthylene reacted directly with activated Sm and Yb metal by samarium or ytterbium in THF to yield the respective C2-symmetric trans-rac-ansa-lanthanocene complexes [(η5-C12H8)2Ln(THF)2] (Ln = Sm, Yb) (Scheme 5).55
 | ||
| Scheme 5 Syntheses of [(η5-C12H8)2Ln(THF)2] (Ln = Sm, Yb).55 | ||
Besides salt metathesis and oxidation of lanthanide metals, a reductive approach is also known. Thus, [(C5H4Me)2Yb(DME)] was obtained by reducing [(C5H4Me)2YbCl] with metallic sodium,56 while reduction of [(C5H4SiMe3)2YbCl]2 with Na/Hg resulted in [(C5H4SiMe3)2Yb].57 Even the highly reactive thulium compound [(1,2,4-C5H2tBu3)2Tm(THF)] is accessible from [(1,2,4-C5H2tBu3)2TmI] with KC8 in toluene.7 Reduction of the corresponding Nd complex [(1,2,4-C5H2tBu3)2NdI] with KC8 in the presence of [18]crown-6 led to the divalent ate-complex [(1,2,4-C5H2tBu3)2Nd(μ-I)(K[18]crown-6)].9 This compound reacts with a methylene group from the (1,2,4-C5H2tBu3) ligand forming a “tuck-in” complex. Metallate complexes of dysprosium [(1,2,4-C5H2tBu3)2Dy(μ-X)(K[18]crown-6)] (X = BH4, Br, I) were obtained in similar way by reduction of the corresponding trivalent precursors (Scheme 6).8
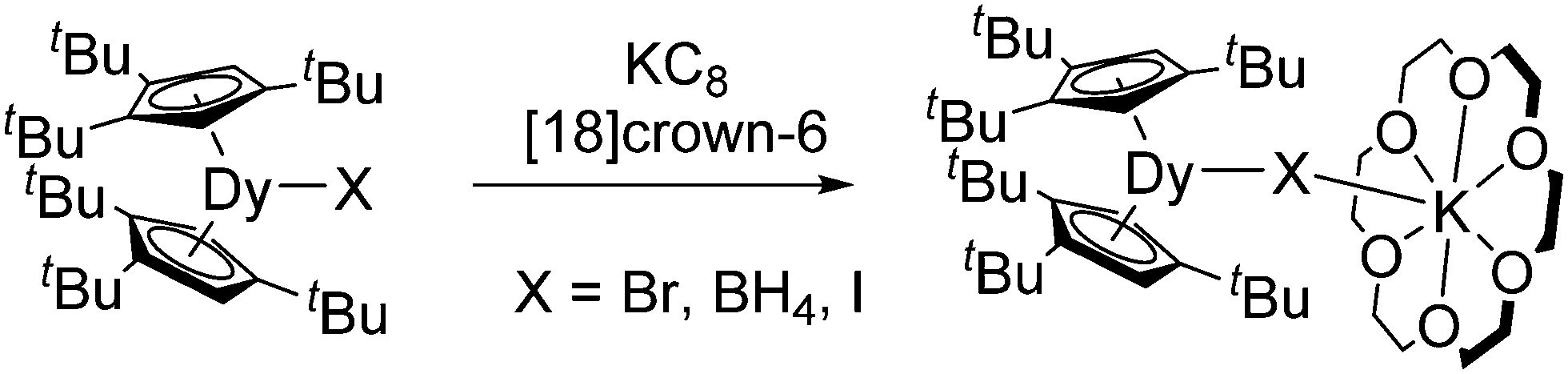 | ||
| Scheme 6 Synthesis of [(1,2,4-C5H2tBu3)2Dy(μ-X)(K[18]crown-6)] (X = BH4, Br, I).8 | ||
By using the bulkier ligand pentaisopropylcyclopentadienyl (CpiPr5) linear divalent metallocenes of almost all rare earth elements [(CpiPr5)2Ln] (Ln = Y, La, Ce, Pr, Nd, Gd, Ho, Er) can be synthesised by potassium graphite reduction of the corresponding iodine precursors [(CpiPr5)2 LnI] (Scheme 7).10,11 The Tm and Lu derivatives were obtained by in situ reduction. Thus, LnI3 was reacted with 2.5 equiv. of NaCpiPr5 first, followed by reduction with KC8 in benzene.11
 | ||
| Scheme 7 Synthesis of [(CpiPr5)2Ln] (Ln = Y, La, Ce, Pr, Nd, Gd, Ho, Er).11 | ||
Another common reductive approach to access donor functionalised metallocenes starts from the trivalent amido complexes [{(Me3Si)2N}3Ln(III)(μ-Cl)Li(THF)3] (Ln = Yb, Eu). These were mostly reacted with functionalised indenes or related ligands, which resulted in deprotonation of the ligand and concurrent reduction of the metal.58–61 As byproduct {(Me3Si)2N}2 is formed. Due to the higher redox potential, this reaction pathway does not work with samarium compounds.
A remarkable reductive approach was reported by Harder and co-workers.62 They reacted the benzyl complexes [(2-Me2N-benzyl)3Ln] (Ln = Sm, Yb) with the perarylated cyclopentadiene (4-nBu-C6H4)5C5H (CpBIGH) to obtain the divalent complexes [(CpBIG)2M] (M = Yb, Sm). The steric bulk of the ligand seems to be a driving force for the reduction process. Although the reaction mechanism could not be fully deduced, the formation of a 2-Me2N-benzyl radical was anticipated since 1,2-di(2-Me2N-phenyl)ethane was found as a major side product in the mother liquors. The ethyl and isopropyl derivatives [{(4-EtC6H4)5C5}2Sm] and [{(4-iPrC6H4)5C5}2Sm] were synthesised in a similar manner from the benzyl compounds [(DMAT)2Sm(THF)2] (DMAT = 2-Me2N-α-Me3Si-benzyl).63
The corresponding europium compound [(CpBIG)2Eu] was prepared in a simple protonation reaction from [Eu(DMAT)2(THF)2] with two equivalents of CpBIGH.64
3. Properties
3.1. Solid state structures of the base free metallocenes
[(C5Me5)2Ln] (Ln = Sm, Eu, Yb) have been structurally characterised in their unsolvated form in the solid state by single crystal X-ray crystallography.19,42,44 Instead of forming a coplanar sandwich structure, a “bent-metallocene” structure like in the corresponding alkaline earth metallocenes is preferred (Fig. 1). This bent structure is also observed in gas phase.65–67 The carbon–Ln distances in [(C5Me5)2Ln] decrease from Sm to Yb in agreement with the lanthanide contraction. However, due to the similarities of the ionic radii in the neighbouring elements Sm and Eu, the structural parameters of [(C5Me5)2Eu] and [(C5Me5)2Sm] are almost identical. For both metallocenes, the average Ln–C distance is 2.79(1) Å. [(C5Me5)2Yb] is obtained in two structural modifications, depending on the crystallization method. Only slight differences in the molecular bond lengths are observed for the two modifications. The corresponding Yb–C average distance is 2.66 Å, around 0.12 Å shorter than in the Sm- or Eu-metallocene. The bend angle centroid–Ln–centroid for [(C5Me5)2Sm] is 140.1° and for [(C5Me5)2Eu] 140.3°.42,44 With 145/146° the bend angle is slightly wider in the ytterbium compound.19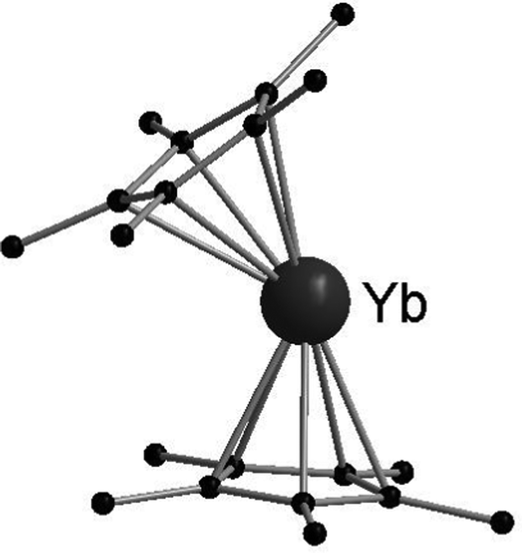 | ||
| Fig. 1 Molecular structure of [(C5Me5)2Yb] in the solid state featuring the bent structure of the metalloecene (Reproduced from the CIF file CCDC: 1242530).19 | ||
Note, that the structures of the pentamethylcyclopentadienyl compounds are closely related to the corresponding metallocenes of calcium and strontium, which also exhibit the bent structural motif. In the [(C5Me5)2Ca], the bending angle is 147° and the Ca–C distances are 2.609(6) Å, which is comparable to the corresponding angles and distances in the Yb-compound.68 [(C5Me5)2Sm] and [(C5Me5)2Eu] most likely exhibit structural resemblances to [(C5Me5)2Sr]. However, no solid state structural data is available for the Sr decamethylmetallocene.
The reason why the bent structures are preferred over the linear geometries both in the lanthanide decamethyl metallocenes and the corresponding alkaline earth metal compounds has been intensely discussed in the literature and investigated by theoretical methods. In principle, the following reasons have been discussed in the literature:
More recent relativistic, gradient-corrected density functional (DFT) calculations performed on [(C5Me5)2Yb] contradict earlier results gained from molecular mechanics force field calculations.72,73 Electrostatic and orbital interactions between the metal and the ring are identified as the main reason for the driving force away from linearity. Using this method, no bending was found for (C5Me5)2 unit with a central dummy atom.
In contrast to [(C5Me5)2Ln] compounds, metallocenes with bulkier substituents such as (C5Ph5) tend to form linear structures due to a steric clash of the substituents. Thus, single-crystal X-ray diffraction studies of [(C5Ph5)2Yb] exhibit a highly symmetric structure with two parallel cyclopentadienyl ligands in a staggered conformation.47 Detailed studies concerning the structure of [(CpBIG)2M] were performed. The propeller-like ligands have opposite chirality and interlock with each other. The ligands are located in a parallel fashion, however, the metals are bound in a nearly perfect η5,η5-fashion (Fig. 2). However, unusually high displacement factors of the metal atoms parallel to the ring planes were observed, resulting in bent Cpcentre-M-Cpcentre units.62 A linear structure with a pseudo-D5d symmetry was also observed for the pentaisopropylcyclopentadienyl compounds [(CpiPr5)2Ln] (Ln = Y, La, Ce, Pr, Nd, Eu, Sm, Gd, Ho, Er, Tm, Yb, Lu).10,11
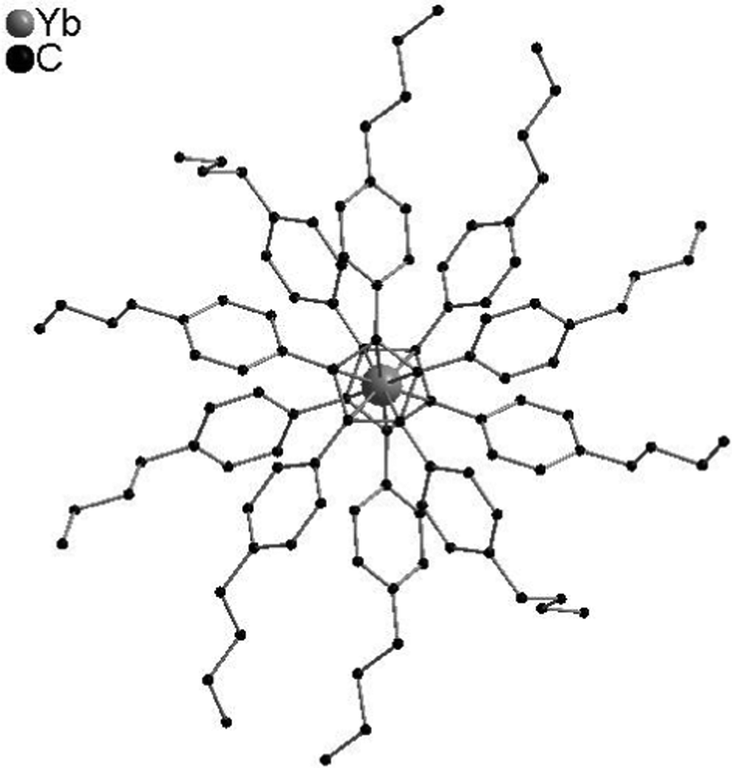 | ||
| Fig. 2 Molecular structure of [(CpBIG)2Yb] in the solid state showing the propeller-like ligand arrangement (Reproduced from the CIF file CCDC 665304).62 | ||
3.2. Bonding
For a long time, no disagreement was found on the bonding in [(C5Me5)2Ln]. As for the metallocenes of the alkaline earth metals, the bonding was supposed to be mainly ionic. No contribution of the f-electrons was observed. Relativistic DFT calculations on [(C5Me5)2Yb] revealed large charge separations between the ligands and the metal indicating significant ionicity in the compounds. The electron population in the f-orbitals were found to be close to 14, confirming no contribution of these orbitals to the binding.73,74 However, a more recent study using quantum chemical methods on the DFT level comparing [(C5Me5)2Sm] with [(C5Me5)2Sr] showed a substantial covalent interaction in the Sm compound. The authors further elaborate that the ligand–metal bond in lanthanide(II) complexes is in general partially covalent.753.3. Electronic structure
The electronic structures of [(CpiPr5)2Ln] (Ln = Y, La, Ce, Pr, Nd, Eu, Sm, Gd, Ho, Er, Tm, Yb, Lu) were investigated, e.g., by ultraviolet–visible (UV-Vis) spectroscopy.10,11 The results support the expected 4fn+1 electron configuration for Ln(II) = Sm, Eu, Tm, Yb and a 4fn5dz21 configuration for the other rare earth compounds ([Kr]4dz21 for Y(II)). EPR spectroscopy showed a significant s–d orbital mixing in the highest occupied molecular orbital and hyperfine coupling constants. Magnetic susceptibilities measured at room temperature suggests that the more pronounced 6s–5d mixing may be associated with weaker 4f–5d spin coupling.The oxidation state of the divalent complexes [(CpBIG)2M] (M = Eu, Yb, Sm) was confirmed by Harder and co-workers using various physical methods.64 Temperature-dependent magnetic susceptibility data of [Yb(CpBIG)2] confirmed the divalent oxidation state by showing diamagnetism. Temperature-dependent 151Eu Mössbauer investigations of [(CpBIG)2Eu] ranging from 93K to 215 K showed an agreement with other EuII species. Detailed analysis of the Mössbauer spectra provided information about the dynamics of the EuII ion within the sandwich complex. X-ray absorbance near edge spectroscopy (XANES) also confirmed the divalent oxidation state of [(CpBIG)2M] (M = Eu, Yb, Sm). Furthermore, [(CpBIG)2Eu] showed extremely bright orange emission under UV excitation at room temperature. The photoluminescence spectra of this compound also confirm the divalent oxidation state.
3.4. Other properties
An interesting feature of [(C5Me5)2Eu] is as mentioned above that Mössbauer spectra of 151Eu can be recorded with a 151Sm source.76 At 4.2 K and 77 K broad absorptions at 13 mm s−1 were observed. The broadness of the signals is explained by spherical paramagnetic relaxation that takes place at a rate comparable to the Mössbauer time scale of 9.7 × 10−9 s.The 171Yb isotope has a nuclear spin of 1/2 and a natural abundance of 14.27%. The gyroscopic moment is 4.712 × 107 rad T−1 s−1. Due to the full 14 f shell, Yb(II) ions are diamagnetic and therefore organometallic Yb(II) species can be well studied by NMR spectroscopy.77 For [(C5Me5)2Yb(Et2O)] a shift of 36 ppm with ω1/2 = 90 Hz was reported. The corresponding THF solvate gives a resonance at 87 ppm with ω1/2 = 24 Hz.
The metal-bond disruption energies were determined for [(C5Me5)2Sm] and [(C5Me5)2Sm(THF)2], which are 69.4(2.4) and 72.7(2.9) kcal mol−1, respectively.78
3.5. Influence of the substituents of the cyclopentadienyl ligand
As already pointed out, most metallocenes were synthesised using the pentamethylcyclopentadienyl ligand. On the one hand, bulkier substituents such as (1,2,4-C5H2tBu3) are needed to stabilise the highly reactive divalent thulium and dysprosium complexes. Bulky substituents therefore also reduce the reactivity. In a detailed study, the reactivity of [{(4-EtC6H4)5C5}2Sm] and [{(4-iPrC6H4)5C5}2Sm] was compared with [(C5Me5)2Sm] and [(C5Me5)2Sm(THF)n]. Not too surprisingly, the strongly shielded Sm(II) compounds showed significantly lower reactivity.63 By using pentaisopropylcyclopentadienyl linear divalent metallocenes of almost all rare earth elements [(CpiPr5)2Ln] (Ln = Y, La, Ce, Pr, Nd, Eu, Sm, Gd, Ho, Er, Tm, Yb, Lu) are accessible.11In general, the ansa-samarocenes feature structures with a stronger bending of the ligands and hence a wider biting angle. As a result, they showed higher activity and selectivity as catalysts for the polymerization of ethylene and 1-olefines.32
4. Lewis acid–base complexes
Due to the large size of the Sm(II), Eu(II) and Yb(II) ions in the (C5Me5)2Ln moieties, all three compounds act as Lewis acids. The reactivity in this case is determined by the ionic radii and is similar for all three compounds. Like their relatives [(C5Me5)2Ca] and [(C5Me5)2Sr], [(C5Me5)2Ln] qualify as hard Lewis acids. The synthesis of Lewis acid–base complexes can be generalised (Scheme 8). Usually, the simple combination of the desired ligand and [(C5Me5)2Ln(solvent)n] in a suitable solvent leads to the formation of the anticipated complex. However, in some cases it is necessary to use the solvent-free reactants, e.g. for the preparation of complexes with ligands containing rather soft donor atoms that form weaker bonds with the hard lanthanide atoms. As for the geometry of the Lewis base adducts, either one or two additional neutral ligands can coordinate to the [(C5Me5)2Ln] fragment, depending on the steric demand of the Lewis bases and the size of the metal. Besides Lewis acid–base complexes also some protolysis and substitution reactions are discussed in the chapter. | ||
| Scheme 8 General synthesis routes to Lewis acid–base adducts of the divalent lanthanocenes. | ||
4.1. Oxygen donor ligands
As hard Lewis's acids, the metallocenes form stable complexes with oxygen donors, e.g. ethers. All three pentametyhlcyclopentadienyl derivatives were first reported as solvates of THF or diethylether.13,14 These solvates are the most commonly used reactants, not only because of their lower reactivity, but also because they can be easily obtained by the aforementioned reactions in the respective ethers. The crystal structure of [(C5Me5)2Sm(DME)] solvated with DME was also reported.79 Selected (C5Me5)2Sm-ether complexes are depicted in Scheme 9 with the corresponding Sm–O bond lengths.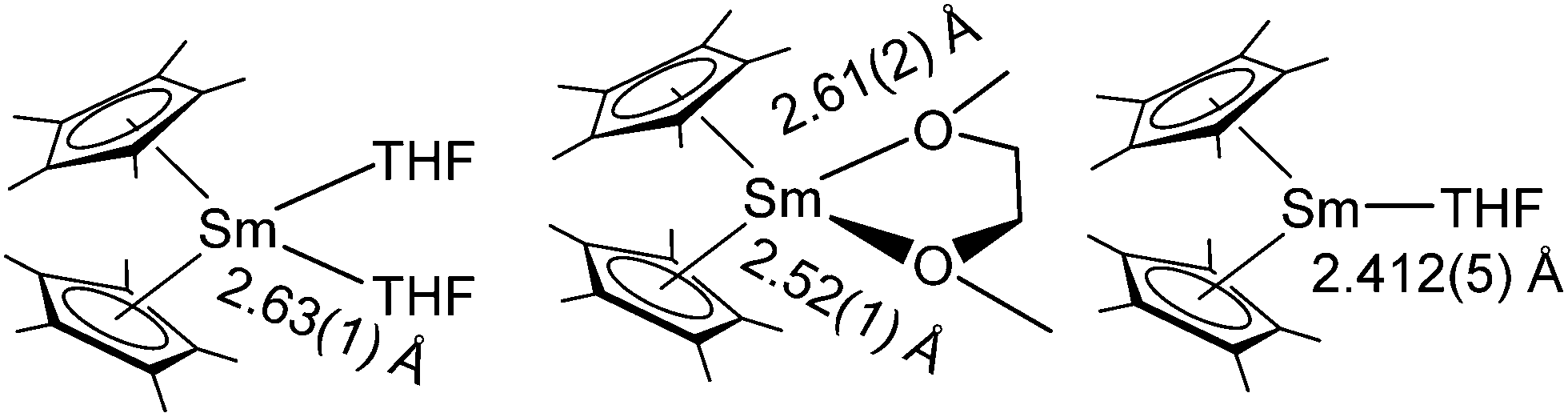 | ||
| Scheme 9 Ethereal adducts of samarocene.14,79,80 | ||
In order to gain more information about the reactivity of samarium(II), a bis(dihydropyran) and a mono(tetrahydropyran) complex were synthesised.80 [(C5Me5)2Sm(OC5H10)] was obtained by dissolving [(C5Me5)2Sm(THF)] in tetrahydropyran (OC5H10). After evaporation, the obtained maroon solid was dissolved in toluene and evaporated again to yield the dark brown solid of [(C5Me5)2Sm(OC5H10)]. [(C5Me5)2Sm(OC5H8)2] (OC5H8 = 3,4-dihydro-2H-pyran) was synthesised by dissolving [(C5Me5)2Sm(THF)2] in toluene and evaporating the solvent until the solid monosolvate [(C5Me5)2Sm(THF)] was present. The solid was dissolved in 3,4-dihydro-2H-pyran. After evaporation, [(C5Me5)2Sm(OC5H8)2] was obtained quantitatively. Investigation of the physical properties of both complexes indicated the presence of samarium(II). The molecular structure determined by X-ray methods revealed Sm–O bond lengths of 2.655(6) and 2.699(7) Å for [(C5Me5)2Sm(OC5H8)2] and 2.630(6) and 2.770(9) Å for [(C5Me5)2Sm(OC5H10)]. Divalent complexes have Sm–O bond lengths in the range of 2.62(2)–2.699(7) Å, whereas trivalent complexes range from 2.44(2) to 2.511(4) Å.
Phosphine oxides were also employed as oxygen donors as ligands. [(C5Me5)2Sm(OPPh3)(THF)] was reported by Evans.81 It was prepared by combining a solution of Ph3PO and [(C5Me5)2Sm(THF)2] and isolated as black crystals. The compound was identified by 1H NMR and IR spectroscopy, magnetic moment measurements and elemental analysis. No solid state structure is available. The syntheses of the phosphine oxide complexes [(C5Me5)2Yb(OPMe3)] (yellow orange) and [(C5Me5)2Yb(OPMe3)2] (orange) were reported as well.82 They were prepared by adding either one or two equivalents of OPMe3 to a toluene solution of [(C5Me5)2Yb]. The solution behaviour was studied by variable temperature 1H and 31P NMR spectroscopy, showing slow intermolecular exchange at room temperature. THF is competitive with phosphine oxide ligands, while diethylether is not. Addition of diethylether to a toluene solution of the monophosphine oxide complex results in precipitation of [(C5Me5)2Yb(OPMe3)2] and concomitant formation of [(C5Me5)2Yb(OEt2)]. The authors explain this by facilitated molecular exchange via intermediate [(C5Me5)2Yb(OPMe3)(OEt2)]. Despite isolated as crystalline solids, no X-ray crystallographic studies are available.
Reactions of [(C5Me5)2Sm(THF)2] with the anionic O-donors KOAr (OAr = OC6H2tBu2-2,6-Me-4, OC6H3iPr2-2,6) in THF resulted in the ionic Sm(II) phenolate complexes [(C5Me5)Sm(THF)2(OAr)(μ-C5Me5)K(THF)2]n, in which a polymeric structure is formed (Scheme 10).83 A similar reaction was seen by using the thiolate SC6H2iPr3-2,4,6 as reagent. The obtained polymeric structural motif, which is also seen by using carbanions as well as N- and P-donors, is frequently observed (see Schemes 14 and 23). Within these polymeric structures “intermolecular” interactions between the potassium atom and the C5Me5 ligands are observed.
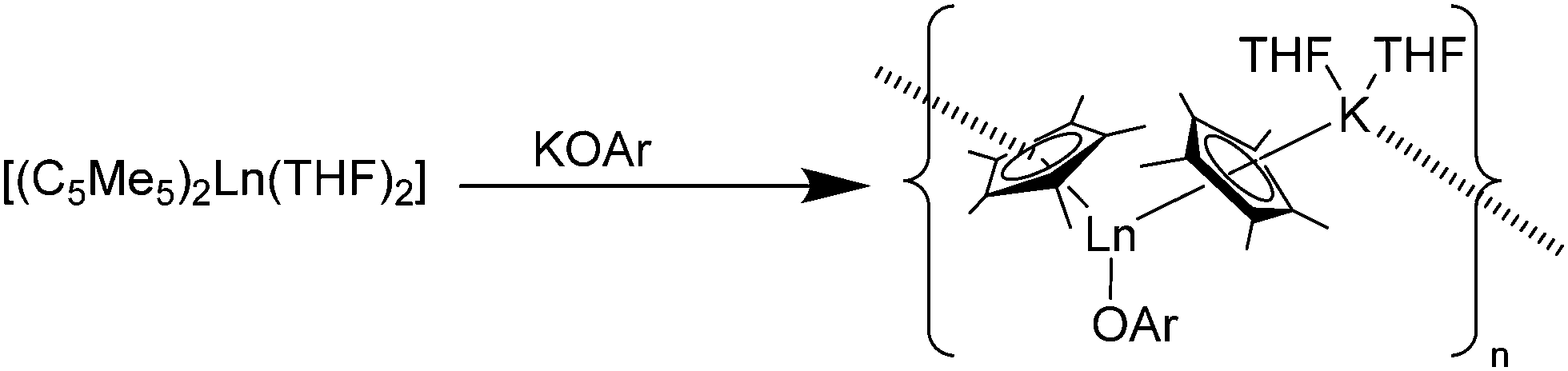 | ||
| Scheme 10 Polymeric chain in [(C5Me5)Sm(THF)2(OR)(μ-C5Me5)K(THF)2]n.83 | ||
Dimeric, divalent lanthanide complexes [(C5Me5)Sm(μ-OAr)]2 (Ar = C6H3tBu2-2,6, C6H2tBu2-2,6-Me-4 and C6H2tBu3-2,4,6)) can be obtained by diffusion of ArOH into a toluene solution of [(C5Me5)2Sm(THF)2] (Scheme 11).84 Interestingly, the samarium atom is not oxidised but one of the C5Me5 ligands is protonated. Single crystal X-ray analysis of [(C5Me5)Sm(μ-OAr)]2 with Ar = C6H2tBu3-2,4,6 showed that it is an unsolvated dimeric samarium(II) complex with mixed ArO and C5Me5 ligands with an Sm(μ-O)2Sm unit, which is exactly planar. However, the μ-OAr bridges are unsymmetric. The bond distance of the Sm(1)–O(1) bond (2.425(5) Å) is significantly shorter than that of the Sm(1)–O(1′) bond (2.512(6) Å). The solubility of these three complexes in toluene follows the order C6H2tBu3-2,4,6 > C6H2tBu2-2,6-Me-4 > C6H3tBu2-2,6. There are two other synthetic routes that yield [(C5Me5)Sm(μ-OAr)]2. In addition to the above-mentioned route, the reaction of [(C5Me5)2Sm(THF)2] and [Sm(OAr)2(THF)3] leads to the same complexes (Scheme 11). Divalent lanthanide complexes with both ArO and C5Me5 ligands are extremely rare. When [(C5Me5)Sm(μ-OAr)]2 (Ar = C6H2tBu3-2,4,6) is exposed to a trace amount of air, the trivalent samarium complex [(C5Me5)2Sm(OAr)] is obtained.
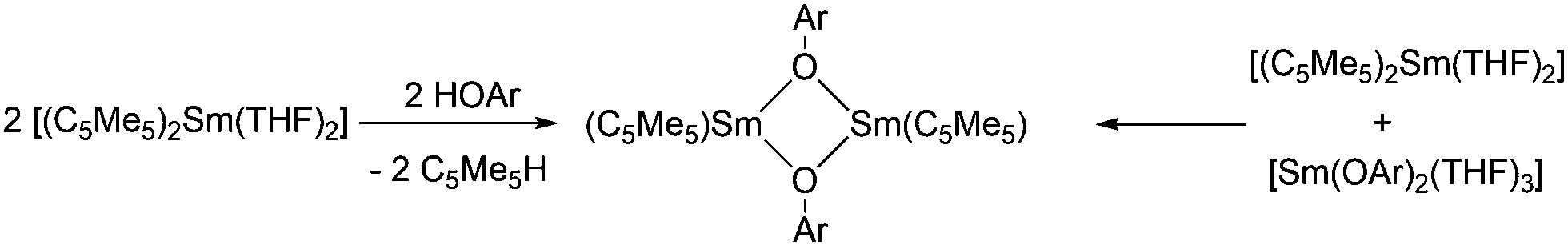 | ||
| Scheme 11 Synthesis of [(C5Me5)Sm(μ-OAr)]2.84 | ||
In addition to the reaction with alcohols, a rather unusual dinuclear samarium compound was reported from a protolysis reaction of [(C5Me5)2Sm(THF)2] with the bulky silanole (tBuO)3SiOH.85 By reacting both compounds in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 stoichiometric ratio in toluene, green crystals of the dinuclear siloxide-bridged species [(C5Me5)Sm{μ-OSi(OtBu)3}3Sm] were isolated in 93% yield (Scheme 12). The two Sm(II) atoms are bridged by three -OSi(OtBu)3 ligands. During the reaction, one pentamethylcyclopentadienyl ligand is cleaved and one samarocene is completely protolysed. Thus, one of the Sm centres is coordinated by a C5Me5 ligand and three bridging OSi(OtBu)3 units, while the second Sm(II) atom is surrounded only by these bridging silanols. The latter Sm(II) atom is additionally coordinated by three of the Si–O–C oxygen atoms of the silanols, creating a six-fold coordination sphere. The bridging Sm–O distances are in the range of 2.432(3)–2.523(3) Å. The reactivity of this complex was further investigated.85
:3 stoichiometric ratio in toluene, green crystals of the dinuclear siloxide-bridged species [(C5Me5)Sm{μ-OSi(OtBu)3}3Sm] were isolated in 93% yield (Scheme 12). The two Sm(II) atoms are bridged by three -OSi(OtBu)3 ligands. During the reaction, one pentamethylcyclopentadienyl ligand is cleaved and one samarocene is completely protolysed. Thus, one of the Sm centres is coordinated by a C5Me5 ligand and three bridging OSi(OtBu)3 units, while the second Sm(II) atom is surrounded only by these bridging silanols. The latter Sm(II) atom is additionally coordinated by three of the Si–O–C oxygen atoms of the silanols, creating a six-fold coordination sphere. The bridging Sm–O distances are in the range of 2.432(3)–2.523(3) Å. The reactivity of this complex was further investigated.85
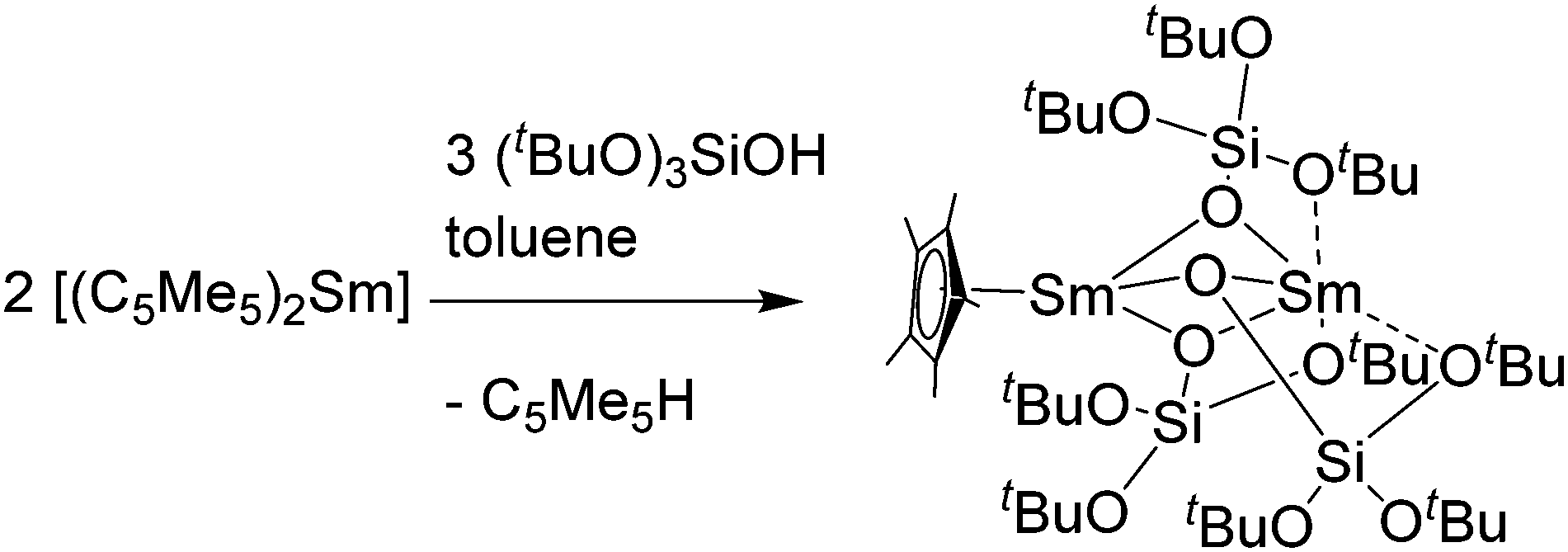 | ||
| Scheme 12 Reaction of [(C5Me5)2Sm(THF)2] with (tBuO)3SiOH.85 | ||
4.2. Monodentate nitrogen ligands
Complexes are also formed with various nitrogen donor containing ligands. However, amine complexes of the metallocenes are rarely reported in the literature.When [(C5Me5)2Yb] is synthesised in ammonia (see above), a mixed ammonia–THF adduct was formed after work up in THF.50 The small ammonia ligand seems capable of coordinating to the small Lewis acidic Yb ion. Interestingly, the analogous reaction with [(C5Me5)2Eu] yields the mono-THF solvate despite the larger ionic radius. Here, the more pronounced Lewis acidic character of the smaller Yb(II) could be the reason for the additional ammonia ligand (Scheme 3).
Green [(C5Me5)2Sm(tBuNH2)] was prepared from [(C5Me5)2Sm] and tert-butylamine in toluene, but not from (C5Me5)2Sm–THF solvates (Scheme 13).86 The amine in this case is a weaker donor than THF. When the reaction was carried out in the amine as solvent, a purple solid was obtained that was most likely [(C5Me5)2Sm(tBuNH2)2], but no conclusive data were collected. The solid state structure of [(C5Me5)2Sm(tBuNH2)] was determined by single crystal X-ray crystallography, which revealed Sm–N distances of 2.804(10) and 2.737(7) Å for the two independent molecules in the asymmetric unit. In the same study the reaction with N-methyl imidazole (N-MeIm) with [(C5Me5)2Sm(THF)2] is reported. This ligand can replace THF by forming the purple double substituted complex [(C5Me5)2Sm(N-MeIm)2] (Scheme 13). The solid state structure reveals Sm–N distances of 2.618(10) and 2.673(10) Å. The authors note the resemblance to the structure of [(C5Me5)2Sm(THF)2].
 | ||
| Scheme 13 Reaction of tert-butyl amine and N-methyl imidazole with samarocene.86 | ||
Although the reaction of samarocene with nitriles usually leads to redox reactions yielding Sm(III) products (see below), it is possible to isolate Lewis acid–base complexes from Sm(II) and tert-butyl nitrile.87 Combining [(C5Me5)2Sm(THF)2] and Me3CCN in THF and subsequent cooling yields brown crystalline [(C5Me5)2Sm(NCCMe3)2(THF)] (Fig. 3). Structural data was obtained by single crystal X-ray crystallography to reveal a rather unusual coordination of three additional neutral ligands to the (C5Me5)2Sm fragment. The N or O donor atoms of THF and Me3CCN create a plane perpendicular to the C5Me5–Sm–C5Me5 axis. The N–Sm distances are 2.735(3) Å and the Sm–O bond length is 2.716(3) Å. If the analogous reaction is carried out in toluene as solvent, green crystals of the THF-free compound [(C5Me5)2Sm(NCCMe3)2] were obtained in addition to other unstable products. Though the connectivity of the atoms was unambiguously determined, the structural data was too poor to discuss metric parameters.
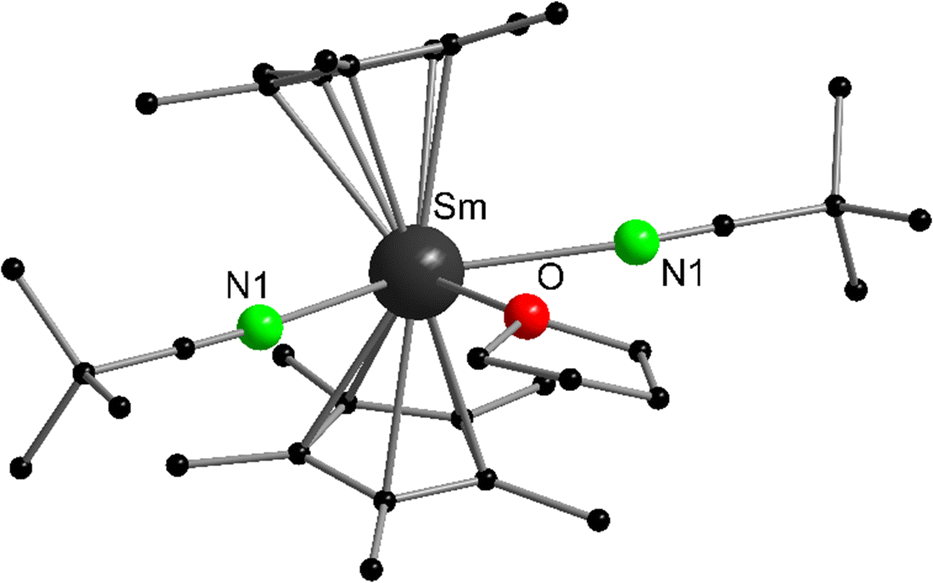 | ||
| Fig. 3 Molecular structure of [(C5Me5)2Sm(NCCMe3)2(THF)] in the solid state emphasizing the unusual coordination of three additional ligands (Reproduced from the CIF file CCDC: 637938).87 | ||
Lewis-acid–base interaction is also possible with phosphine imines; in particular, the interaction of the phosphine imine Et3PNH with [(C5Me5)2Yb] was investigated.82 The ligand behaves similar to the isoelectronic phosphine oxide ligands (see above). The 1:1 complex is formed during the reaction of Et3PNH and [(C5Me5)2Yb] in toluene. The solution behaviour of [(C5Me5)2Yb(Et3PNH)] was studied by variable temperature, multinuclear NMR spectroscopy. The ligand shows slow intermolecular exchange at room temperature. If a second equivalent of Et3PNH is added, the 2:1 complex [(C5Me5)2Yb(Et3PNH)2] precipitates as an orange solid from solution at −80 °C. No X-ray crystallographic data is available.
The first pyridine adduct of the metallocenes is [(C5Me5)2Yb(NC5H6)2].88 It is prepared by reaction of excess pyridine with [(C5Me5)2Yb(OEt2)] in toluene. The complex was isolated as dark green prisms that were investigated with X-ray crystallography. The Yb–N bond lengths are 2.585 and 2.544 Å.
Nocton and co-workers disclosed a number of N-donor adducts of [(1,3-C5H3tBu2)2Sm]. The N-aromatic heterocycles, pyridine, picoline, 4-tert-butyl-pyridine, isoquinoline and quinolone were coordinated to the metal atom via the N-atom.89 As magnetic measurements show, the electronic structure of Sm(II) of these compounds is f6. Thus, the simple coordination adducts were formed and no redox reaction took place.
Reactions of [(C5Me5)2Sm(THF)2] with the anionic N-donors K(NRR′) (NRR′ = NHC6H2tBu3-2,4,6 and N(SiMe3)2) in THF resulted in the ionic Sm(II) complexes [(C5Me5)Sm(THF)x(NRR′)(μ-C5Me5)K(THF)2]n, in which a polymeric structure is formed (Scheme 14).83 In this structure, a potassium cation bridges the complexes by coordinating with one of each of the neighbouring C5Me5 ligands. In a similar reaction of [(C5Me5)2Yb(THF)2] with KN(SiMe3)2 the corresponding Yb(II) complex [(C5Me5)Yb(N(SiMe3)2)(μ-C5Me5)K(THF)2]n was obtained.
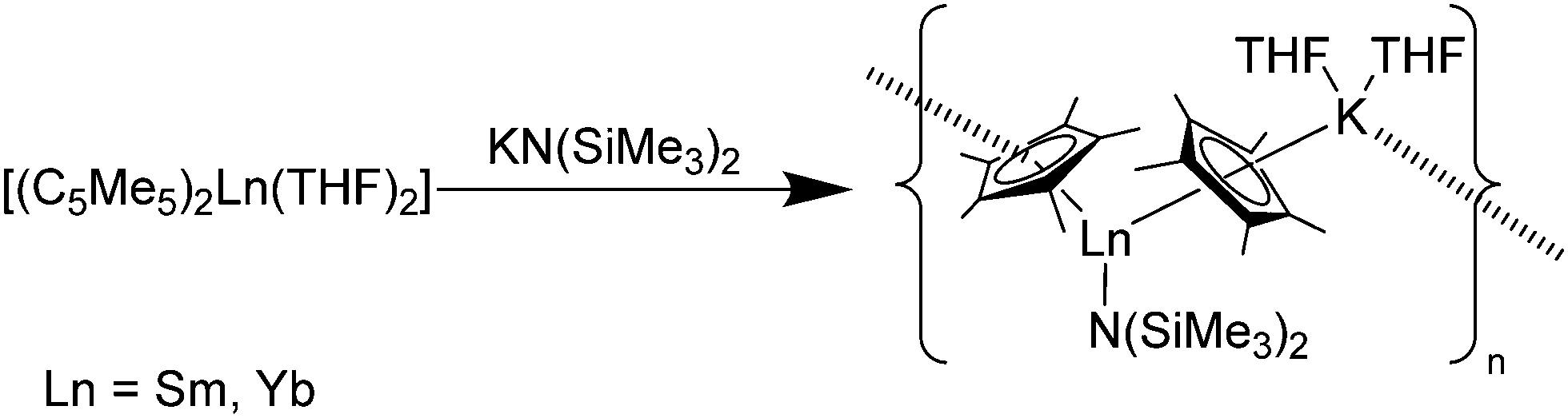 | ||
| Scheme 14 Polymeric chain in [(C5Me5)Ln(N(SiMe3)2)(μ-C5Me5)K(THF)2]n (Ln = Sm, Yb).83 | ||
4.3. Bipyridine and terpyridine, and related systems
Complexes of 2,2’-bipyridine (bipy) with all three classical metallocenes were reported in literature.36,90,91 They are prepared by the combination of the ether containing precursors, i.e. [(C5Me5)2Ln(OEt2)], in a suitable solvent. The Sm and Yb (red-brown) complexes have been structurally characterised. The nitrogen-Yb bond lengths are 2.324 and 2.318(5) Å, while for the larger Sm ion the two distances are 2.427(2) and 2.436(2) Å, respectively. It is worth noting that due to their strong reducing properties, Yb and Sm are able to transfer electron density to the bipyridine, thereby reducing it to the radical ligand. This property is more pronounced in the corresponding samarium complexes, while for ytterbium bipy the concept of intermediate valence tautomerism was coined by Andersen, meaning that the ground state consists of a [(C5Me5)2Yb(II)(bipy)] and a [(C5Me5)2Yb(III)(bipy)˙−] in different ratios.92 The amount of electron density transferred to the corresponding ligand is also determined by the ligand, e.g. phenanthroline is reduced to the corresponding radical ligand,93 while the oxidation state of ytterbium in the bipyridine-complex is between two and three. Bipy-complexes and complexes with bipy-related ligands such as terpyridine94 or phenanthroline93 have been extensively investigated, but a full review would go beyond the scope of this chapter. The novel charge-transfer from Yb(II) to nitrogen-containing aromatic ligands was also studied in 2:1 metal-to-ligand adducts of the type [Yb(II)–(ligand)–Yb(II)(C5Me5)2] (ligand = tetra(2-pyridyl)pyrazine (tppz), 6′,6′′-bis(2-pyridyl)-2,2′:4′,4′′:2′′,2′′′-quaterpyridine and 1,4-di(terpyridyl)-benzene (dtb)).95
Unique complexes were obtained by reaction of different tris(2-pyridyl)stannate derivatives with the Ln(II) metallocenes. Thus, treatment of [LiSn(2-C5H3N-5-Me)3(THF)] with [(C5Me5)2Ln(OEt2)] (Ln = Eu, Yb) resulted in LnII sandwich complexes [Ln{Sn(2-C5H3N-5-Me)3}2] comprising the anionic tris(pyridyl)-stannate as a κ3N-coordinating ligand (Scheme 15).96 Further reaction of [Yb{Sn(2-C5H3N-3-Me)3}2] with another equivalent of [(C5Me)2Yb(OEt2)] resulted in a ligand rearrangement and the mixed complex [(C5Me5)Yb{Sn(2-C5H3N-3-Me)3}] (Scheme 15).97
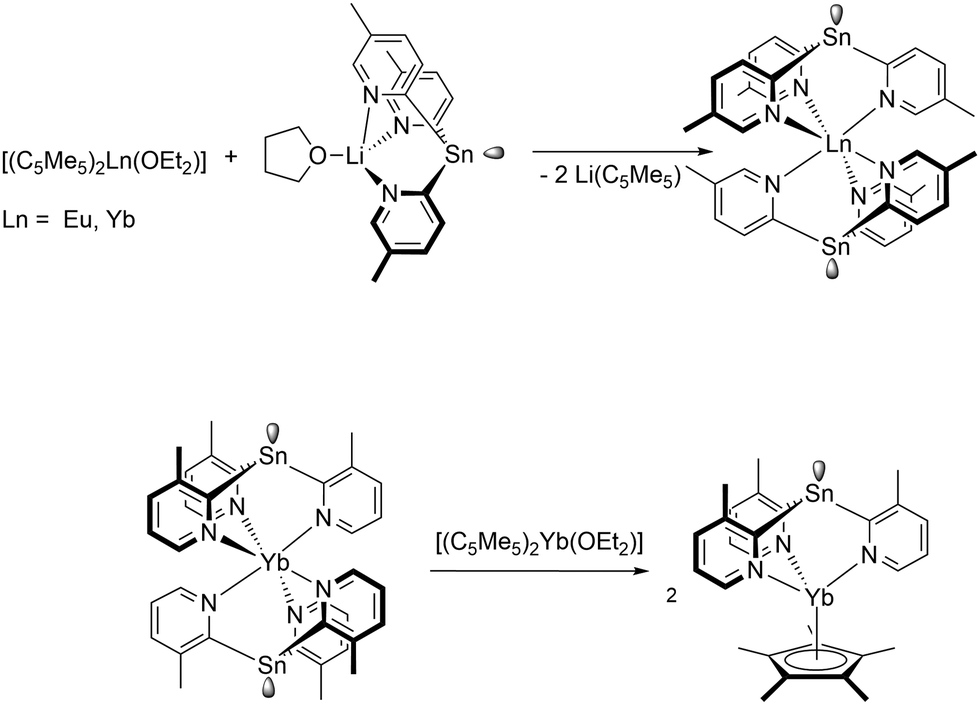 | ||
| Scheme 15 Reaction of a tris(2-pyridyl)stannate derivatives with [(C5Me5)2Ln(OEt2)] (Ln = Eu, Yb).96,97 | ||
The divalent tin atom in the ligand backbone is suitable to coordinate to other Lewis-acid complexes, such as [(C5H5)3La].98 The resulting ligand [(C5H5)3La{Sn(2-py5Me)3Li(THF)}] (py5Me = C5H3N-5-Me) gave in the presence of [(C5Me5)2Yb(OEt2)] via substitution of the (C5Me5)-rings the pentametallic complex [Yb{Sn(2-py5Me)3La(C5H5)3}2], in which the two tris(2-pyridyl)stannate units and two unsupported Sn–La bonds in terminal positions encapsulate the Yb2+ cation (Scheme 16).97 Similarly, the group 13 adducts [{Li(THF)Sn(2-py5Me)3}MEt3] (M = Ga, In), which exhibit long Sn–M bonds, afforded with [(C5Me5)2Eu(OEt2)] the corresponding compounds [Eu{Sn(2-py5Me)3MEt3}2] (Scheme 16).99 A complete different reactivity was seen by using [(C5Me5)2Sm(OEt2)] as precursor. Instead of a (C5Me5)-ring substitution, a redox reaction, which alters the tin ligand was seen and [(C5Me5)2Sm{MEt2(2-py5Me)2}] (M = Ga, In) were formed as products.97
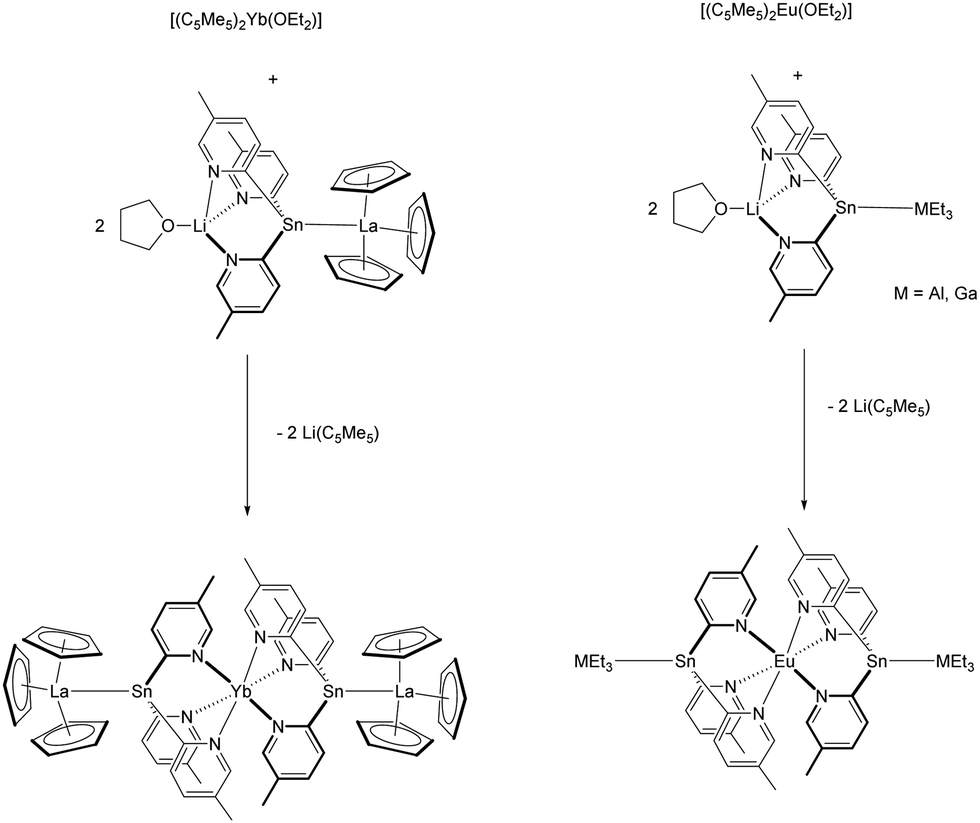 | ||
| Scheme 16 Reaction of a tris(2-pyridyl)stannate adduct with (C5Me5)2Ln(OEt2)] (Ln = Eu, Yb).97,99 | ||
4.4. Ytterbium – transition metal complexes bridged by redox non-innocent N-donor ligands
An elegant method for studying the communication between a divalent organo-lanthanide fragment and a transition metal complex is the use of redox non-innocent N-donor ligands as bridge. Thus, in a related synthetic method as described in the previous chapter [(C5Me5)2Yb(OEt2)] was reacted with palladium complexes, [(bipym)PdMe2] (bipym = bipyrimidine) and [(taphen)PdMe2] (taphen = 4,5,9,10-tetraazaphenanthrene) to give the heterobimetallic complexes [(C5Me5)2Yb(bipym)PdMe2] and [(C5Me5)2Yb(taphen)PdMe2], respectively (Scheme 17). In these complexes, an electron is transferred from the ytterbocene fragment to the ligand, increasing the electron-donating properties of the ligands. As shown by oxidative addition of MeI, this strongly influences the reactivity of the Pd species.100 | ||
| Scheme 17 Synthesis of the heterobimetallic complexes [(C5Me5)2Yb (bipym)PdMe2] and [(C5Me5)2Yb(taphen)PdMe2].100 | ||
In addition to the Pd complex, also the corresponding Ni complex [(bipym)NiMe2] was treated with [(C5Me5)2Yb(OEt2)]. This resulted in the analogue heterobimetallic complex [(C5Me5)2Yb(bipym)NiMe2], which was further reacted with CO to give [(C5Me5)2Yb(bipym)Ni(CO)2] and acetone. By comparison with the reactivity of the parent [(bipym)NiMe2] complex, it was shown that the divalent lanthanide fragment has a strong influence on the reaction kinetics.101 [(C5Me5)2Yb(bipym)NiMe2] was also used as very efficient catalyst for alkene isomerization in the presence of catecholborane.102
In another study, deprotonated 2-pyrimidin-2-yl-1H-benzimidazole (Hbimpm) was coordinated with a NiMe2 fragment. The corresponding ionic complex [K(bimpm)NiMe2] was subsequently reacted with [(C5Me5)2Yb(OEt2)]. Instead of a simple addition product, the bimpm underwent a coupling reaction, which is a result of the single electron transfer process from the {(C5Me5)2Yb} fragment.103
Intramolecular electron transfer was also observed in a complex in which a nickel atom and a {(C5Me5)2Yb} fragment are bridged by a macrocyclic biquinazoline ligand (Mabiq). The mixed Yb–Ni complex, [(C5Me5)2Yb(Mabiq)Ni]B(C6F5)4, was synthesised upon reaction of [Ni(II)(Mabiq)]B(C6F5)4 with [(C5Me5)2Yb(OEt2)] (Scheme 18). As supported by spectroscopic studies the complex is best described as [(C5Me5)2Yb(III)(Mabiq˙)Ni(II)]+ with a ligand-centred radical delocalised over both the diketiminate and bipyrimidine units of the Mabiq ligand.104
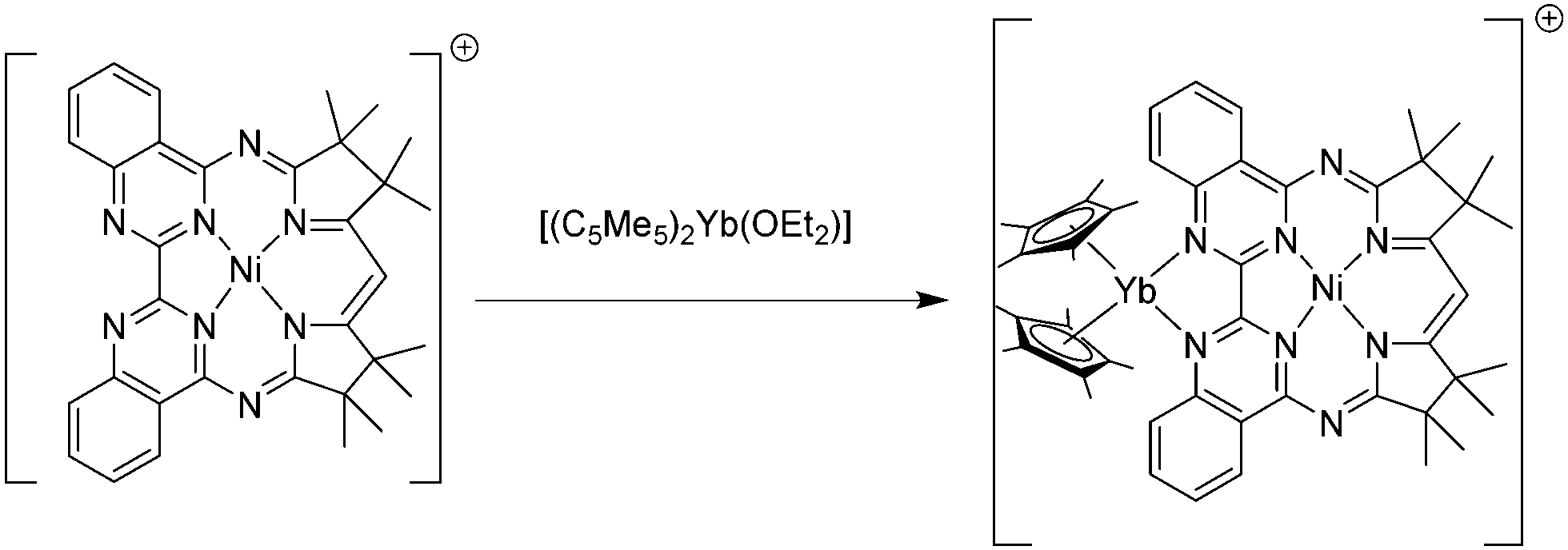 | ||
| Scheme 18 Synthesis of [(C5Me5)2Yb(Mabiq)Ni(II)]+.104 | ||
Electronic and magnetic communication was also investigated between trimetallic mixed actinide–lanthanide molecular complexes. The target compounds [(C5Me5)2An{N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH2C6H5)(tpyYb(C5Me4R)2)}]2 (An = Th, U; R = Me, Et; tpy = terpyridine) were obtained by reacting uranium(IV)- and thorium(IV)-bis(ketimide) complexes with [(C5Me5)2Yb(OEt2)]105 and [(C5Me4Et)2Yb(OEt2)] (Scheme 19).106 As linker between the metals terpyridyl-functionalised ketimides were employed. The Yb-ions show a valence equilibria between the divalent and the trivalent oxidation state and exhibit rich electrochemical behaviour consistent with electronic coupling between the actinide and Yb(II/III)tpy˙− moieties. Magnetic studies of the uranium complex indicate a coupled magnetic state between the U(IV) and Yb(III)tpy˙− groups at low temperatures.
C(CH2C6H5)(tpyYb(C5Me4R)2)}]2 (An = Th, U; R = Me, Et; tpy = terpyridine) were obtained by reacting uranium(IV)- and thorium(IV)-bis(ketimide) complexes with [(C5Me5)2Yb(OEt2)]105 and [(C5Me4Et)2Yb(OEt2)] (Scheme 19).106 As linker between the metals terpyridyl-functionalised ketimides were employed. The Yb-ions show a valence equilibria between the divalent and the trivalent oxidation state and exhibit rich electrochemical behaviour consistent with electronic coupling between the actinide and Yb(II/III)tpy˙− moieties. Magnetic studies of the uranium complex indicate a coupled magnetic state between the U(IV) and Yb(III)tpy˙− groups at low temperatures.
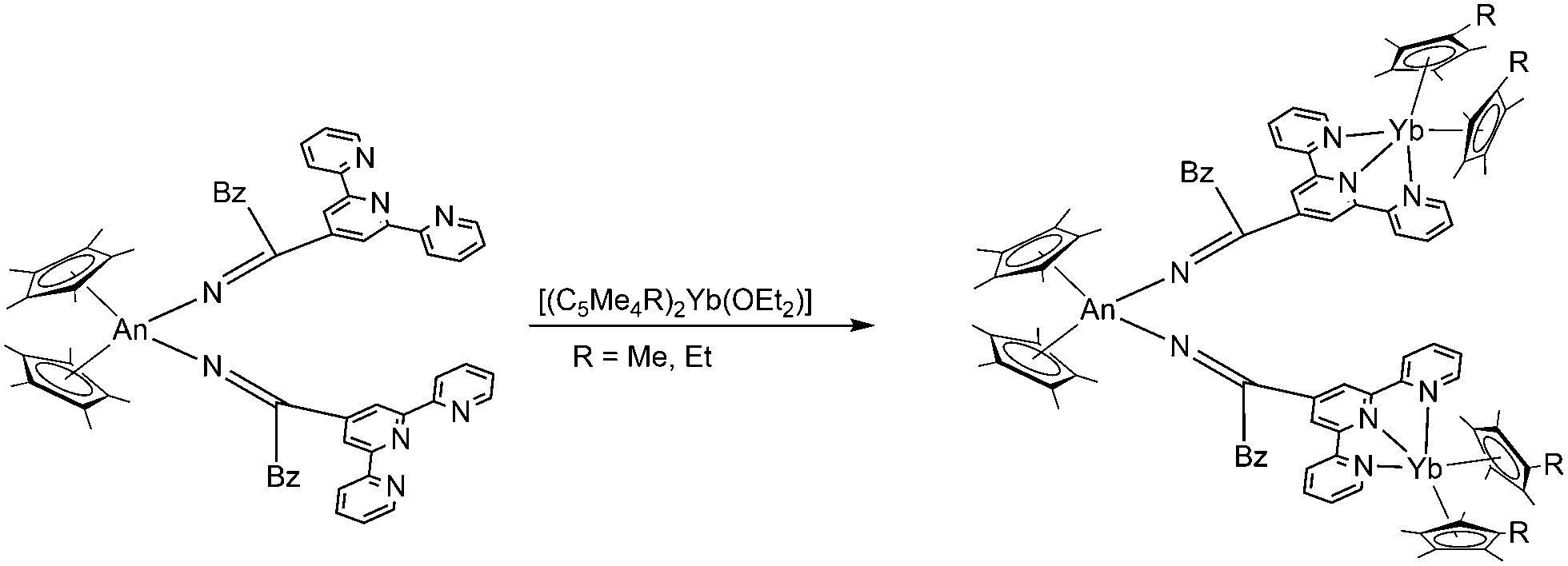 | ||
| Scheme 19 Synthesis of [(C5Me5)2An{NC(CH2C6H5)(tpyYb(C5Me4R)2)}]2.105,106 | ||
In a follow-up study, the corresponding Sm complexes, which were prepared in a similar fashion as the Yb complexes from [(C5Me4Et)2Sm(OEt2)], were synthesised. As seen for the Yb complex, a strong electronic coupling between the metals was observed in the case of the uranium compound [(C5Me5)2U{NC(CH2C6H5)(tpySm(C5Me4Et)2)}]2.106
4.5. Carbon donors
Labile carbon monoxide adducts of the solvent free metallocenes are formed under CO pressure in toluene or methylcyclohexane.107,108 Ethereal solvents are not suitable because the strong oxygen donors prevent carbon monoxide coordination. The formed labile complexes were studied by IR spectroscopy and other spectroscopic methods. The CO stretching frequencies of [(C5Me5)2Sm(CO)] (2153 cm−1) and [(C5Me5)2Eu(CO)] (2150 cm−1) are greater than those of free CO (2134 cm−1), indicating a complete absence of back bonding. In the respective ytterbocene compound, in contrast, somewhat lower CO stretching frequencies were observed. Ytterbocene also forms a CO complex with two CO ligands that dominates at higher pressures. A DFT study carried out in 2002 revealed that the lower back bonding frequency of the Yb bound CO is due to isocarbonyl formation.109The coordination in all carbon monoxide complexes is reversible and CO can be removed in vacuo. Due to their lability, no X-ray crystallographic studies could be carried out on carbon monoxide complexes.
Isocyanides are ligand systems that are very closely related to carbon monoxide. The first 1:2 complex of [(C5Me5)2Yb] and other ytterbocenes with the isocyanide 2,6-Me2C6H3NC was reported by Andersen and co-workers.107 It was synthesised by combining [(C5Me5)2Yb(OEt2)] and 2,6-Me2C6H3NC in toluene. The mean Yb–C bond distance in [(C5Me5)2Yb(2,6-Me2C6H3NC)2] is 2.538 Å. No isocyanide complexes are reported for [(C5Me5)2Eu], whereas the reaction of [(C5Me5)2Sm] with an isocyanide resulted in reductive cleavage of the ligand (see below). However, with 1,2,4-C5H2tBu3 as the ligand of the samarium atom, the crystal structure of an isocyanide complex could be successfully resolved.110
Andersen and co-workers also reported the synthesis of two phosphine–ylidene ytterbium complexes, namely [(C5Me5)2Yb(Me2PhPCHSiMe3)] and [(C5Me5)2Yb(Me2PhPCH2)] (Scheme 20).82 They were prepared by combining the respective ylidene with [(C5Me5)2Yb] in toluene. Extensive, multinuclear, temperature-variable NMR spectroscopic investigations were carried out to elucidate the solution behaviour of the phosphine ylidene complexes as well as the interaction of the carbon with the ytterbium atom. Based on this, a mechanism for the fluxional processes in solution was suggested (please refer to the literature for further details).82 Briefly, the compounds underwent fast intermolecular exchange at room temperature. Dark green crystals of [(C5Me5)2Yb(Me2PhPCHSiMe3)] were investigated by X-ray crystallography to reveal a Yb–C bond distance of 2.69(2) Å.
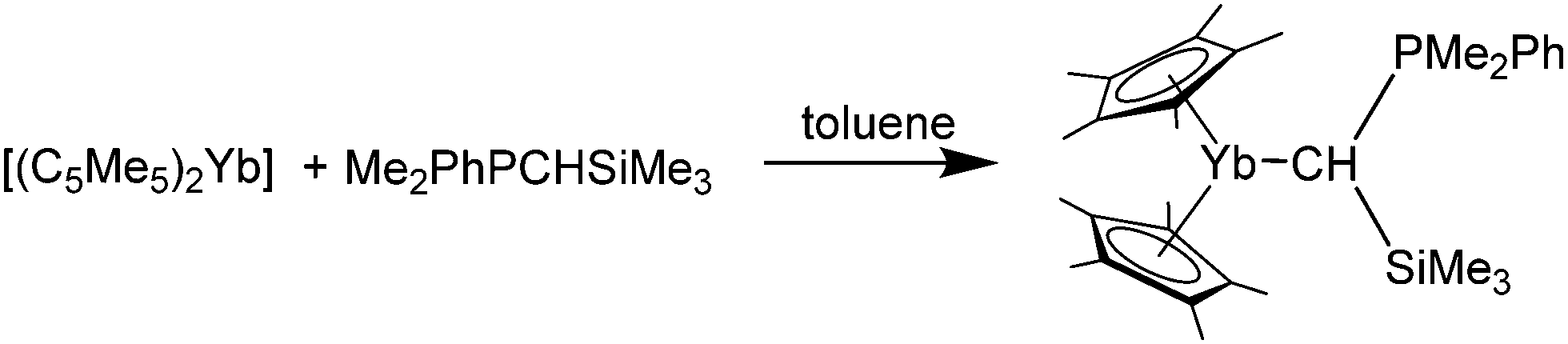 | ||
| Scheme 20 Reaction of a phosphine ylide with [(C5Me5)2Yb].82 | ||
The first two lanthanide N-heterocyclic carbene (NHC) complexes reported in literature were [(C5Me5)2Sm(NHC)] and the corresponding 1:2 complex [(C5Me5)2Sm(NHC)2] (NHC = 1,3,4,5-tetramethylimidazol-2-ylidene) (Scheme 21).111 Despite the rather soft carbon donor atom, [(C5Me5)2Sm(NHC)] was prepared by combining [(C5Me5)2Sm(THF)] with one equivalent of NHC in toluene, the resulting [(C5Me5)2Sm(NHC)] being a dark green, high melting compound. By adding a second equivalent of NHC, the bis-carbene-complex [(C5Me5)2Sm(NHC)2] was obtained. It was possible to structurally investigate [(C5Me5)2Sm(NHC)2] by single crystal X-ray crystallography. The Sm–NHC distances are 2.837(7) and 2.845(7) Å, respectively.
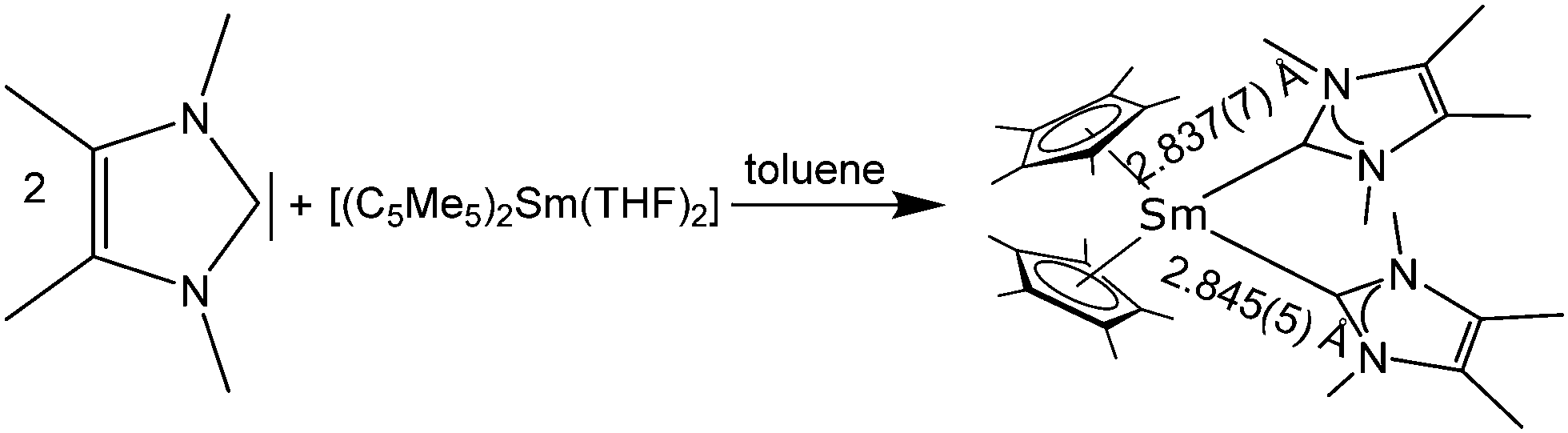 | ||
| Scheme 21 Successive reaction of [(C5Me5)2Sm(THF)2] with two equivalents of 1,3,4,5-tetramethylimidazol-2-ylidene.112 | ||
Furthermore, [(C5Me5)2Sm(C3N2Me2iPr2)] was reported.112 The 1,3-diisopropyl-4,5-dimethylimidazoline-2-ylidene reacts with [(C5Me5)2Sm(THF)2] which results in the carbene-complex. The complex was characterised by single crystal X-ray structure analysis. The Cp–Sm distances are 2.5823(16) and 2.5957(15) Å, and the Sm–C(carbene) distance is 2.782(3) Å, which is considerably shorter than in the prior described compound. Another Sm–NHC complex ligated by 1,3-diisopropyl-4,5-dimethylimidazoline-2-ylidene is similar.112
In addition to the Sm–NHC complexes, Yb–NHC compounds of composition [(C5Me4Et)2Yb(NHC)] (NHC = 1,3,4,5-tetramethylimidazol-2-ylidene and 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) were reported. Due to the smaller ion radius of Yb compared to Sm, only one NHC ligand is bound to the metal atom. The Yb–C bond length for the tetramethyl derivative is 2.669(4) Å.113
Compounds in which the triple bond of alkynes act as an electron donor for a Lewis acidic metal are common for transition metals. Even after thorough literature research, however, we are only aware of one η2-coordinated alkyne complex of the lanthanide metallocenes.114 Combining desolvated [(C5Me5)2Yb] and 2-butyne in pentane, [(C5Me5)2Yb(η2-(H3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH3)] was obtained as dark purple/red needles (Scheme 22). The complex was investigated with IR, 1H and 13C NMR spectroscopy and its solid state structure was elucidated by X-ray crystallography. Based on the investigations carried out, the complex fragments only weakly influence each other and little or no backbonding is observed. The average Yb carbon distance is 2.850 Å.
C–CH3)] was obtained as dark purple/red needles (Scheme 22). The complex was investigated with IR, 1H and 13C NMR spectroscopy and its solid state structure was elucidated by X-ray crystallography. Based on the investigations carried out, the complex fragments only weakly influence each other and little or no backbonding is observed. The average Yb carbon distance is 2.850 Å.
 | ||
| Scheme 22 Reaction of [(C5Me5)2Yb] with 2-butyne and [(η2-C2H4)Pt(PPh3)2].114 | ||
[(C5Me5)2Yb] can initiate the polymerization of ethylene, though no complex with an olefin as a ligand was isolated. However, the reaction of base-free [(C5Me5)2Yb] with the platinum ethylene complex [(η2-C2H4)Pt(PPh3)2] in toluene yielded the ethylene bridged, heterobimetallic complex [(C5Me5)2Yb(μ-C2H4)Pt(PPh3)2] as red crystals (Scheme 22).115 IR and NMR spectroscopy indicate an exchanging system in solution. With respect to the solid structure, the Yb–ethylene–carbon distances are 2.770 and 2.793 Å, respectively. The hydrogen atoms of the ethylene moiety were refined isotropically and are slightly tilted towards the ytterbium atom, resulting in four distinct Yb–H distances of 2.58(5), 2.64(3), 3.09(4) and 3.15 (3) Å. The authors conclude that the interaction between the olefin and the ytterbium centre is rather weak.
Another complex, which was synthesised as polymerization catalyst, is [(C5Me5)Ln{CH(SiMe3)2}(C5Me5)K(THF)2]n (Ln = Sm, Eu, Yb).116 The reaction of [(C5Me5)2Ln(THF)2] (Ln = Sm, Eu, Yb) with KCH(SiMe3)2 in THF resulted in the Ln(II) alkyl complexes in 90–92% isolated yields (Scheme 23). The Sm–C5Me5 average bond distances in the Sm-complex are 2.85(2) and 2.86(2) Å and are therefore in the 2.84(2)–2.97(2) Å bond distance range of those found in the analogous Sm(II) complexes with a heteroatom-containing monodentate anionic ligand. The Sm–CH(SiMe3)2 bond distance is 2.64(1) Å and is hence between the Sm–C bond lengths of Sm(II) (2.787(5) and 2.845(5) Å) and Sm(III) alkyl complexes (2.48(1) Å). The Eu–C5Me5 average bond distances of the Eu-complex are 2.83(2) and 2.91(2) Å and are comparable with those of the Sm–C5Me5 bonds in the Sm-complex due to the similar ion sizes of Eu(III) and Sm(II). The Eu–CH(SiMe3)2 bond distance is 2.65(1) Å. The Yb–C5Me5 average bond distances are 2.74(3) and 2.79(3) Å and are in the range of those found in comparable Yb(II) complexes.
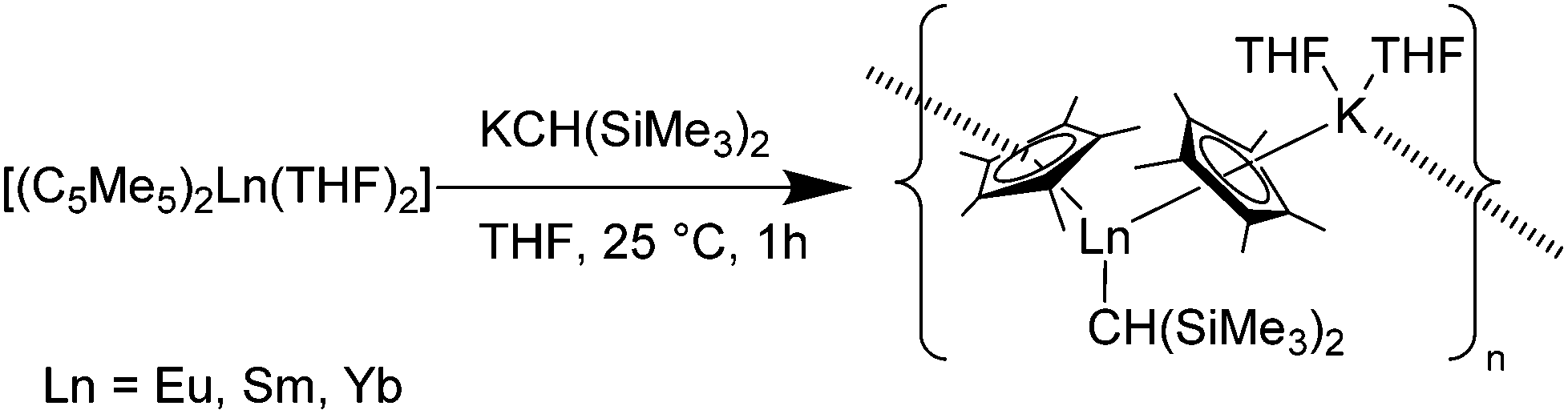 | ||
| Scheme 23 Polymeric chain in [(C5Me5)Ln{CH(SiMe3)2}(C5Me5)K(THF)2]n (Ln = Eu, Sm, Yb).116 | ||
Reaction of unsolvated [(C5Me5)2Yb] and the beryllium piano stool complex [(CH3)Be(C5Me5)] in pentane yields the uncommon methyl bridged complex [(C5Me5)2Yb(μ-CH3)Be(C5Me5)] as dark orange crystals (Scheme 24).117 The interaction in solution proved to be rather weak and a fast exchange process was observed by NMR spectroscopy. The solid state structure reveals a Yb–C distance of 2.766 Å, which is similar to the distance in the bridging olefin complex of platinum and shorter than the Yb–C distance in the Yb-alkyne complex (see above). The hydrogen atoms derived from the Fourier difference map show an average distance of 2.59 ± 0.08 Å to the ytterbium atom. The authors emphasise the complex as a model for methane complexation by ytterbium.
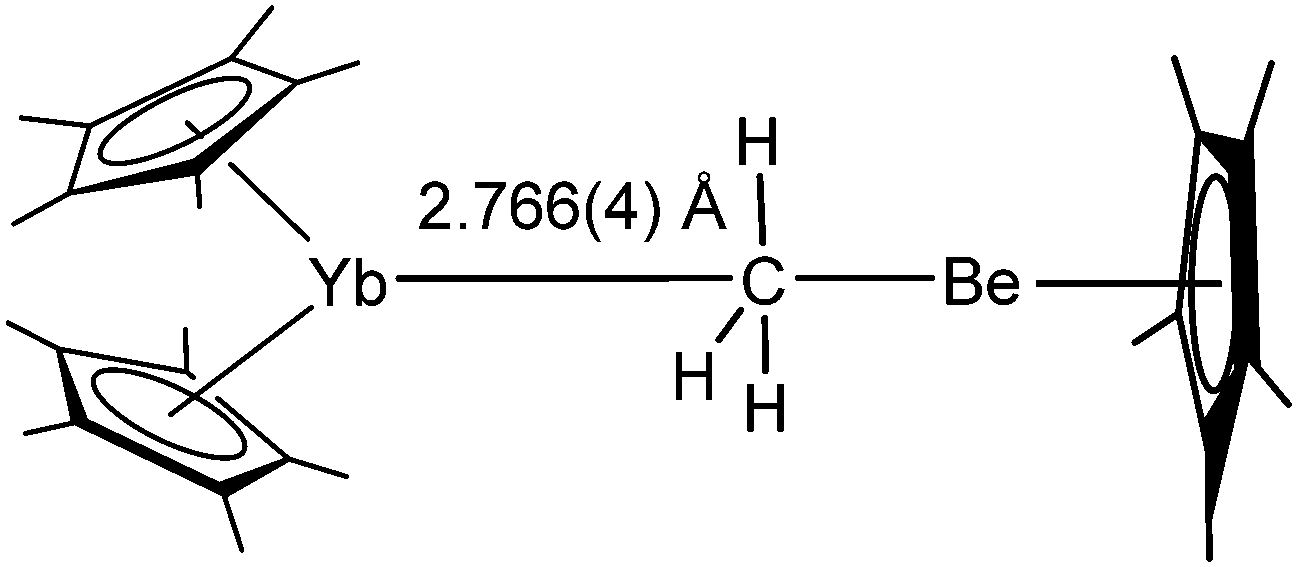 | ||
| Scheme 24 Structure of [(C5Me5)2Yb(μ-CH3)Be(C5Me5)].51 | ||
A related complex is formed by the reaction of [(C5Me5)2Yb(THF)] and AlEt3 in toluene. The ethyl bridged, heterobimetallic complex [(C5Me5)2Yb(μ-Et)AlEt2(THF)] was isolated as green crystals.118 AlMe3 and Al(iC4H9)3 give similar structures. The structure of the ethyl derivative was determined. The Al and Yb centres are linked by the ethyl moiety attached to the aluminium ion. The latter is tetrahedrally coordinated by three ethyl units and one THF molecule. Both carbon atoms of the bridging C2H5 unit bind to the ytterbium ion. The Yb–C distances are 2.854(18) and 2.939(12) Å. The bond is weak and therefore broken by the addition of THF or other donors. The use of the ytterbocene THF solvate as a precursor is still possible because the organoaluminum compound is a stronger Lewis acid and therefore removes THF from the Yb atom.
The solvent free metallocenes [(C5Me5)2Ln] (Ln = Sm, Eu, Yb) react with [Et3NH][BPh4] to form the divalent π-coordinated tetraphenyl borates [(C5Me5)Ln(μ-η6:η1-Ph)2BPh2] (Ln = Sm, Eu, Yb). Two of the phenyl rings of the tetraphenylborate counteranion coordinate η6 to the lanthanide atoms (Scheme 25).119,120 [(1,3-C5H3tBu2)2Eu] reacts in a similar way to give [(1,3-C5H3tBu2)Eu(μ-η6:η1-Ph)2BPh2].121
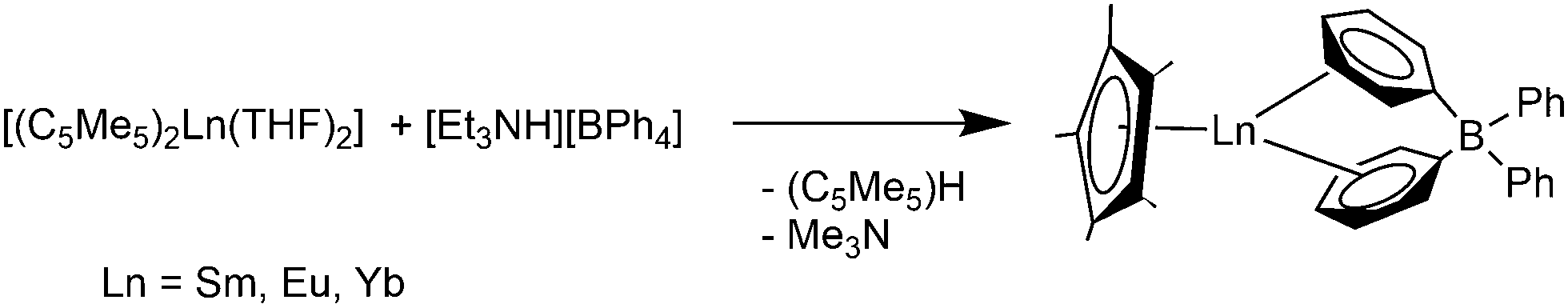 | ||
| Scheme 25 Synthesis of [(C5Me5)Ln(μ-η6:η1-Ph)2BPh2] (Ln = Sm, Eu, Yb).119,120 | ||
4.6. Silicon, phosphorus, and sulphur donors
Complexes containing ligands with soft donor atoms of the third row period of the periodic table are comparatively less common in combination with divalent metallocenes of the lanthanides. Ligands with sulphur, phosphorus or silicon are easily replaced by oxygen containing ligands like THF. Therefore, the solvent free metallocenes or at least the diethylether complexes have to be used as precursor.Recently, lanthanide(II) silyl complexes were reported.116 [(C5Me5)2Ln(THF)2] reacted with KH and H3SiPh yielding [(C5Me5)Ln(SiH3)(THF)(C5Me5)K(THF)]n (Ln = Yb(II), Sm(II), Eu(II)) (Scheme 26). Due to poor crystal quality, the Sm complex could not be characterised, whereas the Eu and Yb-analogues were investigated by crystallographic studies. The Ln–(C5Me5) average bond distances are for Ln = Eu 2.85(1) and 2.86(1) Å, and for the Yb-complex 2.76(1) for both C5Me5 units. The Eu–SiH3 and Yb–SiH3 average bond distances are 3.239(3) and 3.091(3) Å, respectively.
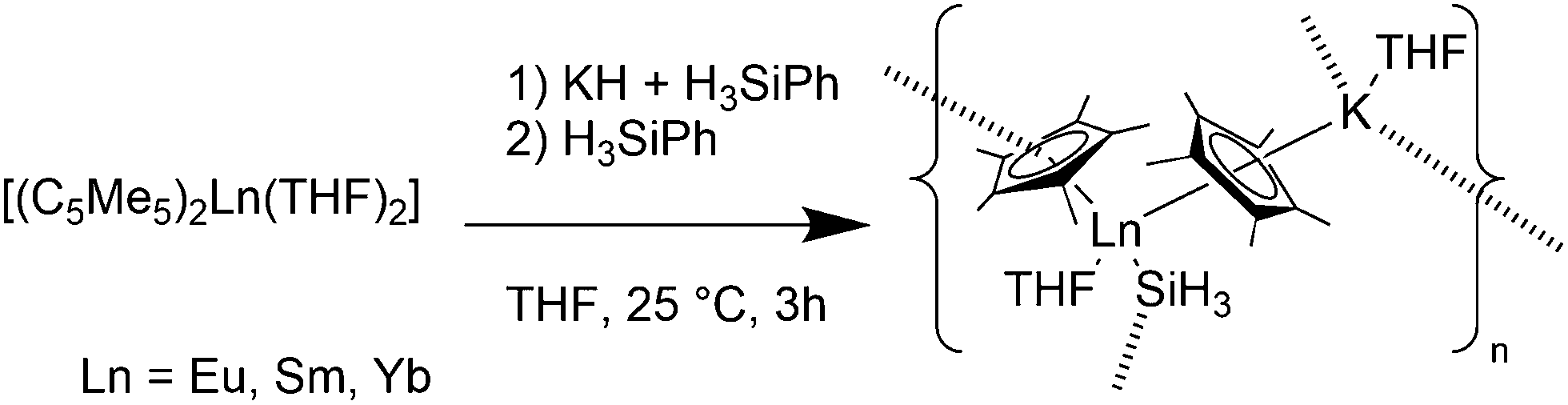 | ||
| Scheme 26 Polymeric chain in [(C5Me5)Ln(SiH3)(THF)(C5Me5)K(THF)]n (Ln = Eu, Sm, Yb).116 | ||
The first [(C5Me5)2Ln-silylene] complex was reported in 2003 by West and Evans.122 [(C5Me5)2Sm-(NHSi)] was prepared by combining solvent-free samarocene and NHSi (NHSi = 1,3-di-tert-butyl-2,3-dihydro-1H-1,3,2-diazasilol-2-ylidene) in toluene. As expected, the bond between the Si(II)-centre and Sm is rather weak. The silylene ligand is immediately replaced by THF if present in solution. The solid structure shows a Sm–Si bond length of 3.1903(10) Å. The silylene coordinates asymmetrically and is slightly tilted to one side, allowing a long-range interaction with one tert-butyl methyl group of the silylene. In the course of renewed interest in low-valent main group donors, two new silylene complexes of decamethylsamarocene, [(C5Me5)2Sm(Si(OC6H4-2-tBu)){(NtBu)2CPh}] and [(C5Me5)2Sm(Si(OtBu)){(NtBu)2CPh}] were reported in 2015.123 They were obtained from the reaction of [(C5Me5)2Sm(OEt2)] and the respective four-membered N-heterocyclic silylenes {PhC(NtBu)2}SiOtBu and {PhC(NtBu)2}SiO(2-tBu-C6H4) in toluene and isolated as emerald green solids. The solid state structures of both complexes were determined, whereby long Sm–Si distances of 3.4396(15) Å ({PhC(NtBu)2}SiOtBu) and 3.3142(18) Å in {PhC(NtBu)2}SiO(2-tBu-C6H4) indicate weak interactions. The compounds were extensively characterised and investigated by quantum chemical calculations on the DFT level confirming the weak bond without covalent character.
Among the phosphorus compounds, the reactions of the etherates [(C5Me5)2Ln(OEt2)] (Ln = Eu, Yb) and the bidentate phosphines Me2PCH2CH2PMe2 and Me2PCH2PMe2 were investigated.124 In the first case, insoluble coordination polymers are formed with Me2PCH2CH2PMe2. In the second case, the sterically less flexible Me2PCH2PMe2 yields the soluble complexes [(C5Me5)2Ln(Me2PCH2PMe2)] (Eu: red, Yb: green). According to NMR investigations of the Yb compound, the phosphine coordinates in a bidentate fashion. No X-ray data is available for these complexes.
Complexes of monodentate phosphines were reported for ytterbocene. Due to the reduced donor strength of the phosphines, the solvent-free complex [(C5Me5)2Yb] was used as precusor.82 Upon reaction with PMe3 in toluene, the green 1:2 complex [(C5Me5)2Yb(PMe3)2] was formed and isolated from toluene at −80 °C. A similar protocol with PEt3 yielded the blue 1:1 complex [(C5Me5)2Yb(PEt3)]. The solution behaviour of the complexes was investigated and NMR spectroscopy shows that the interaction between phosphine and ytterbium is very weak in solution. Although isolated as crystals, no X-ray crystallographic data is available.82
As with the lanthanide(II) silyl complexes, a phosphide complex was also synthesised (Scheme 27). Reaction of [(C5Me5)2Sm(THF)2] with KPH(C6H2tBu3-2,4,6) resulted in decomplexion of one C5Me5 ligand from the Sm(II) atom and the formation of the polymeric species [(C5Me5)Sm(THF)(μ-PHAr)K(C5Me5)(THF)]∞ (Ar = C6H2tBu3-2,4,6) (Scheme 27).83 Within this compound a “C5Me5K” unit is bonded to the phosphide site with its K atom. The Sm–P bond (3.234(2) Å) is significantly longer than those found in samarium(II) bis(phosphide) complexes. A similar reaction was seen by using the thiolate SC6H2iPr3-2,4,6 as reagent.83
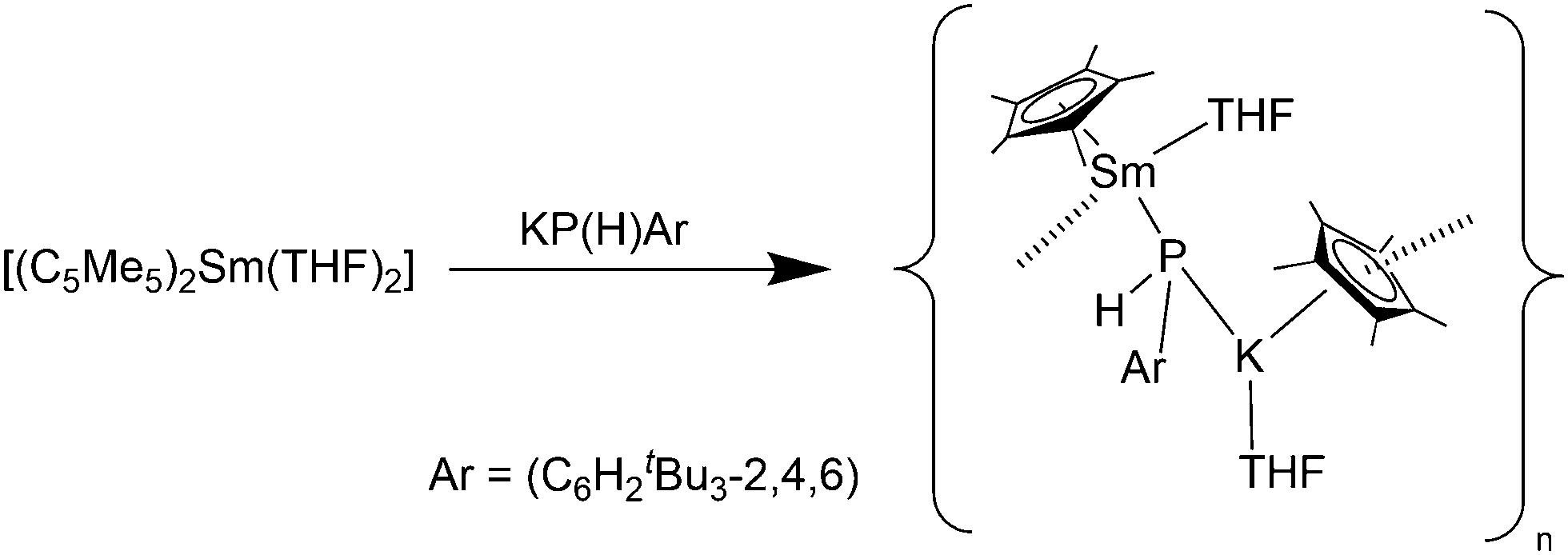 | ||
| Scheme 27 Synthesis of [(C5Me5)Sm(THF)(μ-PHAr)K(C5Me5)(THF)]∞.83 | ||
More recently, the reactions between metallocenes of ytterbium or samarium with tetramethylbiphosphinine (tmbp), a bipyridine analogue with phosphorus donors, were reported.125 In this case, the diethyl solvates were employed for the synthesis of [(C5Me5)2Ln(tmbp)] (Ln = Sm, Yb) in toluene as dark brown crystals. The solid state structures were determined by X-ray diffraction (Fig. 4). The Ln–P bond lengths in the solid state are 2.909(2) and 2.972(2) Å for the Sm compound, and 2.872(2) Å and 2.983(2) Å for Yb one respectively. The complexes were extensively investigated by NMR spectroscopy and theoretical calculations to assess the electron density transferred from the ligand to the lanthanide ion. The authors aimed to investigate whether the metals are in a divalent oxidation state or in a trivalent state with a reduced tmbp ligand. In the case of Yb, the oxidation state +II was found to be the most accurate, while in the samarocene complex the Sm ion is best described as trivalent.
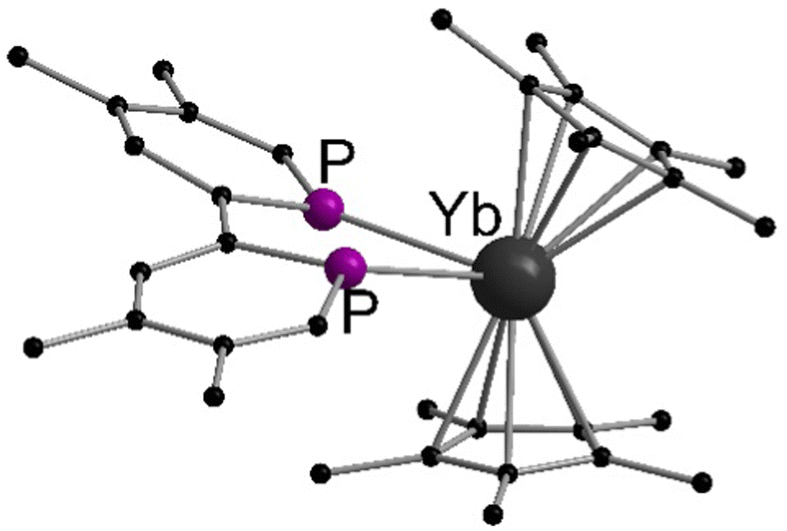 | ||
| Fig. 4 Molecular structures of tetramethylbiphosphinine comlexes of ytterbocene and samarocenene in the solid state (Reproduced from the CIF file CCDC 1452181).125 | ||
It should be noted that, to the best of our knowledge, no Lewis acid–base adduct with neutral sulphur donor ligands has been reported in the literature, probably due to the very weak coordination abilities of the soft sulphur donors with regard to the hard character of the lanthanide ions.
4.7. Aluminium and gallium compounds
The low valent aluminium compound [(C5Me5)Al]4 can act as a Lewis base due to its free electron pair. Heating [(C5Me5)2Eu] or [(C5Me5)2Yb] and [(C5Me5)Al]4 in an evacuated ampoule resulted in red (Eu) or green (Yb) crystals of [(η5-C5Me5)2Ln-Al(η5-C5Me5)], respectively.126 These compounds were the first compounds with an aluminium-4f metal bond. Both compounds were structurally investigated by single crystal X-ray crystallography (Fig. 5). The Eu–Al bond is 3.3652(10) Å long, whereas the Yb–Al bond is slightly shorter with 3.1981(11) Å. The compounds even decompose in hydrocarbon solvents. However, they were thoroughly characterised using standard analytical methods and special care was taken to ensure that there was no hydride species between aluminium and the lanthanide that would lead to an Ln(III) complex. In addition, extensive quantum chemical calculations were carried out. According to these calculations, the interaction between the lanthanide and the aluminium atom is mainly electrostatic, resulting in very low binding energies, which is consistent with the low stability of both compounds in solution.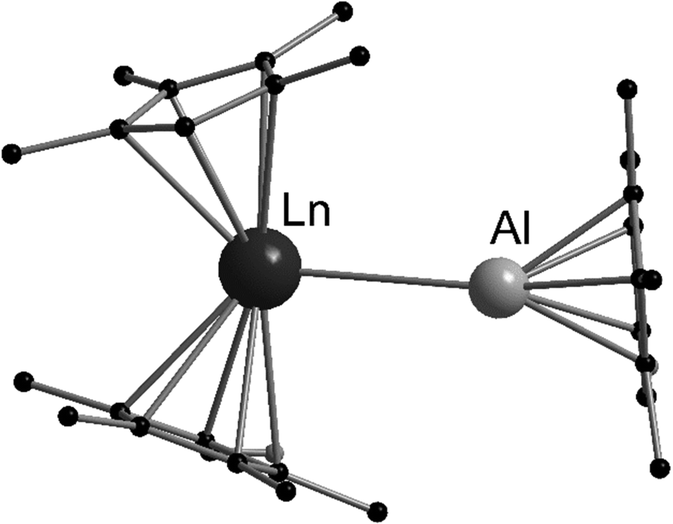 | ||
| Fig. 5 Molecular structure of [(η5-C5Me5)2Ln-Al(η5-C5Me5)] (Ln = Eu, Yb) in the solid state (based on the CIF file CCDC 296933).126 | ||
Complexes of [(C5Me5)2Eu] and [(C5Me5)2Yb] and the heavier Ga congener of [(C5Me5)Al]4, namely [(C5Me5)Ga], have also been prepared.127 The reaction of solvent free [(C5Me5)2Eu] and THF deficient [(C5Me5)2Yb(THF)1−n] with [(C5Me5)Ga] in toluene in the correct stoichiometric ratio yielded [(η5-C5Me5)2Eu–Ga(η5-C5Me5)2)] and [(η5-C5Me5)2(THF)Yb-Ga(η5-C5Me5)2], respectively. In contrast to the Al compounds, the two Ga complexes can be prepared and are stable in solution. Both compounds were characterised by standard analytical methods including X-ray crystallography. The Eu–Ga contacts in [(η5-C5Me5)2Eu–Ga(η5-C5Me5)2] are 3.2499(6) Å and 3.3907(6) Å. In [(η5-C5Me5)2(THF)Yb–Ga(η5-C5Me5)2], the Yb–Ga distance is 3.2872(4) Å. The Yb–O distance from the THF–oxygen atom is 2.418(2) Å. The most striking difference between [(η5-C5Me5)2Eu–Ga(η5-C5Me5)2] and [(η5-C5Me5)2Eu–Al(η5-C5Me5)] is that two Ga(C5Me5) fragments coordinate to the europium atom while in the analogue Al compound only one Al(C5Me5) donor coordinates to the europium centre. This is rationalised by the longer Eu–Ga bonds compared to the Eu–Al bonds, allowing the coordination of two ligands.
5. Reactivity as a reducing agent
As early as 1984, Deacon et al. reported that [(C5H5)2Yb(DME)] can be oxidised with thallous, mercuric, argentic and cuprous salts to give [(C5H5)2Yb-X] (X = O2CMe, O2CC6F5, O2CC5H4N, Cl, Br, I, CCPh, C6F5, (MeCO)2CH, (PhCO)2CH).6 In recent years, the focus has been on substituted metallocenes, mainly the decamethyl derivatives. In general, [(C5Me5)2Ln] (Ln = Sm, Eu, Yb) or its solvates can act as reducing agents according to the reaction shown in Scheme 28 (i.e. Eu(III)/Eu(II) = −0.35 V, Yb(III)/Yb(II) = −1.1 V, Sm(III)/Sm(II) = −1.5 V vs. normal hydrogen electrode).128 While europocene is a weak reductant and quite stable in the oxidation state +II, ytterbocene is more strongly reducing.
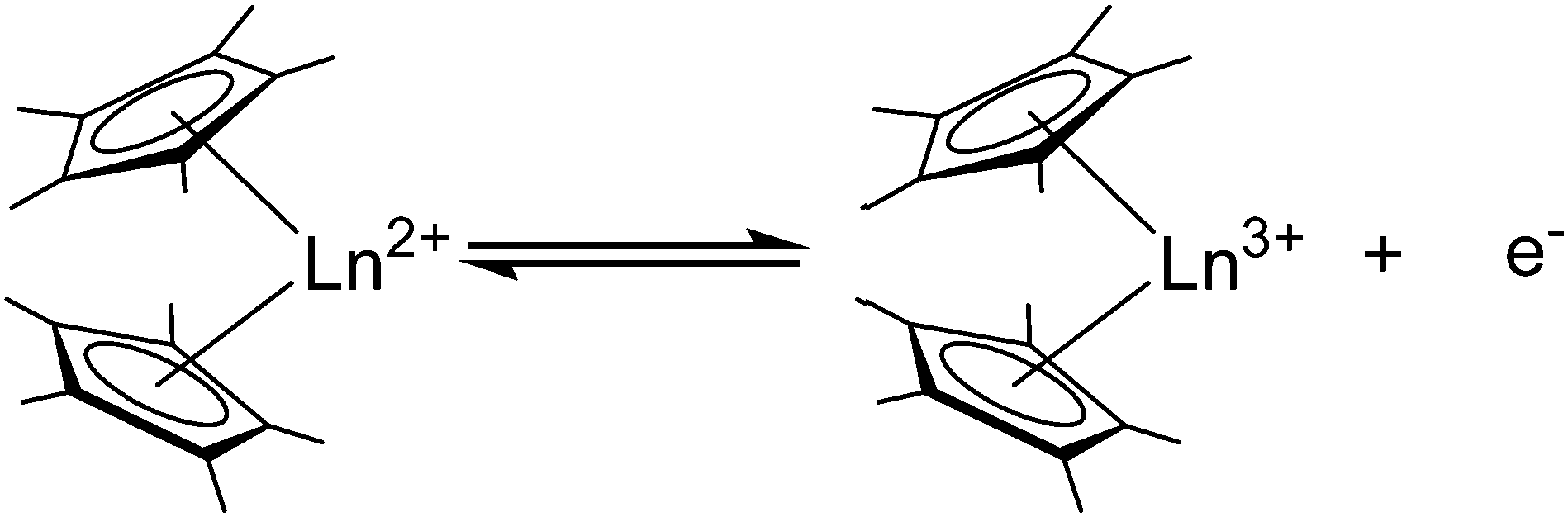 | ||
| Scheme 28 Redox reactivity of the three pentametyhlcyclopentadienyl derivatives (Ln = Eu, Sm, Yb). | ||
Samarocene is the strongest reducing agent in this series being able to react with dinitrogen (see below, Scheme 72). Its reactivity has been compared with the reactivity of alkaline metals, which is why most of the work on redox reaction involving the title compounds revolves around Sm. The reduction strength can thus be ranked in the order (C5Me5)2Eu < (C5Me5)2Yb < (C5Me5)2Sm.129 In addition, divalent thulium metallocenes, which have a very strong reduction potential, are known. However, due to their high reactivity these compounds are difficult to handle and only a few reactions were reported.41 Note, [(C5Me5)2Tm(solvate)n] was not isolated. Only derivatives with bulkier substituents are known. The following section provides an overview of the reduction reactivity patterns of the divalent metallocenes.
5.1. Reactivity towards organic reagents
As early as in the first publication of the synthesis of [(C5Me5)2Sm(THF)2], Evans mentioned the ability of the molecule to enhance the hydrogenation of 3-hexyne to cis-3-hexene.14 The ability of the samarocene to reduce carbon–carbon triple bonds was later demonstrated by the reaction of Ph–CC–Ph and [(C5Me5)2Sm(THF)2] (Scheme 29).130,131 According to elemental analysis the isolated black material has the empirical formula [(C5Me5)2SmCC6H5]. By further spectroscopic analysis, the structure of a bis-samarium complex with a bridging enediyl-ligand [(C5Me5)2Sm(C6H5)CC(C6H5)Sm(C5Me5)2] was identified, and the oxidation state of +III was confirmed. Hydrolysis yielded pure trans-stilbene. Interestingly, by adding THF to the complex, divalent [(C5Me5)2Sm(THF)2] was formed again. Reaction of the enediyl complex with molecular hydrogen yields the hydride-bridged Sm(III) complex [(C5Me5)2Sm(μ-H)]2.
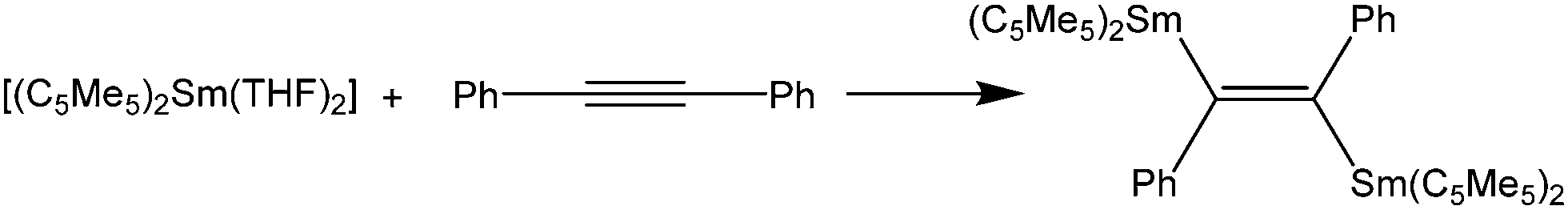 | ||
| Scheme 29 Reaction of [(C5Me5)2Sm(THF)2] with diphenyl acetylene.130,131 | ||
The chemistry of [(C5Me5)2Sm(solvate)n] and terminal alkynes as well as those of the products obtained has been widely studied.132,133 Samarocene reacts with terminal alkynes HCC–R to form trivalent products of the type [(C5Me5)2Sm(CC–R)(THF)] (R = –Ph, –CH2CH2Ph, –CH2NEt2, –CH2CH2CHMe2, –CHMe2, –CMe3) when the THF solvate of the metallocene was used as reactant (Scheme 30).132,133 Similar reactivity was observed in the reaction of [(1,3-C5H3tBu2)2Sm] with phenylacetylene.134 The complex [(C5Me5)2Sm(CC–Ph)(THF)] was structurally characterised, however from the reaction of trivalent [(C5Me5)2Sm(THF)2(BPh4)] and the corresponding potassium alkynide.133 The carbon Sm–C(alkyne) bond length is 2.50(2) Å.
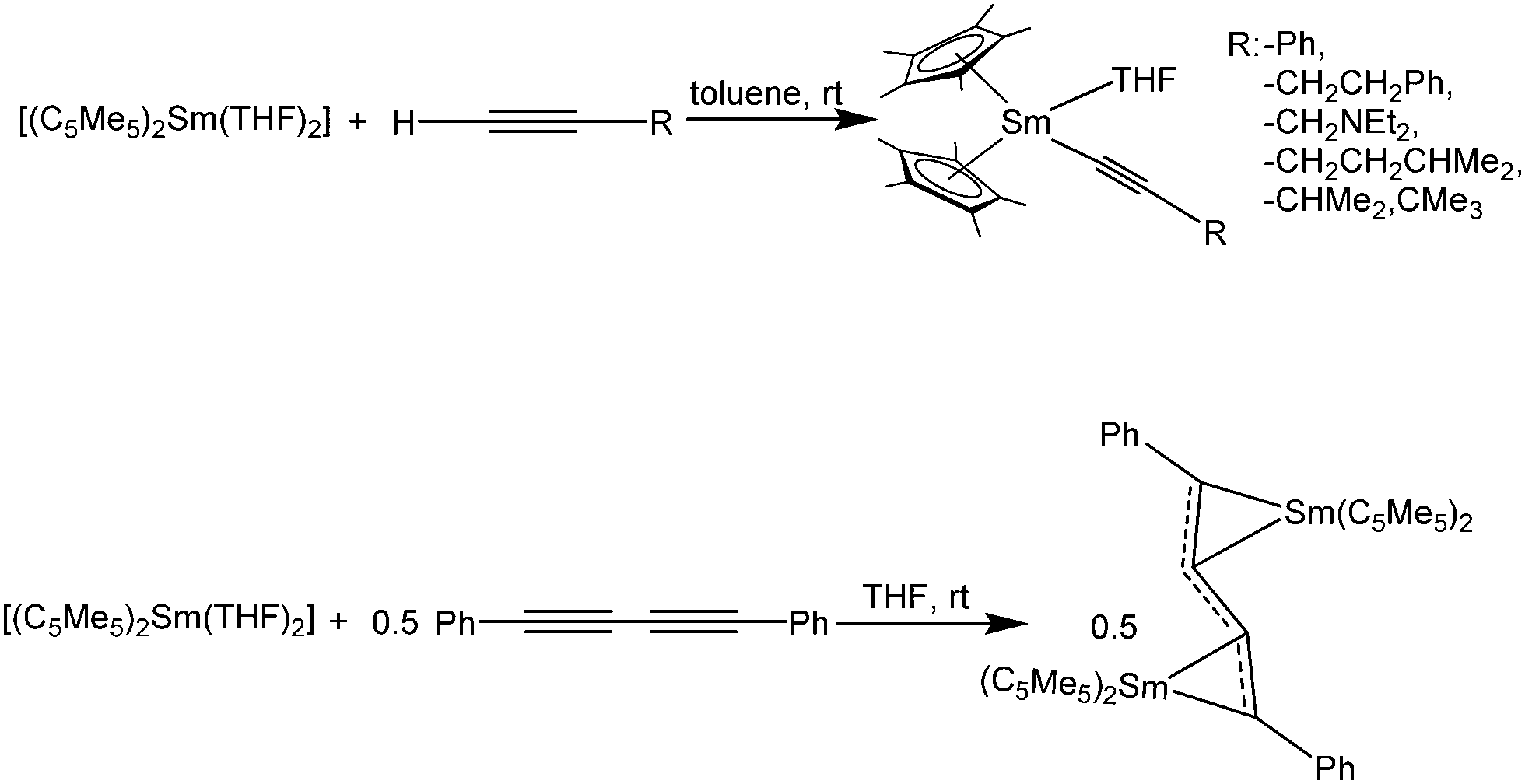 | ||
| Scheme 30 Reactions of [(C5Me5)2Sm(THF)2] with various alkynes.132,133 | ||
The reactivity of these alkynide complexes, regarding the coupling of the alkynide moieties was investigated in depth.132 If the reaction between solvent-free [(C5Me5)2Sm] and terminal alkynes was carried out in the absence of THF, coupling of two alkynide moieties occurs to yield structurally diverse Sm complexes (Scheme 31). The structure of the resulting bridging ligand strongly depends on the nature of the electronic and steric factors of the substituents at the alkynide ligand. Coupling does not occur for the bulky CC-CMe3 moiety (Scheme 31). For example, [(C5Me5)2Sm(CC–Ph)] yields the coupled trienediyl complex [{(C5Me5)2Sm}2(μ-η2:η2-Ph-CCCC–Ph)], which is also obtained from the reaction of [(C5Me5)2Sm(THF)2] and the butadiyne Ph–CC–CC–Ph (Scheme 30).135,136 It is also possible to synthesise the coupled complex by thermolysis of the solvated THF complex [(C5Me5)2Sm(CC–Ph)(THF)] or from the divalent [(C5Me5)2Sm] and PhCCH (Scheme 31).136 The use of unsolvated [(C5Me5)2Sm] also yields the coupled products for RCCH (R = (CH2)2Ph, CH2NEt2, (CH2)2CHMe2 and CHMe2). It should be noted that this synthesis route does not work for any other substituent on the alkynide unit. The authors point out that the reactivity of these compounds has to be evaluated on a case-to-case basis, considering the properties of the solvent and the steric and electronic nature of the alkynide ligands.
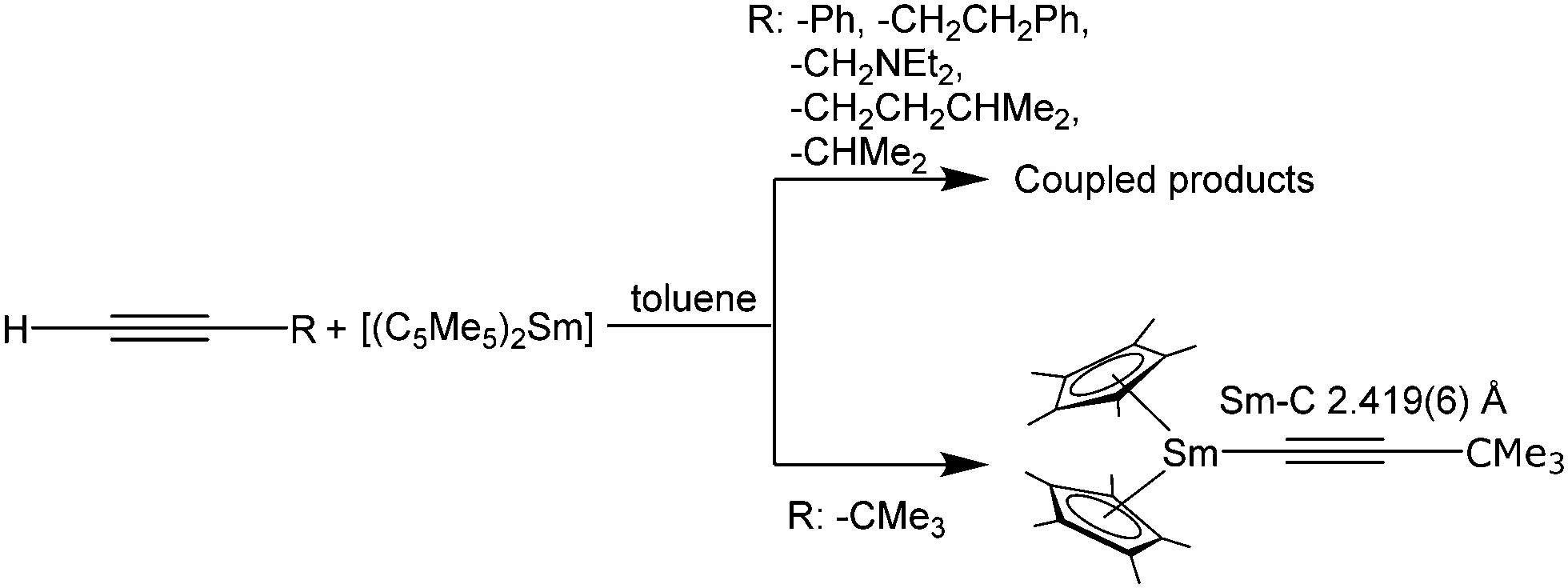 | ||
| Scheme 31 Reactions of unsolvated [(C5Me5)2Sm] with various alkynes.136 | ||
In contrast, decamethylytterbocene reacts with PhCCH to form the red trinuclear, mixed valence compound [(C5Me5)2Yb(μ-CCPh)2Yb(μ-CCPh)2Yb(C5Me5)2] (Scheme 32).137 In this case, a redox reaction and a concomitant acid–base elimination of two HC5Me5 molecules formally take place. However, no closer elucidation of the mechanism is reported. The two outer Yb atoms, which are coordinated by two C5Me5-ligands and two bridging phenylacetylene moieties, are in the oxidation state +III as confirmed by the averaged Yb–C pentamethylcyclopentadienyl-distances of 2.61(2) Å. The central Yb atom remains in the divalent oxidation state and is surrounded by four bridging phenylacetylide ligands in a distorted tetrahedral fashion (average Yb–C distance 2.52 Å). Furthermore, four close distances to the C–Ph atoms of the respective ligands are found in the solid state structure, saturating the coordination sphere of the divalent Yb atom. An electron exchange between the di- and the trivalent Yb centres was excluded by magnetic susceptibility studies.
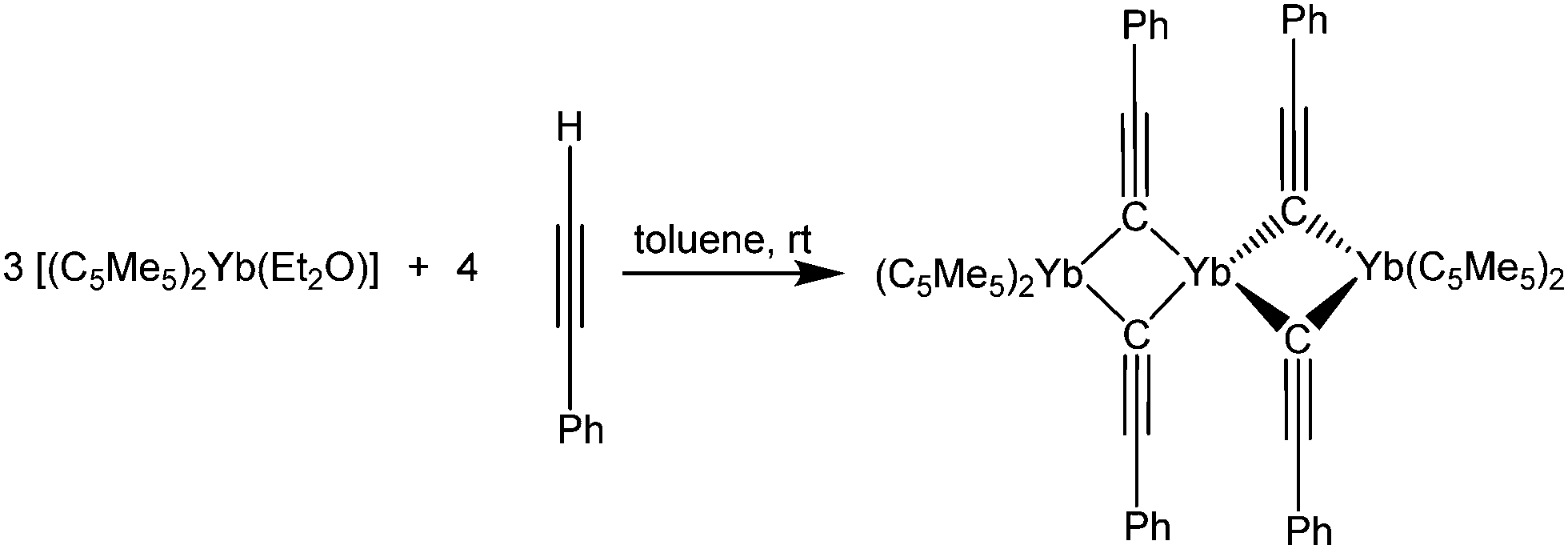 | ||
| Scheme 32 Reaction of with phenylacetylene.137 | ||
Interestingly, [(C5Me5)2Sm(THF)2] reacts in THF with acetylene to yield the binuclear acetylide-bridged complex [{(C5Me5)2Sm(THF)}2(μ-η1:η1-C2)] with concomitant H2 evolution (Scheme 33).138 When the reaction was carried out with desolvated samarocene, no clear product was isolated. The increased reactivity of [(C5Me5)2Sm] resulted in an intractable mixture of products. A comparable reactivity was observed by carrying out the reaction of [(C5Me5)2Sm(THF)2] and acetylene in toluene, which is a weakly coordinating solvent (Scheme 33). The formation of by-products was, to a lesser extent, also observed with the use of THF as a solvent in NMR studies. The X-ray crystallographic analysis of [{(C5Me5)2Sm(THF)}2(μ-η1:η1-C2)] reveals a rare bridging motive of the carbon ligand and an almost linear arrangement of the metals and the carbon atoms. In addition to the acetylide ligand, a THF molecule is bound to each of the samarium atoms. The Sm–C bond lengths are 2.438(7) and 2.448(8) Å. Note that the C–C bond length is 1.21(1) Å, meaning only a slight deviation from acetylene. Hence, the alkyne is not activated.
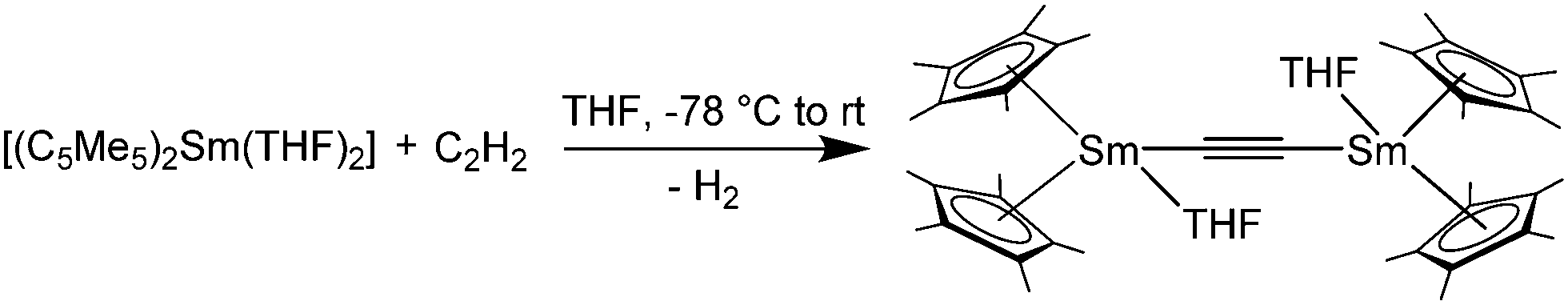 | ||
| Scheme 33 Reaction of [(C5Me5)2Sm(THF)2] and acetylene.138 | ||
Like the triple bonds of alkynes, the double bonds of alkenes can also be reduced by [(C5Me5)2Sm]. The simplest alkene ethylene is polymerised to polyethylene by the solvated as well as the desolvated complex.139 With substituted alkenes like propylene, trans-butene and allylbenzene Sm(III) allyl-complexes of composition [(C5Me5)2Sm(allyl)] are formed. In some cases, the THF solvate shows no reactivity, pronouncing once more the ability of THF to block the reactive site of the [(C5Me5)2Sm] fragment. The allyl ligand is in all cases in a η3-coordination mode in the solid state. With 1,3-butadiene, a coupling reaction of two allyl ligands occurred to give a bis-samarocene complex with a bridging bis-allyl ligand consisting of eight CHn-moieties. Depending on the substituents, the three different Sm–Callyl distances vary in the range of 2.551(17)–2.730(17) Å. Furthermore, [(C5Me5)2Sm] isomerises cis-stilbene to the trans isomer.139 The dark maroon-purple, dinuclear complex [{(C5Me5)2Sm}2(μ-η2:η4-PhCHCHPh)] was isolated from the stoichiometric reaction of decamethylsamarocene and cis-stilbene (Scheme 34). Here, two [(C5Me5)2Sm] fragments are bridged by the double bond of stilbene. In addition, both stilbene phenyl rings exhibit close contacts of the ortho carbon atom with one of the samarium centres. This results in the asymmetric tilt of the phenyl moieties towards one samarium centre. A similar dinuclear product, [{(C5Me5)2Sm}2(μ-η2:η4-PhCHCH2)], was obtained from the reaction of decamethylsamarocene with styrene (Scheme 34). The [(C5Me5)2Sm] fragments coordinate on opposite sides to the styrene double bond. Both the ipso as well as the ortho carbon of the phenyl ring coordinate to one of the Sm ions which results in asymmetric η4:η2 coordination and unequal bond Sm–C double bond distances. Distances in the range of 2.537(15) to 2.732(15) Å were determined. Shorter distances belong to the η2-bound samarium fragment. The phenyl-C-Sm contacts are 2.85(2) and 2.77(2) Å, respectively.
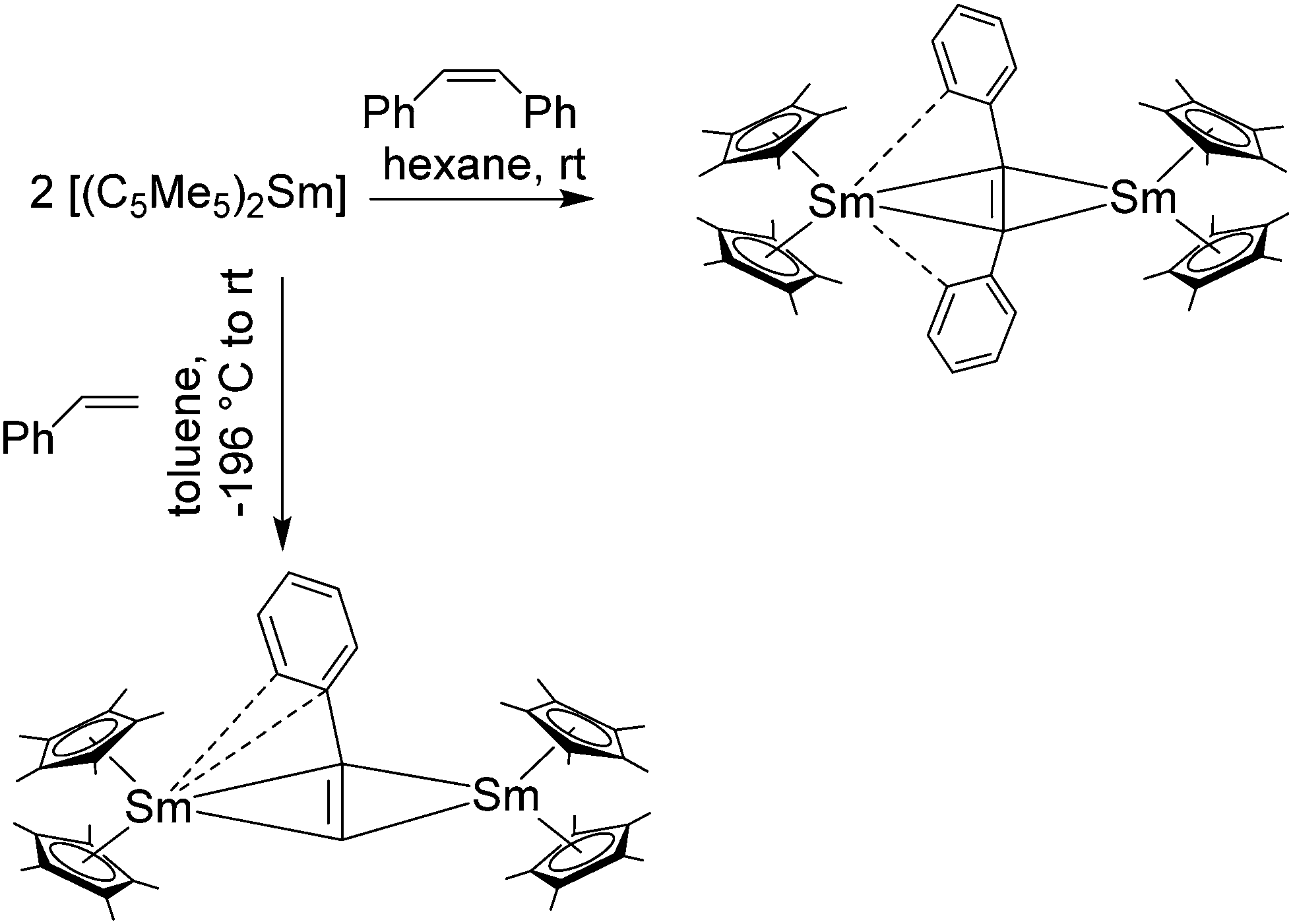 | ||
| Scheme 34 Reactions of [(C5Me5)2Sm] with styrene and stilbene.136 | ||
In contrast to the chemistry described above, the reaction of [(C5Me5)2Sm] with the diene monomers isoprene (C5H8) and myrcene (C10H16) resulted in weakly coordinated hydrocarbon complexes.131 The isoprene complex [{(C5Me5)2Sm}2(μ-η2:η4-CH2CHC(Me)CH2)] was formed at −36 °C in toluene (Scheme 35). The addition of THF to this compound quantitatively resulted in the divalent species [(C5Me5)2Sm(THF)2]. The reaction of myrcene with [(C5Me5)2Sm] was analogous to that of isoprene. The olefin complex [{(C5Me5)2Sm}2(μ-η2:η4-CH2CHC(CH2)CH2CH2CHC(Me)2] was obtained as product. The addition of THF reversed the reaction and myrcene as well as [(C5Me5)2Sm(THF)2] was formed.131
 | ||
| Scheme 35 Reactions of [(C5Me5)2Sm] with isoprene.131 | ||
The reaction of samarocene and cyclopentadiene results in trivalent [(C5Me5)2Sm(C5H5)] with the concomitant formation of molecular hydrogen.140 The authors emphasise the similarity to the reactivity of the alkaline metals. All three cyclopentadienyl ligands coordinate in a η5 mode. Interestingly, the addition of a further equivalent of solvent-free [(C5Me5)2Sm] yields the mixed valent, dinuclear complex [{(C5Me5)2Sm(III)}(μ-C5H5)Sm(II)(C5Me5)2]. The addition of THF to a toluene solution of this dimer resulted in the reformation of [(C5Me5)2Sm(C5H5)] and [(C5Me5)2Sm(THF)2].
Various substituted samarocenes and ansa-samarocenes have been used as catalysts for the polymerisation of ethylene and 1-olefines. In general, ansa-samarocenes showed higher activity and selectivity.32 A highly regio- and diastereoselective cross-coupling of allyl/propargyl ethers and δ-ketoesters to functionalised δ-lactones was observed by using various samarocenes as reagents. In dependence on the nature of the cyclopentadienyl ligand, high regio and diastereocontrol were reported in some cases. In contrast, SmI2 gave unsatisfactory results in the transformation indicating the influence of the cyclopentadienyl ligand.141
Another parallel between the chemistry of samarocene and that of alkaline metals is the ability to transfer electrons to the aromatic system of polycyclic aromatic hydrocarbons (PAH).142 The intensely coloured reaction products of solvent-free [(C5Me5)2Sm] and PAH such as anthracene, pyrene, acenaphtylene, 2,3-benzanthracene and 9-methylanthracene were investigated. No reaction occurred with naphthalene and coronene. In all other cases, 2:1 complexes of the general formula [{(C5Me5)2Sm}2(μ-PAH)] were isolated (Scheme 36). The solid structures of the anthracene, pyrene and 2,3-benzanthracene complexes were determined by single crystal X-ray crystallography. In general, samarium atoms coordinate to three PAH C-atoms resulting in a η3-coordination mode on opposing sides of the ligand, one above the ligand plane and one below. Slight differences are observed in terms of the positions of the (C5Me5)2Sm fragments relative to each other. While for the anthracene ligand both metallocenes coordinate at the opposite middle carbons but at the opposite outer benzannulated rings, in the complex with the 2,3-benzanthracene ligand the samarium moieties are located directly opposite each other. In the case of pyrene, the metallocenes are located on the same end of the molecule. In contrast to PAH complexes of alkaline metals the PAH retains its planarity upon reduction in [(C5Me5)2Sm–PAH] complexes. The Sm–C(PAH) bond lengths are in the range of 2.595(4) to 2.840(4) Å and are comparable to the allyl complexes described above. No single crystals of 6-methylanthracene and acenaphthylene for structural studies were obtained, however, the 2:1 stoichiometry was confirmed via elemental analyses. No reaction of samarocene with corocene was observed, which was explained by its low solubility. The interactions of [(C5Me5)2Sm] and the PAH appears to be rather weak, as THF addition yields [(C5Me5)2Sm(THF)2] and the free hydrocarbon.
 | ||
| Scheme 36 Reaction products of samarocene with the polycyclic aromatic hydrocarbons anthracene, 2,3-benzanthracene and pyrene.142 | ||
The reaction with cyclooctatetraene (COT) proceeded differently and yielded after a reaction time of 12 hours the Sm(III) complexes [(C5Me5)Sm(COT)] and [(C5Me5)3Sm] via rearrangement and ligand exchange.143,144 The corresponding diindenyl complex [(C9H7)2Sm(THF)x] shows a similar reactivity.18 With shorter reaction times of 15 min, [(C5Me5)2Sm(THF)2] and COT not only yielded [(C5Me5)Sm(COT)(THF)] and the oxidatively coupled dimer [(C5Me5)2] with toluene as solvent, but also [(C5Me5)2Sm{O(CH2)4(C5Me5)}(THF)] and [(C5Me5)3Sm].145 The complex [(C5Me5)2Eu] shows no reactivity towards these substrates.
The C5Me5 ligands in decamethyleuropocene undergo coupling reactions with the organic moieties of the respective alkyl or aryl halides such as chlorides and bromides, yielding C5Me5R and EuX2.146
The reactions of the ytterbocene and samarocene with various organic chlorides, bromides and iodides were broadly investigated in terms of kinetics and the reaction mechanism. Organic substrates such as benzylchloride, –bromide and fluoride, butylchlorides, isopropylchloride, methyl iodide, phenyliodide and 1,2-diiodoethane were used.129,146–148 The corresponding diindenyl complex [(C9H7)2Sm(THF)x] shows a similar reactivity.18 The products observed in these reactions with [(C5Me5)2Sm(solvent)n] and [(C5Me5)2Yb(solvent)n] are [(C5Me5)2LnX], [(C5Me5)2LnR], [(C5Me5)LnX2] together with radical coupling products of R (Scheme 37). The reactivity of RX follows the order I > Br > Cl ≫ F and benzyl ≈ tertiary > secondary > primary > phenyl corresponding to an atomic abstraction and concomitant generation of an organic radical. As a result of the reduction potential, samarocene reacts more readily than ytterbocene.
 | ||
| Scheme 37 General reactivity of the metallocenes towards halogenated hydrocarbons (Ln = Sm, Yb; X = F, Cl, Br, I). Not all products shown are formed in each case. Sometimes other byproducts were found.129,146–148 | ||
Even unreactive carbon–fluorine bonds of certain perfluoro-olefins were reductively cleaved by the ethereal adducts of all three metallocenes.149 In general, these reactions yielded [(C5Me5)2LnF] with a terminal Ln–F bond as well as complexes with [(C5Me5)LnF2] fragments. For example, [Yb5(C5Me5)6(μ4-F)(μ3-F)2(μ-F)6] was crystallised as a minor reaction product from the reaction of [(C5Me5)2Yb(OEt2)] and a perfluoro olefin. Structural data for diethylether and THF adducts of [(C5Me5)2LnF] for Sm, Eu and Yb is available.
As expected, the reactivity of the metallocenes with perfluoro-olefins also follows the order of the reduction potential, i.e. [(C5Me5)2Sm] > [(C5Me5)2Yb] > [(C5Me5)2Eu].149 The reaction proceeds fast and rather clean if the substrate can form an allylic perfluoro-radical as an intermediate, which was shown by the reaction with perfluoro-2,4-dimethyl-3-ethylpent-2-ene and perfluoro-2,3-dimethylpent-2-ene. Up to four fluorine atoms were abstracted to ultimately yield perfluorotrienes. Perfluorocyclohexene reacts much slower by initial abstraction of an olefinic fluorine atom. In this case, hydrogen containing products were observed that most likely form via hydrogen abstraction from a C5Me5 ligand or a solvent. Successive defluoronization yields perfluorobenzene. Interestingly, visible light enhances the reaction rate. C6F10 does not react with [(C5Me5)2Eu] in the dark, but when illuminated with a tungsten lamp, conversions have been observed in the NMR spectra. It should also be noted that reactions in toluene proceed faster than in diethylether, which is in accordance with the blocking of the reactive, inner sphere reaction site at the metal centre.
Finally, hexafluorobenzene was reported to react with [(C5Me5)2Yb] in hexane to yield the mixed valent complexes [(C5Me5)6Yb4(μ-F)4] and [(C5Me5)4Yb2(μ-F)] (Scheme 38).150 The latter one can be considered as a Lewis acid–base complex between the Yb(III) complex [(C5Me5)2YbF], in which the fluorine ligand is acting as the electron donor and the divalent [(C5Me5)2Yb], which acts as Lewis-base. This is reflected by the unequal Yb-F bond lengths of 2.317(2) Å (Yb(II)–F) and 2.084(2) Å (Yb(III)–F) in its crystal structure. The organometallic [(C5Me5)2Yb(C6F5)] compound is a by-product of the reaction. The mixed valent complex can also be obtained by reaction with various fluorinated olefins and aryls, namely CFHCH2, CF2CH2, C2F4, PhF and PhCF3. No reaction occurred with the fluorinated alkyls like C2F6 or 1,1,1-CF3CH3.149 According to the authors, the strength of the carbon–fluorine bond is not the decisive factor for the reaction of ytterbocene with fluorinated hydrocarbons, since the C–F bond in C6F6 is stronger than in C2F6 (154 kJ mol−1vs. 127 kJ mol−1), but rather a polarisable group on the fluorine atom together with a free metal coordination site appear to be the necessary requirements for C–F activation.
 | ||
| Scheme 38 Reaction of [(C5Me5)2Yb] with C6F6.150 | ||
In contrast, to these methods, the organolanthanide fluoride [(η5-C5H4tBu)2Sm(μ-F)]3 was prepared by oxidation of [(η5-C5H4tBu)2Sm(THF)2] with Me3SnF.151
 | ||
| Fig. 6 Molecular structure of trimeric [(C5Me5)2Sm(NCtBu)(μ-CN)]3 in the solid state (the rear C5Me5 and the hydrogen atoms are omitted for clarity) (Reproduced from the CIF file CCDC 638959).87 | ||
The reactivity of the two cyano-group bearing molecules 7,7,8,8-tetracyanoquinodimethane (TCNQ) and 1,2,4,5-tetracyanobenzene (TCNB) towards [(C5Me5)2Yb(THF)2] was investigated by Trifonov et al.153 The reaction with an equimolar amount of TCNQ in acetonitrile gave the dinuclear symmetric complex [(C5Me5)2Yb(CH3CN){(μ-CN)2C(C6H4)C(CN)2(C5Me5)}]2 (Fig. 7). The dark red-brown complex was investigated by single crystal X-ray crystallography. During the reaction, a C5Me5-moiety of another [(C5Me5)2Yb] molecule adds to one methylidene moiety, creating a monoanionic bridging ligand. The bond lengths observed in the solid structure point to a Yb(III) compound, as do magnetic measurements. When [(C5Me5)2Yb(THF)2] was reacted with TCNB in DMF, the green complex [(C5Me5)2Yb{(μ-CN)2(C6H4)(CN)2}]2 was formed. In this case, the TCNB moiety stays intact. Each of the dianionic ligands coordinates with both Yb-atoms with opposing cyano groups. The Yb–N bond lengths are in the range of 2.338(5) to 2.363(4) Å. The two aromatic rings of the bridging ligands show a parallel, almost eclipsed orientation indicating an interaction of their π-systems.
 | ||
| Fig. 7 Molecular structure of dinuclear [(C5Me5)2Yb(CH3CN){(μ-CN)2C(C6H4)C(CN)2(C5Me5)}]2 in the solid state (Reproduced from the CIF file CCDC: 707131).153 | ||
For a comparison with the activation of dinitrogen (see below, Scheme 72), the reactivity of samarocene towards azobenzenes and hydrazines was extensively studied. One molar equivalent of [(C5Me5)2Sm(THF)2] reacted with azobenzene in toluene to form the green complex [(C5Me5)2Sm(N2Ph2)(THF)] (Scheme 39).154,155 X-Ray crystallographic data shows the coordination or the ligand in a η2-fashion. The N–N distances of the crystallographically independent molecules in the asymmetric units are 2.390(10) and 2.450(10) Å, meaning significant lengthening upon reduction. The reverse addition of azobenzene to two equivalents samarocene in toluene yielded the blue complex [{(C5Me5)2Sm}2(N2Ph2)] including the azobenzene as a bridging ligand (Scheme 39). The solid state structure shows that each of the samarium atoms coordinates only one of the nitrogen atoms in contrast to the η2-mode in the 1:1 compound. The N–N bond however does not change significantly upon coordination. Instead, a lengthening of N–C bonds was observed. This allows the samarium metal to interact with one of the ortho hydrogen atoms of the phenyl ring. The azobenzene ligand in this molecule can be considered the PhNNPh2−-anion.
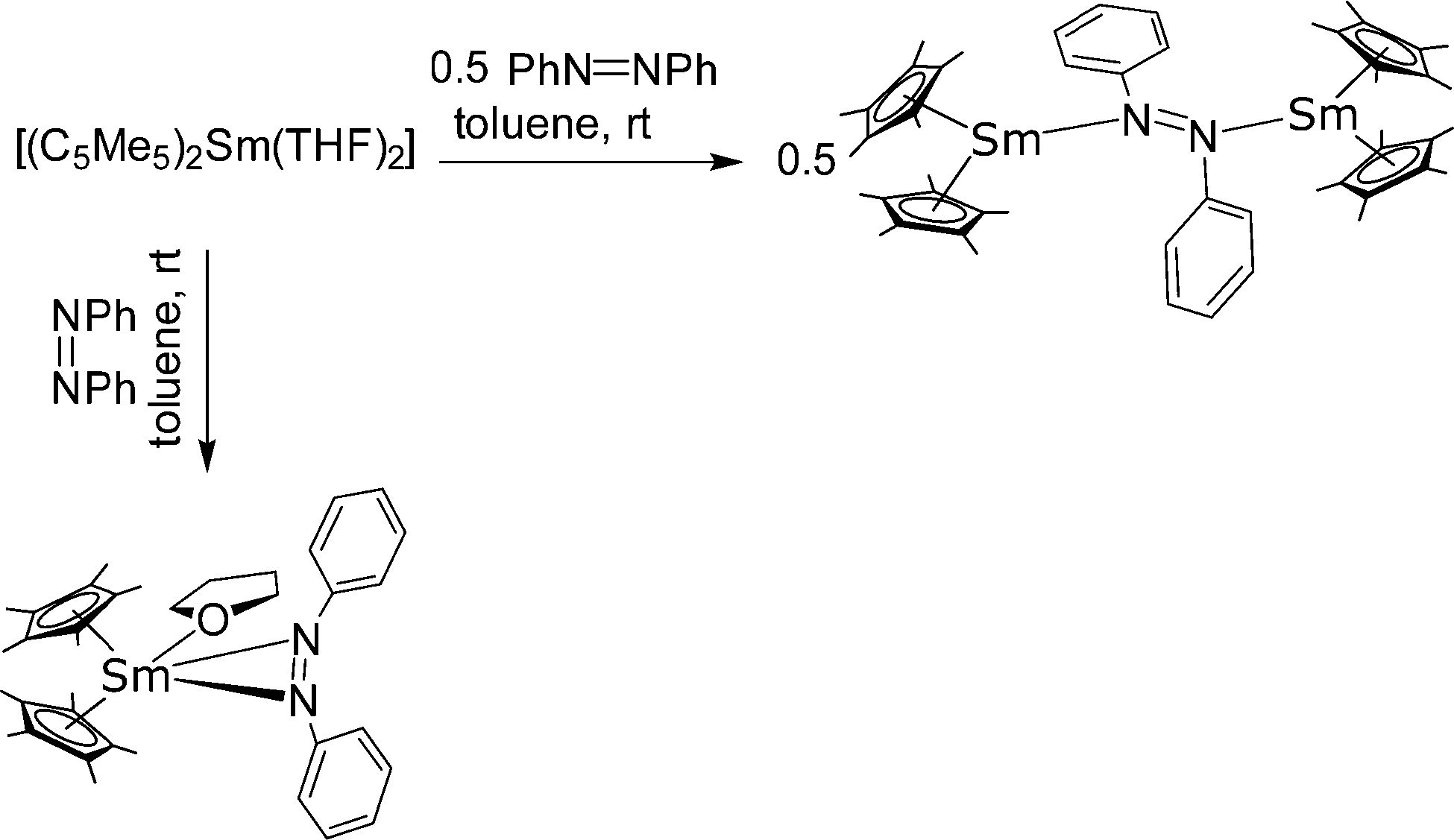 | ||
| Scheme 39 Reactivity of [(C5Me5)2Sm(THF)2] towards different amounts of PhNNPh.154,155 | ||
The reaction of [(C5Me5)2Sm(THF)2] with the analogous hydrazine compound PhHNNHPh in hexane leads to the formation of yellow [(C5Me5)2Sm(NHPh)]xvia N–N bond cleavage.35 The precipitate was recrystallised from THF to give [(C5Me5)2Sm(NHPh)(THF)]. The same product was also observed when [(C5Me5)2Sm(THF)2] was reacted with aniline or phenylhydrazine (Scheme 40). However, unidentified by-products occur and yields are low. The solid state structure of [(C5Me5)2Sm(NHPh)(THF)] reveals a terminal coordinated NHPh-amide. A different reaction pathway was observed when desolvated [(C5Me5)2Sm] was reacted with diphenylhydrazine in toluene. As product [(C5Me5)2Sm(PhNHNPh)] was obtained, which forms the solvate [(C5Me5)2Sm(PhNHNPh)(THF)] after crystallization from THF. In this compound single deprotonated hydrazine acts as a ligand, which coordinates in a η2-fashion to the samarium atom. Further treatment of [(C5Me5)2Sm(PhNHNPh)] with [(C5Me5)2Sm(THF)2] resulted again in [(C5Me5)2Sm(NHPh)(THF)] (Scheme 40).
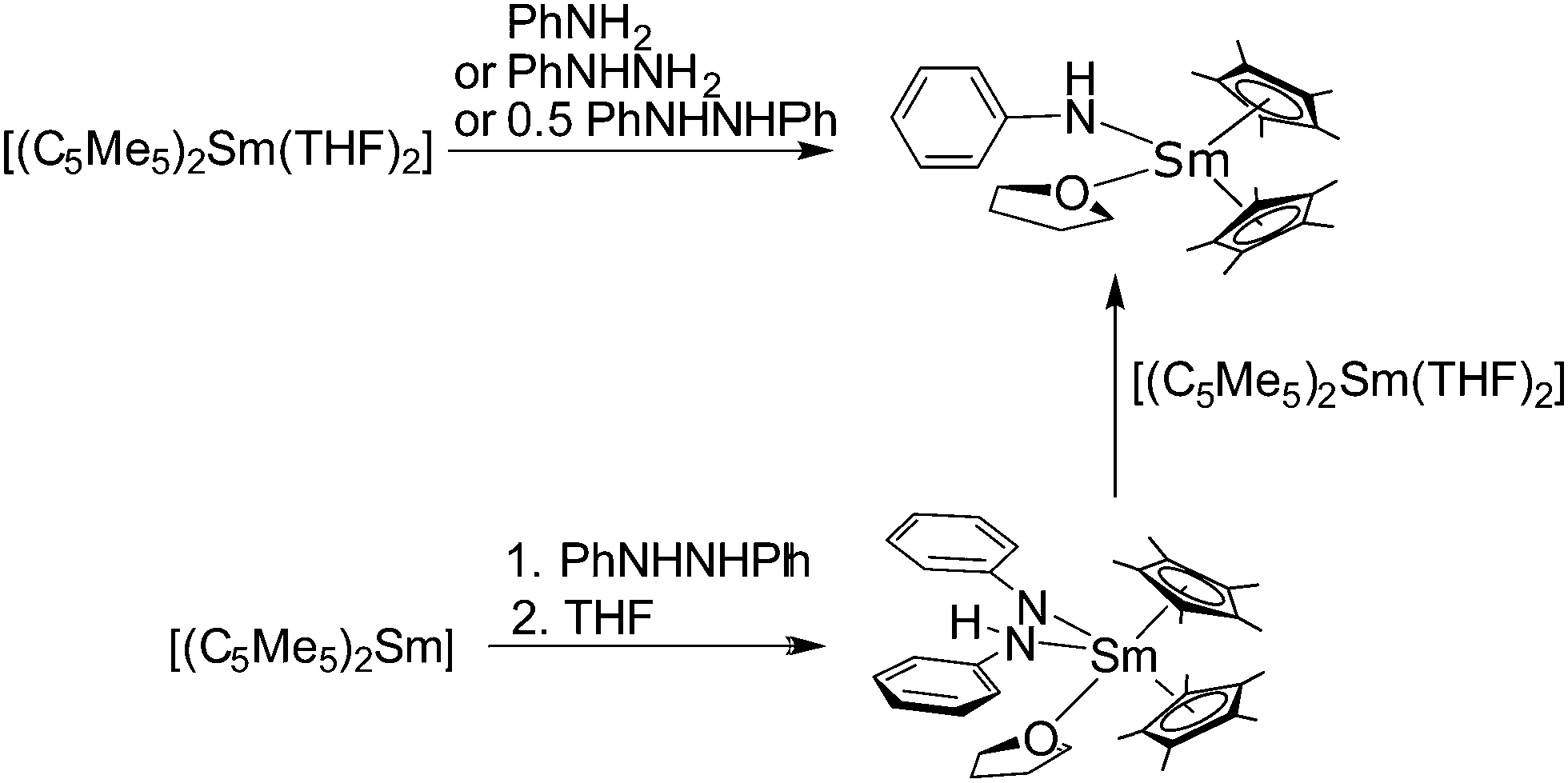 | ||
| Scheme 40 Reactivity of [(C5Me5)2Sm(THF)2] towards aniline, diphenyl hydrazine and phenyl hydrazine and of [(C5Me5)2Sm] towards phenylhydrazine.35 | ||
The parent molecule hydrazine (H2NNH2) reacted with solvent-free samarocene to form the doubly deprotonated dinuclear product [(C5Me5)2Sm(μ-η2:η2-HNNH)Sm(C5Me5)2] as red crystals (Scheme 41). The solid state structure revealed an unsymmetrically bridging mode of the ligand. The hydrazido anion in [(C5Me5)2Sm(μ-η2:η2-HNNH)Sm(C5Me5)2] shows a lengthening in the nitrogen–nitrogen bond compared to hydrazine.
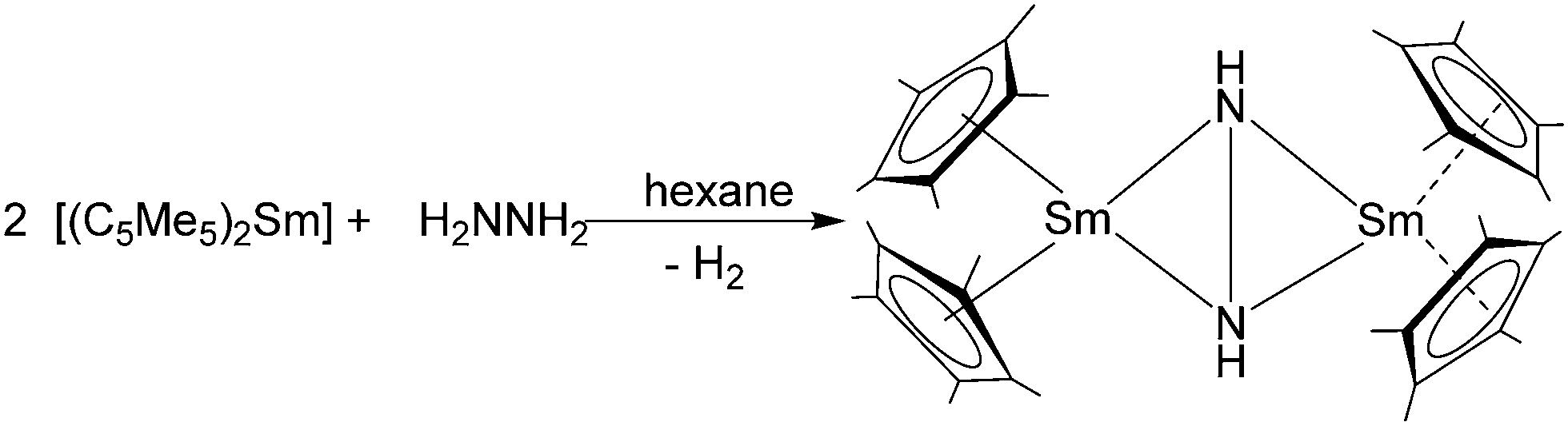 | ||
| Scheme 41 Reaction of [(C5Me5)2Sm] and hydrazine.35 | ||
When desolvated samarocene was reacted with excess hydrazine in benzene, the yellow tetranuclear compound [(C5Me5)4Sm4(N2H2)2(N2H3)4(NH3)2] was isolated.156 The Sm atoms are arranged in a tetrahedral fashion and are bridged by deprotonated hydrazine molecules. During the reaction, one pentamethylcyclopentadienyl ligand of each samarocene unit was protonated. Both HNNH and HNNH2 are present in the molecule as ligands.
A deprotonation was also observed by reacting the neutral formazan ligand L2H (L2 = {PhNNC(4-tBuPh)NNPh} with [(C5Me5)2Sm(THF)2] (Scheme 42). As shown by single crystal X-ray diffraction, the bonding in the six-membered core is delocalised.157
 | ||
| Scheme 42 Reaction of [(C5Me5)2Sm(THF)2] with a formazan.157 | ||
The reaction of [(C5Me5)2Sm(THF)2] with benzaldehyde azine (PhHCNNCHPh) as a further compound with a nitrogen-nitrogen bond resulted in the dinuclear coupling product [{(C5Me5)2Sm}2{μ,η4-(PhCHNNCHPh)2}] (Scheme 43).91 The nitrogen moieties bind in a η2-fashion to the {(C5Me5)2Sm} units. In contrast to the previously described reactions containing C–N bonds, no reduction of the multiple bonds occurs but instead a reductive coupling of the substrate to the bridging ligand. Most likely, the coupling involves some radical intermediates.
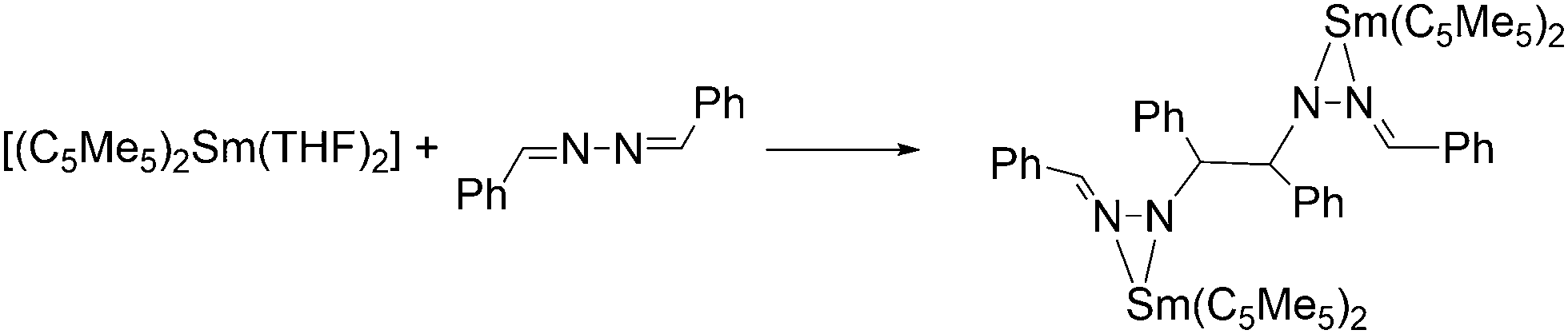 | ||
| Scheme 43 Reaction of [(C5Me5)2Sm(THF)2] with PhCNNCPh.91 | ||
The reaction of either one or two equivalents of benzophenone imine with [(C5Me5)2Sm(THF)2] gave orange red-crystals of [(C5Me5)2Sm(NCPh2)(NH2CHPh2)] (Scheme 44).158 The mechanism was investigated by deuteration experiments. Most likely the reaction proceeds via an electron transfer to an imine, which deprotonates a second molecule present in the reaction mixture.
 | ||
| Scheme 44 Reactions of [(C5Me5)2Sm(THF)2] with different amounts of benzophenone imine.158 | ||
The reduction of [(1,2,4-C5H2tBu3)2Tm] with pyridine resulted in a coupling of the pyridine in para-position to give [{(1,2,4-C5H2tBu3)2Tm}2{μ-(NC5H5-C5H5N)}], in which the metal atoms are bridged by a 1,1′-bis(1,4-dihydropyridylamide) ligand (Scheme 45).39
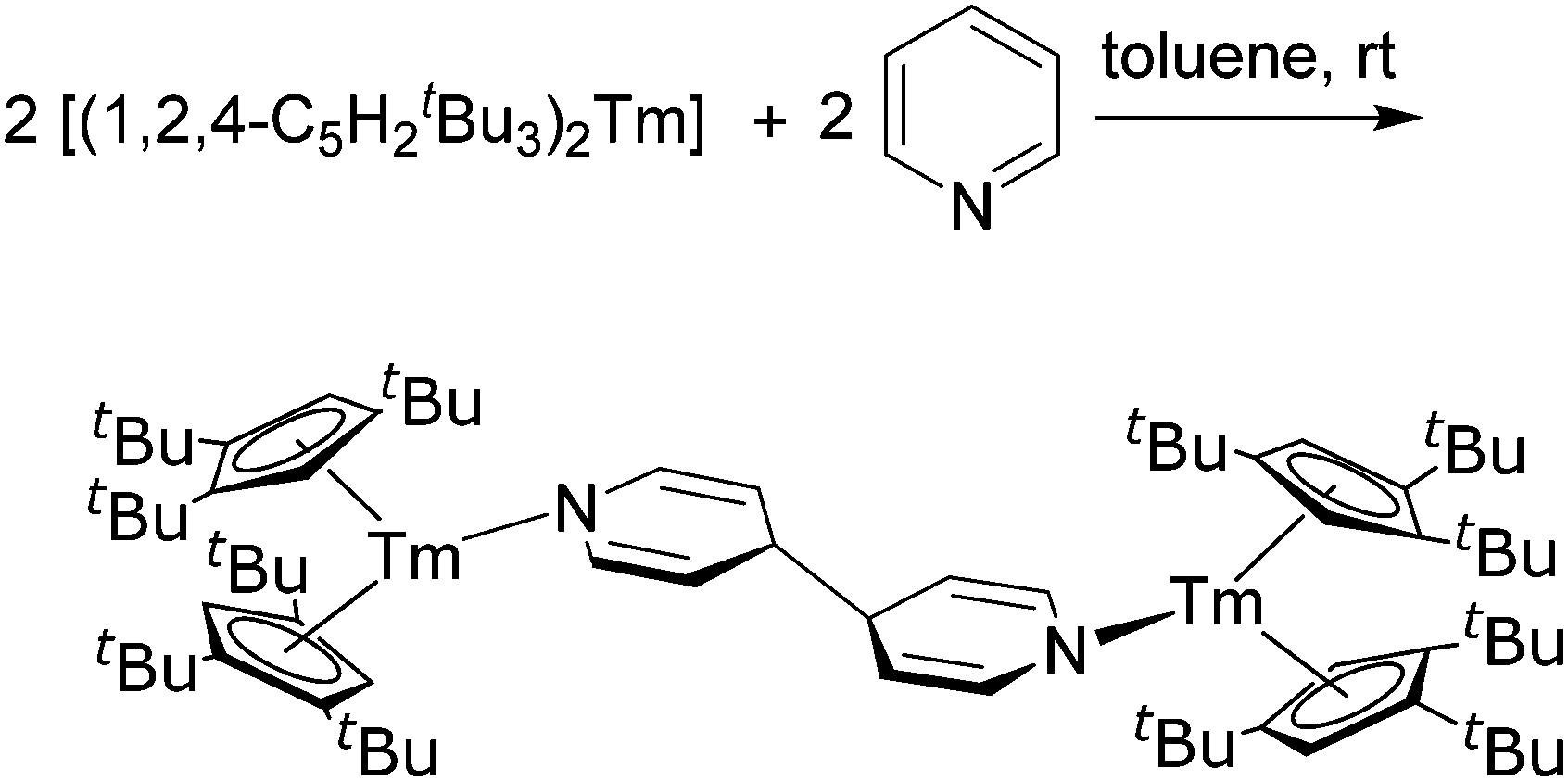 | ||
| Scheme 45 Reactions of [(1,2,4-C5H2tBu3)2Tm] with pyridine.39 | ||
A similar reaction of [(C5Me5)2Sm(THF)2] with pyridazine in toluene resulted in the transfer of one electron and coupling of two complexes to give yellow-orange [{(C5Me5)2Sm(THF)}2{μ-(N2C4H4)2}] (Scheme 46).91 The product was studied by single crystal X-ray crystallography. The neighbouring N atoms are coordinating the samarium atoms in a η2 coordination mode. Coupling occurs via one of the carbon atoms not directly adjacent to the nitrogen atoms.
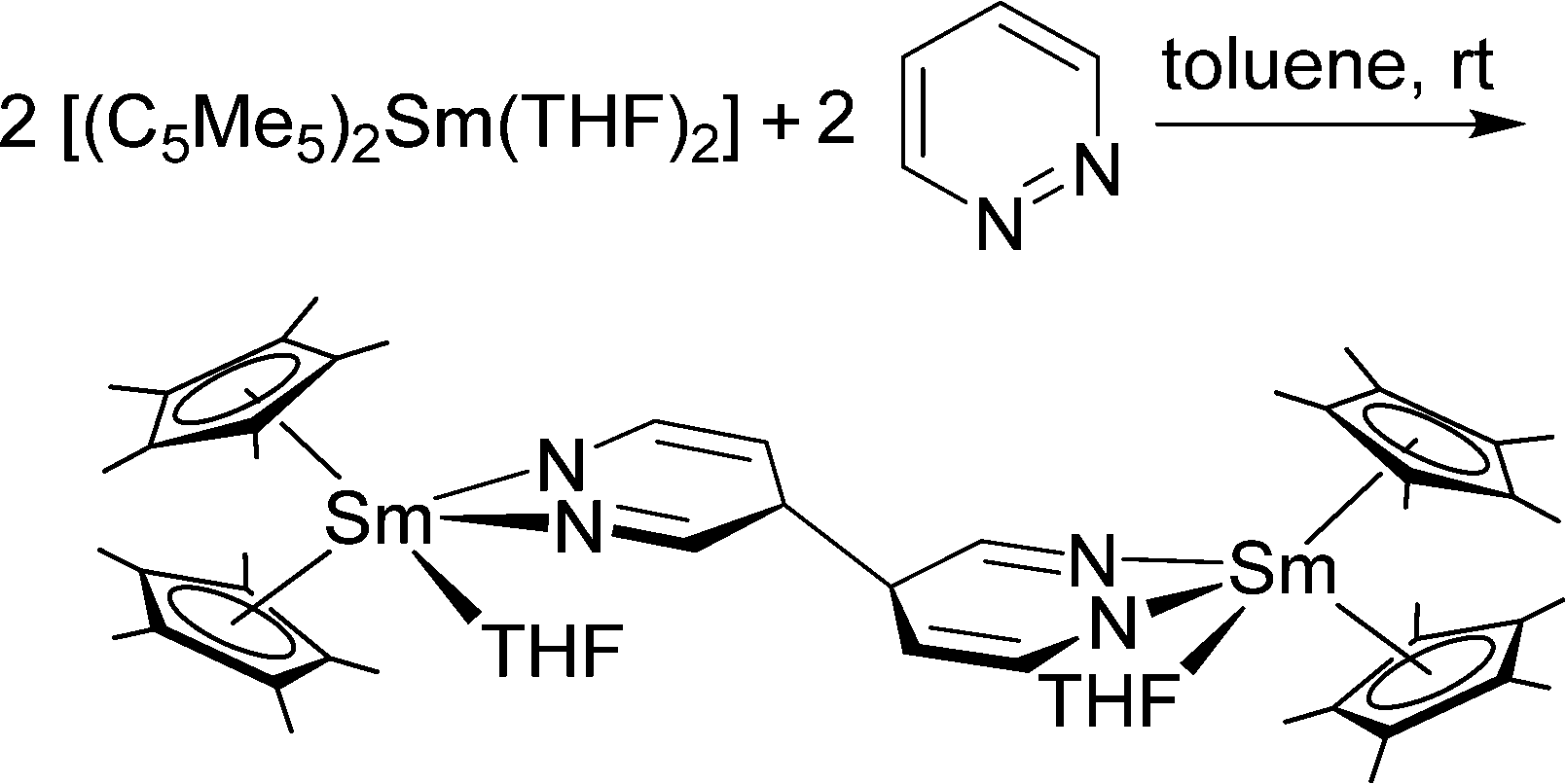 | ||
| Scheme 46 Reaction of [(C5Me5)2Sm(THF)2] and pyridazine.91 | ||
Typically, [(C5Me5)2Sm(THF)2] reacts with multiple bonds to give either [{(C5Me5)2Sm}2(substrate)] with a doubly-reduced substrate, or reductive coupling occurs to form [{(C5Me5)2Sm}2(substrate–substrate)] species with a dianionic, coupled-substrate moiety.159 When [(C5Me5)2Sm(THF)2] reacts with 1,2-bis(2-pyridyl)ethene (py), both types of trivalent Sm-complexes are formed at the same time to yield [{(C5Me5)2Sm}2(μ-η2:η2-pyCHCHpy)] and [{(C5Me5)2Sm}2(μ-η3:η3-1,2,3,4-(py)4C4H4)]. Spectroscopic data was consistent with a trivalent samarium complex. However, the crystallographic data was not sufficient for a detailed bond analysis.
As mentioned above, the reaction of [(C5Me5)2Yb(OEt2)] with 1,10-phenanthroline leads to oxidation of the metal and a reduced radical ligand,93 which is in a monomer/dimer equilibrium. A similar coupling of 1,10-phenanthroline was observed by using the stronger reducing agents [(C5Me5)2Sm(OEt2)] or the tert-butyl substituted metallocenes [(1,3-C5H3tBu2)2Sm], [(1,2,4-C5H2tBu3)2Sm], and [(1,2,4-C5H2tBu3)2Tm].46 In all cases a C–C bond formation on the 4-position of the phenanthroline ring was observed in the solid state. Upon analysis of the solution structure, the authors suggest a thermally reversible C–C coupling in all cases.
An interesting behaviour is found in the reaction of [(C5Me5)2Yb(OEt2)] with 4,5-diazafluorene (Scheme 47).160 The reaction in toluene gave the green intermediate-valent species [(C5Me5)2Yb(N2C11H8)], which could not be structurally characterised. The green compound transformed into the red trivalent diazafluorenyl compound [(C5Me5)2Yb(N2C11H7)] via hydrogen abstraction. This product was structurally characterised with X-ray crystallography. The Yb–N distances are 2.372 ± 0.001 Å, which are in the range of ionic, trivalent [(C5Me5)2Yb(bipy)] complex. Extensive mechanical investigations on the formation of the complex as well as kinetic measurements were carried out. It was shown that [(C5Me5)2Yb(N2C11H7)] is formed via an isolable dimeric complex, formed by the coupling of two diazafluorene ligands and concomitant H2 abstraction.
 | ||
| Scheme 47 Reaction of [(C5Me5)2Yb(OEt2)] with diazafluorene.160 | ||
No C–C coupling occurs by using redox-active ligands such as diazabutadienes. Decamethylytterbocene reacted with N,N-di-tert-butyl-1,4-diazabutadiene to give the oxidised complex [(C5Me5)2Yb(N2(tBu)2C2H2)] via one electron reduction of the diene (Scheme 48).161,162 Similarly, [(C5Me5)2Yb(THF)2] reacts with the diazabutadiene 2-MeC6H4NC(Me)C(Me)NC6H4Me-2 to the corresponding complex [(C5Me5)2Yb(2-MeC6H4NC(Me)C(Me)NC6H4Me-2)] ligated by a diazabutadiene anion.163 Similar studies were performed with other ytterbocenes or other diazabutadienes.162–164 Reaction with samarocene gave the analogous Sm(III) complex.143 The redox potential of [(C5Me5)2Eu(Et2O)] is too low to reduce the N,N-di-tert-butyl-1,4-diazabutadiene and the respective Lewis-acid base complex was formed.165 In contrast, when N,N-di-pentafluorophenyl-1,4-diazabutadiene derivative was reacted with a [(C5Me5)Eu(Et2O)], the corresponding Eu(III) complex [(C5Me5)2Eu(N2(C6F5)2(C(Me))2)] with the reduced ligand as a radical anion was obtained (Scheme 48). The pentafluorophenyl groups induce a higher electron affinity of this ligand.
 | ||
| Scheme 48 Reactions of ytterbocene and europocene with differently substituted diazabutadienes.161,165 | ||
Another redox-active ligand is 1,2-bis(imino)acenaphthene (BIAN), which consists of both a naphthalene ring and a 1,4-diaza-1,3-butadiene moiety. The reaction of [(C5Me5)2Ln(OEt2)] (Ln = Sm, Eu) with an equimolar quantity of the corresponding R-BIAN ligand (R = mesityl, tert-butyl, p-methoxyphenyl) in toluene solution at ambient temperature resulted in [(C5Me5)2Sm(mes-BIAN)], [(C5Me5)2Eu(tBu-BIAN)] and [(C5Me5)2Eu(p-MeO-BIAN)]. Magnetic measurements indicate a trivalent oxidation state of the lanthanide metal (Scheme 49).166 Thus, the BIAN ligand is reduced by one electron. In contrast, the reaction of [(C5Me5)2Sm(OEt2)] with one equivalent of the sterically encumbered ligand, dpp-BIAN (dpp = 2,6-diisopropylphenyl) in THF solution resulted in the loss of a C5Me5 group and formation of [(C5Me5)Sm(dpp-BIAN)(THF)].166 Here a two-electron reduction of the dpp-BIAN ligand has taken place, which is also supported by the corresponding bonding parameters.
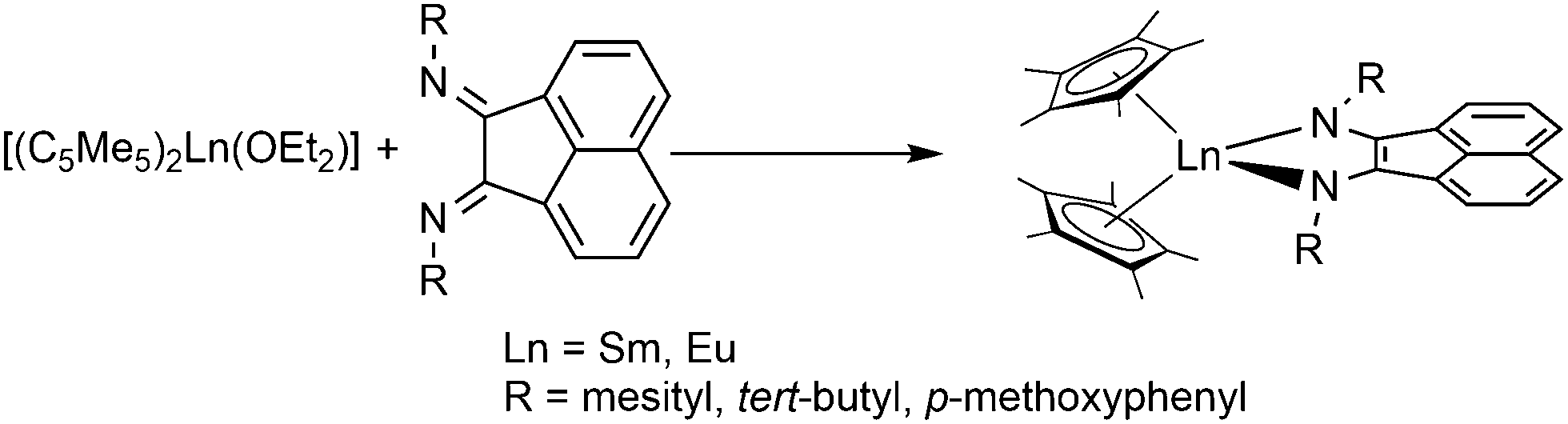 | ||
| Scheme 49 Reactions of samarocene and europocene with differently substituted 1,2-bis(imino)acenaphthenes (BIAN).166 | ||
A metal-induced oxidation of the metallocenes was observed by reacting [(C5Me5)2Yb] with thallium compounds. In these cases, the thallium atom is reduced upon reaction and the corresponding anion is transferred to the lanthanide metal. Following this strategy, [(C5Me5)2Yb] reacts with [Tl(Ph2pz)] and [Tl(azin)] (Ph2pz = 3,5-diphenylpyrazolate, azin = 7-azaindolate) to give [(C5Me5)2Yb(Ph2pz)] and [(C5Me5)2Yb(azin)]. In both cases, the N-donor ligands coordinate with both nitrogen atoms to the Yb-atom.167
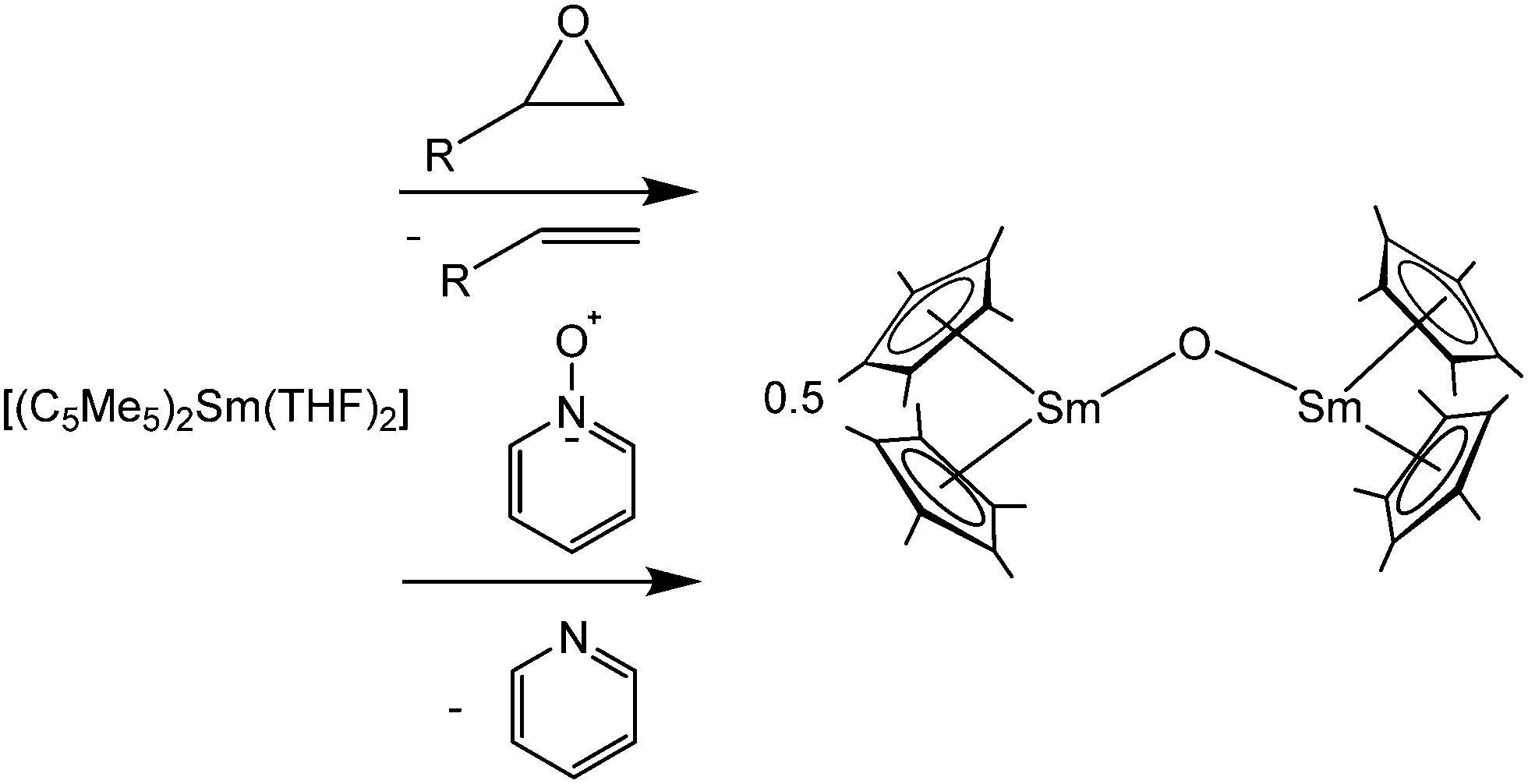 | ||
| Scheme 50 Oxidation of [(C5Me5)2Sm(THF)2] with epoxides and pyridine N-oxide.81 | ||
The cleavage of allylic-, propargylic and vinylic ethers by [(C5Me5)2Sm(THF)n] was investigated via NMR.169–171 Allylic ethers are cleaved by [(C5Me5)2Sm(THF)2], as evidenced by various examples, e.g. the reaction of desolvated [(C5Me5)2Sm] and allyl benzyl ether gave the allyl compound [(C5Me5)2Sm(C3H5)] and the alkoxide compound [(C5Me5)2Sm(OBn)] (Scheme 51).
 | ||
| Scheme 51 Reactivity of [(C5Me5)2Sm(THF)2] towards allylic ethers to yield allyl complexes.169 | ||
Alkoxide formation was seen upon the reaction of [(C5Me5)2Sm(THF)2] with 2,3,5,6-tetramethylphenol in toluene (Scheme 52).172 Concomitant with the release of hydrogen, an orange crystalline solid was isolated. It was identified as the phenolate complex [(C5Me5)2Sm(OC6HMe4)] by X-ray crystallography. As expected, the phenolate ligand binds via the oxygen atom to the Sm centre with a Sm–O bond of 2.13(1) Å. The observed results are in contrast to the reaction of [(C5Me5)2Sm(THF)2] with very bulky phenols (see above, Scheme 11).84 In these reactions the samarium atom is not oxidised but one of the C5Me5 ligands is protonated.
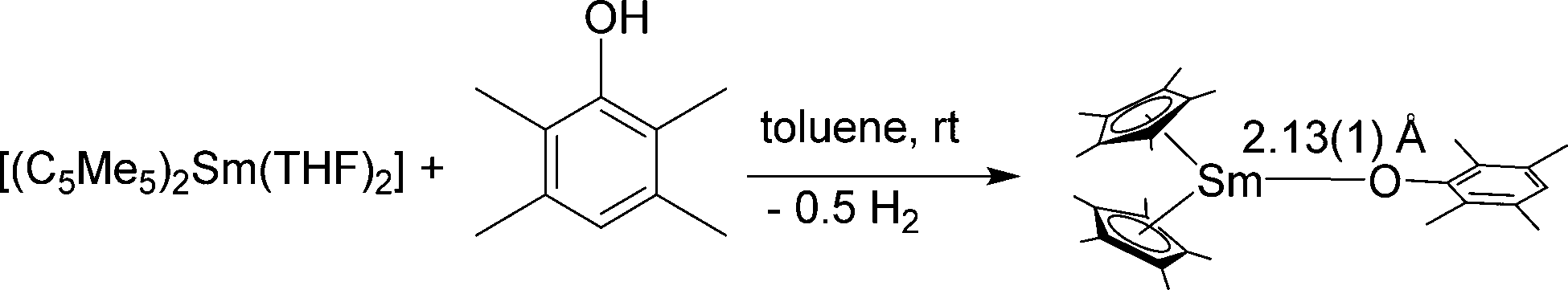 | ||
| Scheme 52 Reaction of [(C5Me5)2Sm(THF)2] with 2,3,5,6-tetramethylphenol.172 | ||
Trivalent alkoxide complexes were obtained by the reaction with diorganoperoxides of the type R-OO-Ar.173 The reaction of [(C5Me5)2Yb(NH3)2] and R-OO-Ar in toluene (R = CMe3, SiMe3) yields products of the composition [(C5Me5)2Yb(OR)(NH3)] as orange solids (Scheme 53). No structural data exist; however, the products were characterised by NMR spectroscopy, elemental analysis, IR spectroscopy and mass spectrometry. The corresponding chalcogenide compounds of S, Se and Te are similarly accessible (see below, Scheme 59).
 | ||
| Scheme 53 Reactivity of decamethylytterbocenes towards aryl substituted organic peroxides.173 | ||
Note, the reaction of [(C5Me5)2Sm(THF)2] with bulky phenols does not give Sm(III) compounds, but dimeric, divalent complexes [(C5Me5)Sm(μ-OAr)]2 (Ar = C6H3tBu2-2,6, C6H2tBu2-2,6-Me-4 and C6H2tBu3-2,4,6)) (see above, Scheme 11).84
Benzoate complexes are accessible by the oxidation of divalent Yb with thallium reagents.6,174 Thus, [(C5H4Me)2Yb(O2CPh)]2 was obtained by reaction of [(C5H4Me)2Yb] with Tl(O2CPh) in THF.174
Multinuclear complexes are observed if the divalent metallocenes of the lanthanides were reacted with 3,6-di-tert-butyl-o-benzoquinone (3,6-dbbq).175 For [(C5Me5)2Sm(THF)2] and [(C5Me5)2Yb(THF)] the reaction with 3,6-dbbq in hexane resulted in the dinuclear compounds [{(C5Me5)Ln}2(dbcat)2] (Fig. 8), in which the former quinone ligand has been reduced to its catecholate (dbcat) form. During the reaction, each lanthanide ion loses one pentamethylcyclopentadienyl ligand. The crystal structure was determined for both compounds. The structure is centrosymmetric, with one of the oxygen atoms of each catecholate ligand bridging the two lanthanide ions. The bond length for the non-bridging oxygen atoms are 2.3127(18) and 2.2269(17) Å (Sm) and 2.2264(19) and 2.1285(18) Å for the Yb complex. The Ln–O bond to the neighbouring Ln ion is slightly longer (0.09 Å for Sm, 0.04 Å for Yb). The lesser reducing europocene yields a different product. The trinuclear mixed-valent compound [(C5Me5Eu)(Eu(THF))2(dbcat)3] is obtained (Fig. 8), in which only one Eu ion is in +III oxidation state. One europium ion has retained one cyclopentadienyl ligand, while the two others have lost both cyclopentadienyl ligands. The structure is rather complex, and the Eu ions have different ligand environments. Every oxygen-catechol atom has a bridging function. The different catecholate bonds are in the wide range from 2.280(2) to 2.504(2) Å, due to the different oxidation states of the three Eu ions.
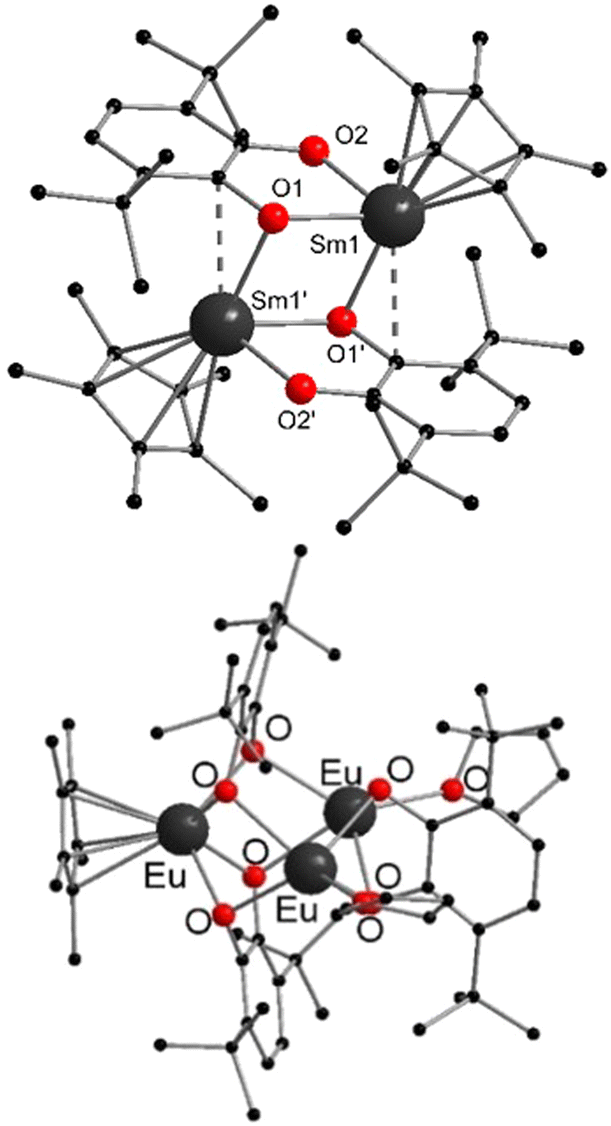 | ||
| Fig. 8 Molecular structures of [{(C5Me5)Sm}2(dbcat)2] (top) and [(C5Me5Eu)(Eu(THF))2(dbcat)3] (bottom) in the solid state (Reproduced from the CIF file CCDC: 1409389, 1409391).175 | ||
As an example for a ketone, the reactivity of fluorenone was investigated in the reaction with ytterbocene and samarocene.176 In both cases, the THF solvates reacted in THF with one equivalent of fluorenone to yield the corresponding radical anion ketyl complexes, [(C5Me5)2Ln(III)(˙OC13H8)(THF)] (Scheme 54). Further reaction with hexamethylphosphoramide (HMPA) led to the replacement of the THF molecule, giving rise to [(C5Me5)2Ln(III)(˙OC13H8)(HMPA)]. The structures of [(C5Me5)2Sm(˙OC13H8)(THF)] and [(C5Me5)2Yb(˙OC13H8)(HMPA)] were determined. Ln–O distances of 2.234(7) Å (Sm) and 2.108(7) Å (Yb) were measured.
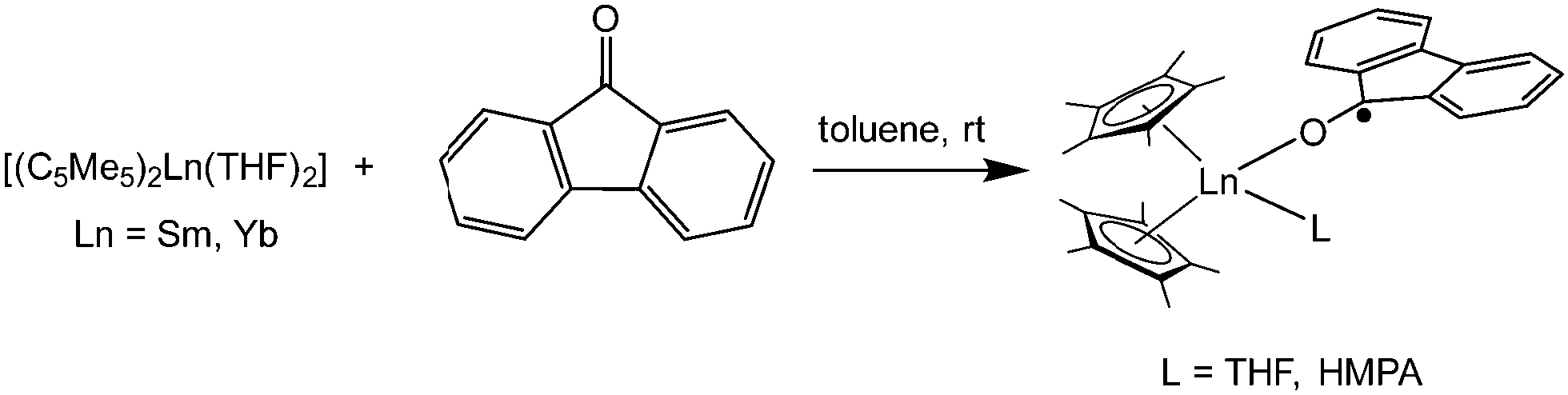 | ||
| Scheme 54 Reaction of decamethylsamarocene and decamethylytterbocene with fluorenone.176 | ||
A dihydroindenoindene diolate complex was obtained by reacting [(C5Me5)2Sm(THF)2] successively with C6H5CCC6H5 and CO to yield the trivalent seven-coordinate samarium complex [{(C5Me5)2Sm}2(OC16H10)] (Scheme 55).177 [{(C5Me5)2Sm}2(OC16H10)] can be recrystallised from THF which results in the eight coordinated THF-solvate [{(C5Me5)2(THF)Sm}2(OC16H10)]. The average Sm–C(ring) length is in the seven-coordinate complex 2.70(3) Å and thus shorter than those of the eight-coordinate complex with a bond length of 2.75(2) Å. The Sm–O(O2C16H10) bond lengths are 2.08(2) and 2.099(9) Å, respectively. As expected, the bond lengths of the seven-coordinate complex are shorter than of the eight-coordinate complex.
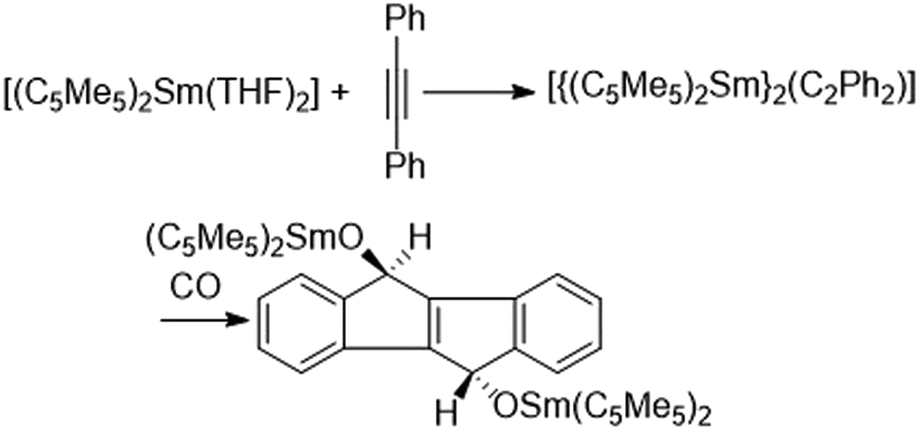 | ||
| Scheme 55 Reaction of [(C5Me5)2Sm(THF)] with diphenylacetylene and CO.177 | ||
:1 stoichiometry of Sm to Bi (Scheme 56).179 The crystal structure of dark red [(C5Me5)2Sm(μ-η2:η2-Bi2)Sm(C5Me5)2] was determined revealing a bridging Bi22− moiety side-on coordinated with the (C5Me5)2Sm units. The Bi-Bi distance is 2.851(1) Å and thus shorter than typical Bi-Bi single bond distances, which range from 2.990(2) to 3.092(2) Å.180–184 For the Sm-Bi distances values ranging from 3.265(1) Å to 3.311(1) Å were determined. The C5Me5–Sm–C5Me5 axes in [(C5Me5)2Sm(μ-η2:η2-Bi2)Sm(C5Me5)2] are reminiscent of the prominent N2 complexes, but are arranged coplanar and not perpendicular as in the corresponding N2 complex (Scheme 72).202
 | ||
| Scheme 56 Reaction of [(C5Me5)2Sm(THF)2] with Ph3Bi.179 | ||
Though no isolable products were obtained by treatment of [(C5Me5)2Sm] with SbPh3, the reaction with the aliphatic stibine SbnBu3 resulted in a complex mixture, containing the isolable product [{(C5Me5)2Sm}3(μ-η2:η2:η1-Sb3)].185 In contrast to the dinuclear pnictogenic complexes of N and Bi, a trinuclear Zintl type anion Sb33− was formed. X-Ray crystallographic studies showed Sb–Sb distances of 2.689(1) and 2.686(1) Å. Two of the (C5Me5)2Sm moieties are bound to two Sb anions of the triangular anion, while one moiety binds only to one and has an additional THF molecule bound to satisfy the coordination sphere.
When [(C5Me5)2Sm] was reacted with Ph2PPPh2 or Ph2AsAsPh2, reductive cleavage of the E–E bond occurred and the trivalent pnictogenide compounds [(C5Me5)2Sm(EPh2)] were formed (Scheme 57).178,186 When THF was present either as a solvent or in the reactant, the corresponding THF-complexes [(C5Me5)2Sm(EPh2)(THF)] were isolated. However, these compounds were unstable and ether cleavage of the coordinated THF took place, yielding a diphenylpnicgtogenyl-functionalised butoxide ligand. However, the phosphide [(1,3-C5H3tBu2)2Sm(PPh2)], which was prepared from [(1,3-C5H3tBu2)2Sm] and Ph2PPPh2, is not prone to ring-opening owing to insufficient space in the Sm coordination sphere for a THF ligand.186
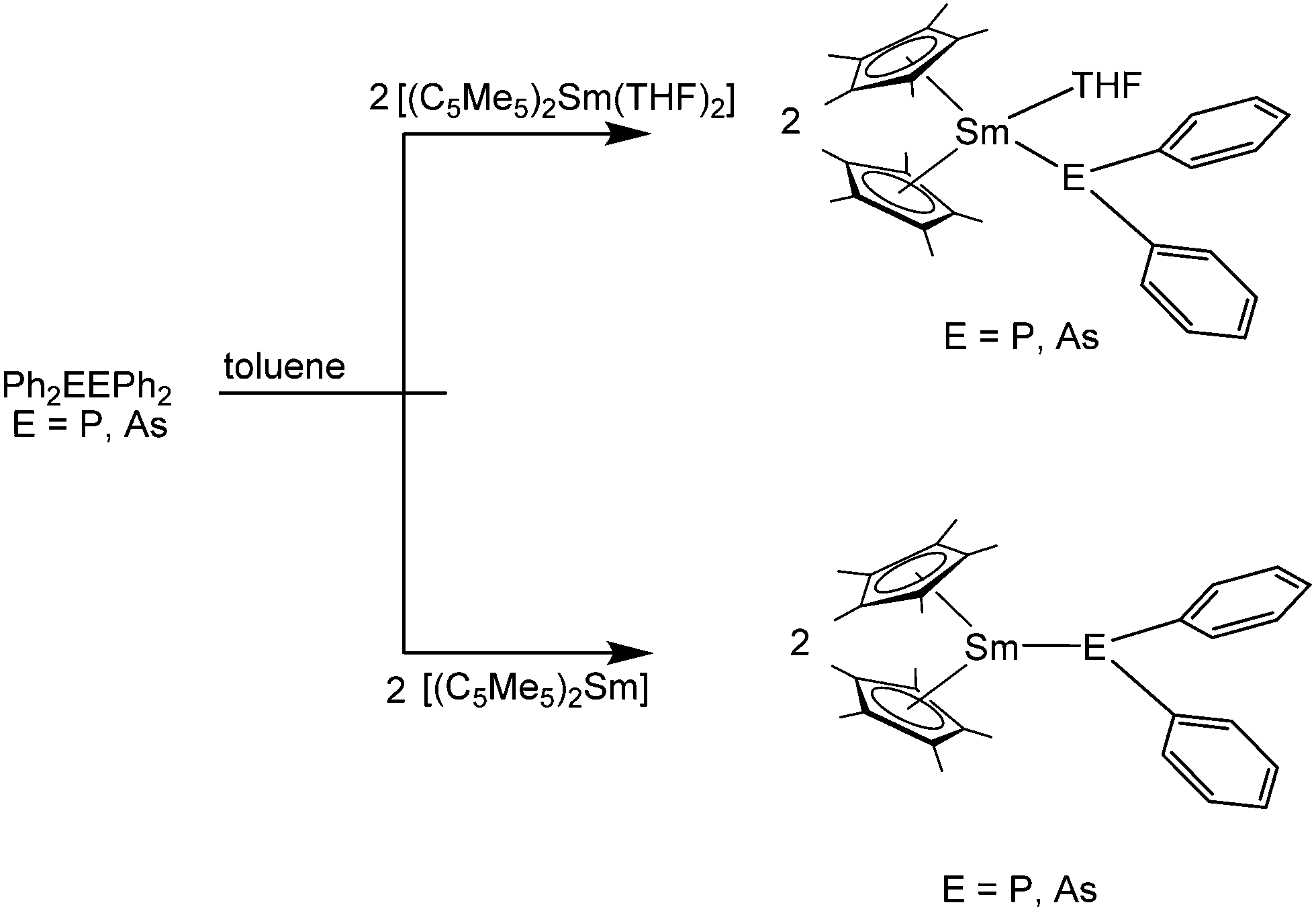 | ||
| Scheme 57 Reactivity of decamethylsamarocene towards diphosphides and diarsenides.180,188 | ||
The crystal structure of [(C5Me5)2Sm(AsPh2)] shows an unsymmetrically coordinated diphenylarsenide ligand that is tilted in a way to enable an η2-interaction of a phenyl ring with the samarium atom, emphasizing the high Lewis acidity of the large Sm(III) ion. The geometry around the arsenic atom strongly deviates from pyramidal or planar geometry. The Sm–As bond lengths of the two crystallographically independent molecules are 2.973(3) and 2.966(3) Å. In contrast, the AsPh2 ligand in [(C5Me5)2Sm(AsPh2)(THF)] is considerably less distorted and features an Sm–As longer bond (3.049(3) Å).
For the corresponding phosphide, only the ring-opened THF product was structurally characterised. In contrast to the arsenic compound, the complex crystallises either as a dimer [{(C5Me5)2Sm}(μ-OC4H8PPh2)]2 or as a polymer [{(C5Me5)2Sm}(μ-OC4H8PPh2)]∞. The Sm atoms are linked by the butoxide ligands via oxo functions and neutral phosphorus donors.
Using a 4:1 stoichiometry of desolvated [(C5Me5)2Sm] to Ph2EEPh2 in non-polar solvents the reaction resulted in mixed valent complexes of the type [(C5Me5)2Sm(μ-EPh2)Sm(C5Me5)2]. The arsenic compound [(C5Me5)2Sm(μ-AsPh2)Sm(C5Me5)2] was only detected by NMR spectroscopy as it quickly decomposes. The phosphorus compound [(C5Me5)2Sm(μ-PPh2)Sm(C5Me5)2] was structurally characterised; however, X-ray data was not of sufficient quality to determine bond distances. The connectivity of all atoms was confirmed, showing a bridging phosphide between the samarium atoms. The reactions of [(C5Me5)2Sm] or [(C5Me5)2Sm(THF)2] with either Ph2SbSbPh2 or Ph2BiBiPh2 did not yield any products with Sm–E bonds. Instead [(C5Me5)2Sm(Ph)] was isolated.
In a different series, phospholyl and arsolyl ligands, such as [(C4Me4P)2], [(C4H2Me2P)2], [(C4-tBu2H2P)2], and [(C4H2Me2As)2] were reacted with [(C5Me5)2Sm(Et2O)] to obtain the samarium(III) complexes [{(C5Me5)2Sm}(C4Me4P)], [{(C5Me5)2Sm}(C4H2Me2P)], [{(C5Me5)2Sm}(C4-tBu2H2P)] and [{(C5Me5)2Sm}(C4H2Me2As)], respectively.187 The reaction of [(C5Me5)2Sm(Et2O)] with [Tl(C4H4P)] yielded [{(C5Me5)2Sm}(C4H4P)].
The reactivity of 2-tert-butyl-1-phosphaethyne tBuCP towards [(C5Me5)2Sm(THF)2] in toluene was investigated (Scheme 58).188 Bright red crystals of [{(C5Me5)2Sm}2(μ-tBuCP–PC-tBu)] were isolated. The reductive coupling of two molecules phospha-alkyne took place to form a P–P bond concomitant with the formation of double bonds between the phosphorus and the carbon atoms. The newly formed ligand bridges two (C5Me5)2Sm(III) moieties on opposing sides of the ligand via one P and one C atom. The Sm–P distances are 2.952(2) and 2.945(2) Å and the Sm–C bond lengths are almost equidistant with 2.557(6) and 2.556(6) Å.
 | ||
| Scheme 58 Reaction of [(C5Me5)2Sm(THF)2] with tBuCP.188 | ||
 | ||
| Scheme 59 Reactivity of decamethylytterbocene towards organic disulfides, diselenides and ditellurides.173,189 | ||
Analogous reactions occurred when either [(C5Me5)2Yb(NH3)2] or [(C5Me5)2Yb(Et2O)] was reacted with organodichalcogenides with different organic moieties such as phenyl or mesityl. Red compounds of the type [(C5Me5)2Yb(ER)(L)] were isolated. The crystal structures of [(C5Me5)2Yb(TePh)(NH3)] and [(C5Me5)2Yb(SPh)(NH3)] (R = phenyl or substituted phenyl group) were determined and revealed Yb–Te bonding distance of 3.039(1) Å and the Yb–S bond length of 2.670(3) Å.
A similar reactivity was observed by using metallocenes with bulkier substituents on the five-membered ring. However, no solvent is coordinated with the metal atoms of these products. Thus, the reaction of [(CpBz5)2Sm] with Ph2E2 (E = Se, Te) at room temperature in THF gave the monometallic compounds [(CpBz5)2Sm(SePh)] and [(CpBz5)2Sm(TePh)].48
Pentavalent phosphine chalcogens readily oxidise samarocene to yield chalcogenide-bridged dinuclear compounds.190 [(C5Me5)2Sm(THF)2] was reacted with SPPh3 or SePPh3 in THF and [{(C5Me5)2Sm(THF)}2(μ-S)] and [{(C5Me5)2Sm(THF)}2(μ-Se)] were obtained as yellow (S) or orange (Se) crystals (Scheme 60). The structures are comparable to the oxygen-bridged product described above. Hence, two samarium atoms are bridged by one E2− ligand. One additional THF molecule is bound to each metal centre to saturate the coordination sphere. The two Sm–E distances are of almost equal length with values of 2.783(1) and 2.779(1) Å for the selenium complex and 2.663(1) and 2.665(1) Å for the sulphur analogue in accordance with the smaller atomic radius of S.
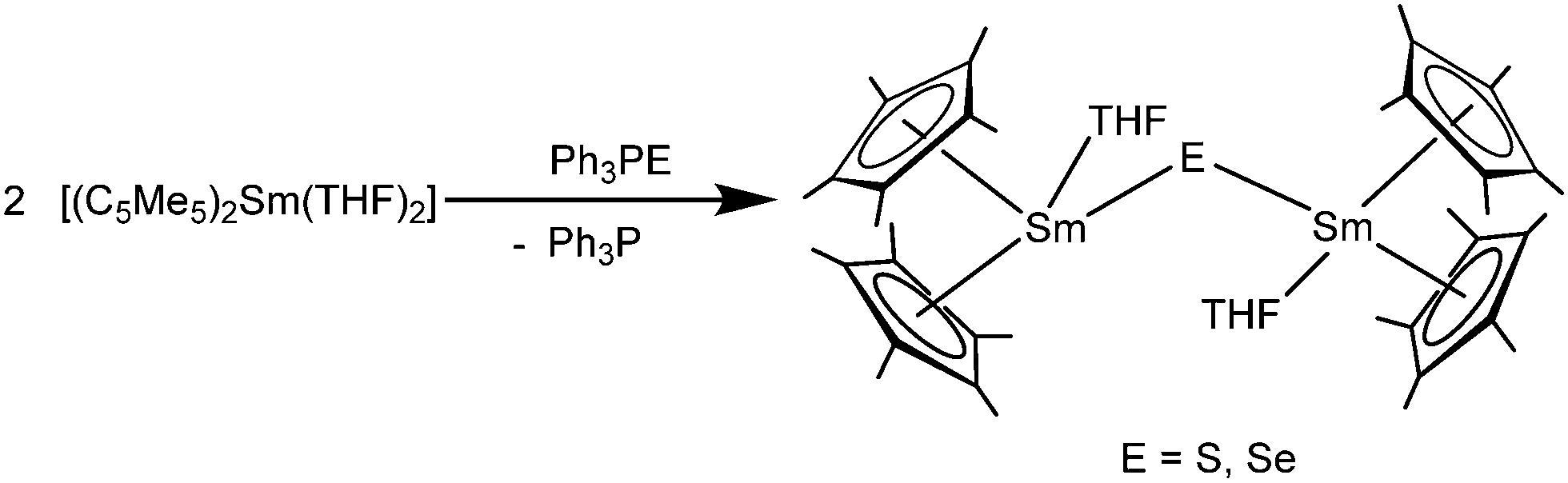 | ||
| Scheme 60 Reaction of [(C5Me5)2Sm(THF)2] with phosphine selenides and tellurides.190 | ||
Ytterbocene was also oxidised by SPPh3 or SePPh3 to give the corresponding chalcogenide complexes, in which the E2− bridges the two metal centres (Scheme 61).191 However, due to the smaller ionic radius of Yb3+, no additional THF ligands are bound to the metal. The crystal structure of purple [{(C5Me5)2Yb}2(μ-Se)] reveals a centrosymmetric molecule with equal Yb–Se distances of 2.621(1) Å. The tellurium analogue was prepared from [(C5Me5)2Yb(OEt2)] and tri-n-butylphosphine telluride in hexane (Scheme 61).
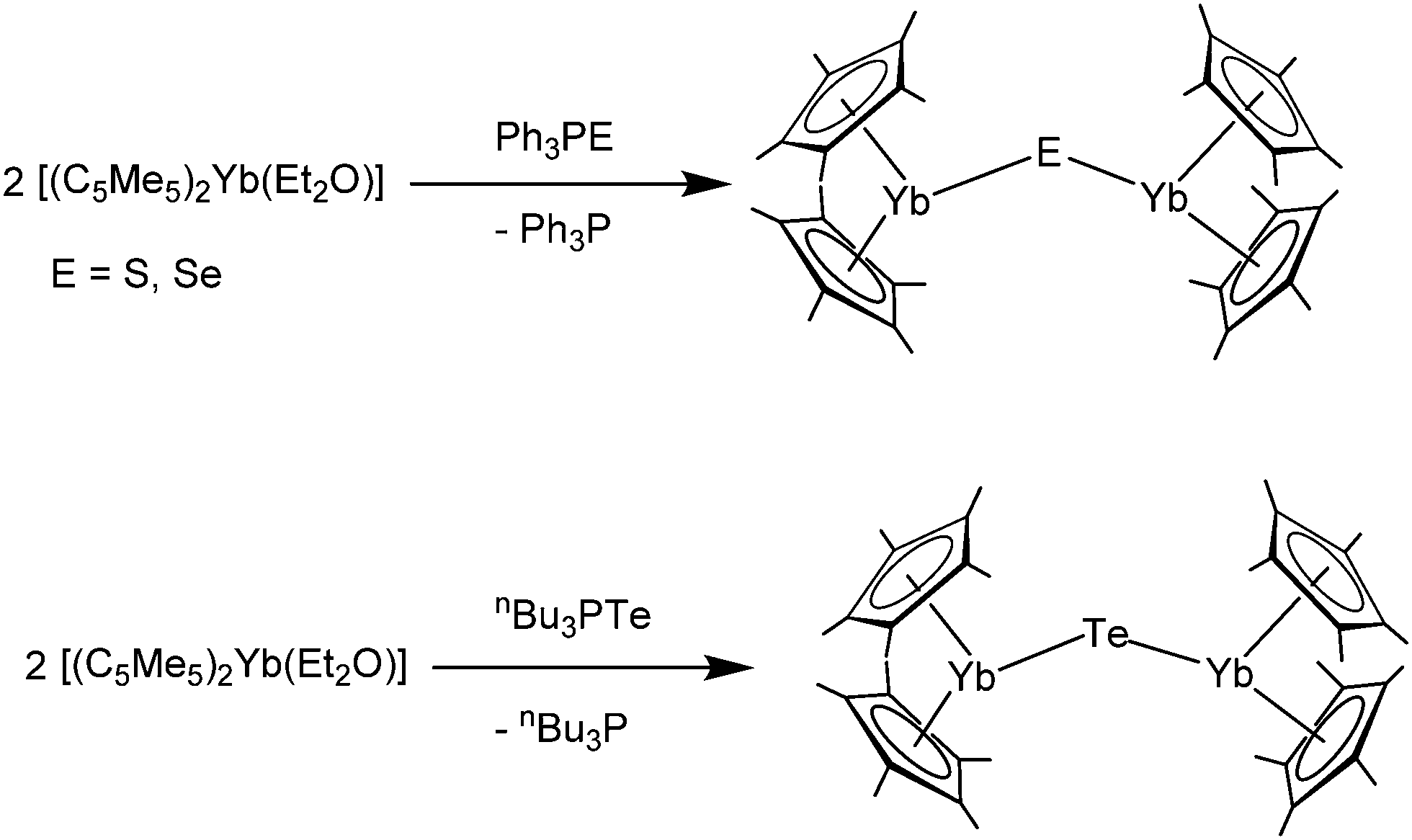 | ||
| Scheme 61 Reactivity of [(C5Me5)2Yb(OEt2)] towards phopshine chalcogenides.191 | ||
The reactivity of tetramethylthiuram disulfide (S2NCMe2)2 towards [(C5Me5)2Sm(THF)2] was investigated by Edelmann and co-workers.189 The sulphur–sulphur bond was reductively cleaved resulting in a chelating dithiocarbamate ligand, which was found in the orange product [(C5Me5)2Sm(S2NCMe2)].
The related redox active sulphurdiimine [(Me3SiN)2S] was investigated as a ligand for [(C5Me5)2Ln(THF)2] (Ln = Sm, Eu, Yb).192 The reaction of [(C5Me5)2Ln(THF)2] with [(Me3SiN)2S] in Et2O yielded in all three cases [(C5Me5)2Ln(Me3SiN)2S] (Scheme 62). The compounds were investigated with single-crystal X-ray diffraction and revealed symmetrical coordination of the diimide ligand. The N–Ln bond distances decrease from Sm to Yb in accordance with the decrease of the ionic radii (Sm–N: 2.458(2)/2.456(2) Å; Eu–N 2.449(2)/2.437(2) Å; Yb–N 2.352(8)/2.361(8) Å). The S–N distances are between those for single and double S–N bonds and are best described as a radical anion. The products were extensively characterised and investigated with DFT calculations with regard to their electronic nature.
 | ||
| Scheme 62 Synthesis of [(C5Me5)2Ln(Me3SiN)2S].192 | ||
5.2. Reactivity towards inorganic molecules
The reactions of [(C5Me5)2Sm(THF)2] with strongly oxidizing nitric oxide NO or nitrous oxide N2O resulted in a mixture of oxidised products, accompanied by a colour change from purple to orange.81 Identification of all the products was not possible. One of the products was samarocene oxide [{(C5Me5)2Sm(THF)2}2(μ-O)]. The corresponding diindenyl complex [(C9H7)2Sm(THF)x] shows a similar reactivity.18Evans investigated the reactivity of [(C5Me5)2Sm(THF)2] towards H2O under controlled conditions, a generally undesired reaction in organosamarium chemistry.193 By exposing a samarocene THF solution to a nitrogen atmosphere containing water vapour, brown crystals of the hexanuclear samarium cluster [{(C5Me5)Sm}6(O9H6)] were formed over a longer period. Two types of reaction take place. First, hydrolysis of a cyclopentadienyl ligand and second, water reduction by Sm(II). The six samarium atoms build an axially elongated octahedron having an oxygen atom in the centre. The remaining oxygen atoms bridge three Sm atoms of each plane of the octahedral Sm6 core. Due to the distorted geometry of the octahedron, the Sm–O distances vary between 2.499(4) and 2.504(4) Å, depending on the position of the Sm atom in the octahedron.
In contrast, no clusters but the organolanthanide(III) hydroxide complexes [{1,3-C5H3(SiMe3)2}2Sm(μ-OH)]2 and [{C5H4(SiMe3)}2Yb(μ-OH)]2 were obtained, when the corresponding metallocene precursors [{1,3-C5H3(SiMe3)2}2Sm(THF)] and [{C5H4(SiMe3)}2Yb(OEt2)] were reacted with water in an ethereal solution. Both complexes are dimeric in the solid state with bridging hydroxide groups, while NMR data indicate that these structures persist in aprotic media.194
Evans et al. reported that [(C5Me5)2Sm(THF)2] is able to reduce CO in THF under a pressure of 6 bar to form the dark orange, tetranuclear complex [({C5Me5)2Sm}2(μ-O2CCCO)(THF)]2 in low yield.195 The bridging ligands can be regarded as ketenecarboxylates and are formally constituted of three carbon monoxide molecules. A schematic drawing of the structure can be found in Scheme 63. Each of the ligands binds one Sm atom at each end of the ligand. Additionally, a kind of dimeric structure is formed, in which the oxygen atoms of the carboxylic acid units are bridging both subunits. However, studies by Andersen et al. on carbon monoxide complexes did not show any redox reactivity of desolvated decamethyl samarocene under a CO atmosphere (see above).108
 | ||
| Scheme 63 Schematic drawing of [{(C5Me5)2Sm}2(μ-O2CCCO)(THF)]2.195 | ||
The divalent thulium complex [(1,2,4-C5H2tBu3)2Tm] reacted with CO to give selective CO reductive dimerization and trimerization in dependence of the stoichiometric ratio (Scheme 64).41 By using an equimolar ratio, the ethynediolate complex [{(1,2,4-C5H2tBu3)2Tm}2(μ-κ(O):κ(O′)-C2O2)] was formed. During the reaction, a dimerization of CO to a ethynediolate (C2O2)2− fragment bridging the two Tm(III) ions was observed. In contrast, treatment of [(1,2,4-C5H2tBu3)2Tm] with an excess of CO led to [{(1,2,4-C5H2tBu3)2Tm}2(μ-O2CCCO)]. This reaction is similar to the one observed of [(C5Me5)2Sm(THF)2] with CO (Scheme 63).195
 | ||
| Scheme 64 Reaction of [(1,2,4-C5H2tBu3)2Tm] with CO.41 | ||
CO2 can be reduced by [(C5Me5)2Sm(THF)2] and related samarocenes (Scheme 65).23,196 When [(C5Me5)2Sm(THF)2] is exposed to an atmosphere of carbon dioxide in toluene or hexane, an intractable mixture of products was obtained according to NMR spectroscopy. Even at −78 °C, no single product was isolated. However, when carried out in THF, [(C5Me5)2Sm(μ-η2:η2-O2C2O2)Sm(C5Me5)2] is cleanly formed and isolated in >90% yield. As shown by X-ray crystallography, reductive coupling of two carbon dioxides gave an oxalate ligand, which bridges two (C5Me5)2Sm moieties. Reductive coupling is a typical reaction pattern of decamethylsamarocene. The structural data was not of sufficient quality for the determination of bond distances and angles. However, the structure was unequivocally established. In contrast to reductive coupling, the related COS undergoes disproportionation when reacted with [(C5Me5)2Sm(THF)2] to give [(C5Me5)2Sm(μ-η2:η1-S2CO)Sm(C5Me5)2(THF)] (Scheme 65). However, according to NMR spectroscopy, another unidentified product is present in this reaction. By adjusting reaction conditions, the complex can be obtained in 90% yield. COS is transformed into a dithiocarbonate ligand, that bridges one samarocene fragment in a bidentate way with both sulphur donors, and the other with the single oxygen atom. Orange crystals were investigated with X-ray crystallography. The dithiocarbonate is planar, the Sm–S bonds are similar with 2.773(2) and 2.821(2) Å while the Sm–O bond is expectedly shorter (2.270(5) Å). Mechanisms for the formation of both CO2 and COS-derived compounds were discussed by the authors.196 Interestingly, though no experimental details are given, [(C5Me5)2Yb(THF)2] forms the sulphur bridged dimer [(C5Me5)2YbSYb(C5Me5)2].191
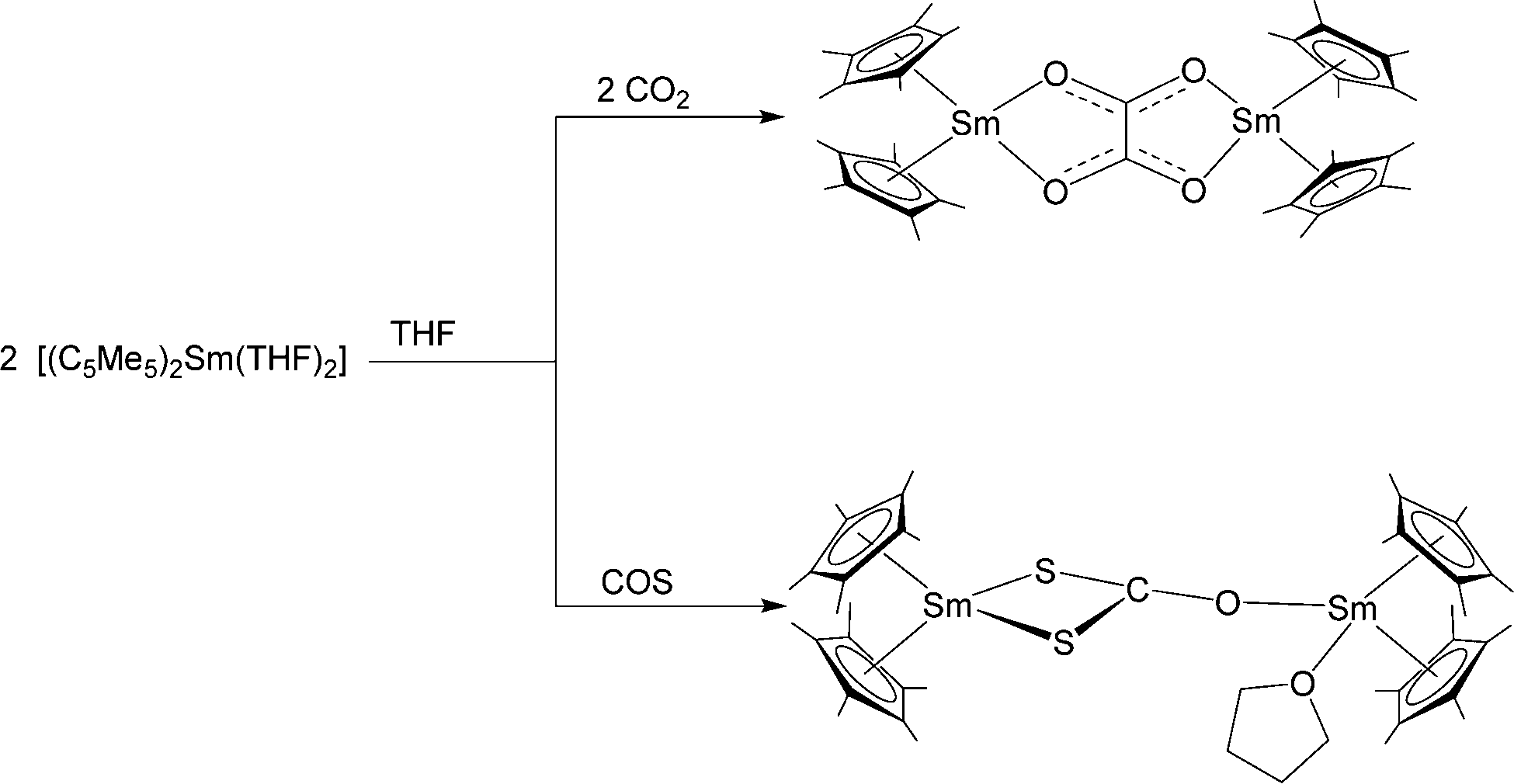 | ||
| Scheme 65 Reactivity of [(C5Me5)2Sm(THF)2] towards CO2 and COS.196 | ||
A different reactivity of CO2 was seen by using the base free and bulky substituted samarocenes [(1,3-C5H3tBu2)2Sm] and [(1,2,4-C5H2tBu3)2Sm].31 A clean formation of bridged carbonate samarium dimers [{(1,3-C5H3tBu2)2Sm}2(μ-CO3)] and [{(1,2,4-C5H2tBu3)2Sm}2(μ-CO3)] was seen. Apparently, a reductive disproportionation of CO2 with the release of CO must have taken place. This contrasts with the formation of the oxalate-bridged samarium dimer shown in Scheme 65. The structures of both compounds are slightly different. While in [{(1,3-C5H3tBu2)2Sm}2(μ-CO3)] a μ-η2:η2-coordination mode of the carbonate ligand was observed, a μ-η1:η2-coordination mode was seen in [{(1,2,4-C5H2tBu3)2Sm}2(μ-CO3)].31
Recently, the reactivity of SO2 towards some of the divalent metallocenes of the lanthanides was extensively investigated.197,198 Each compound was dissolved in THF. SO2 was subsequently condensed onto the solution at −50 °C. In the case of [(C5Me5)2Eu(THF)2], the dinuclear complex [{(C5Me5)Eu}2(μ,κO,κO′-C5Me5SO2)4] was isolated as the only product (Scheme 66). A sulfinate ligand was formed via nucleophilic attack of one Eu(C5Me5) ligand to SO2 In total, four C5Me5SO2 ligands bridge two Eu(C5Me5)-fragments. Thereby, a cage-like structure was created. The O-Eu distances are in the range of 2.309(3) to 2.354(2) Å.
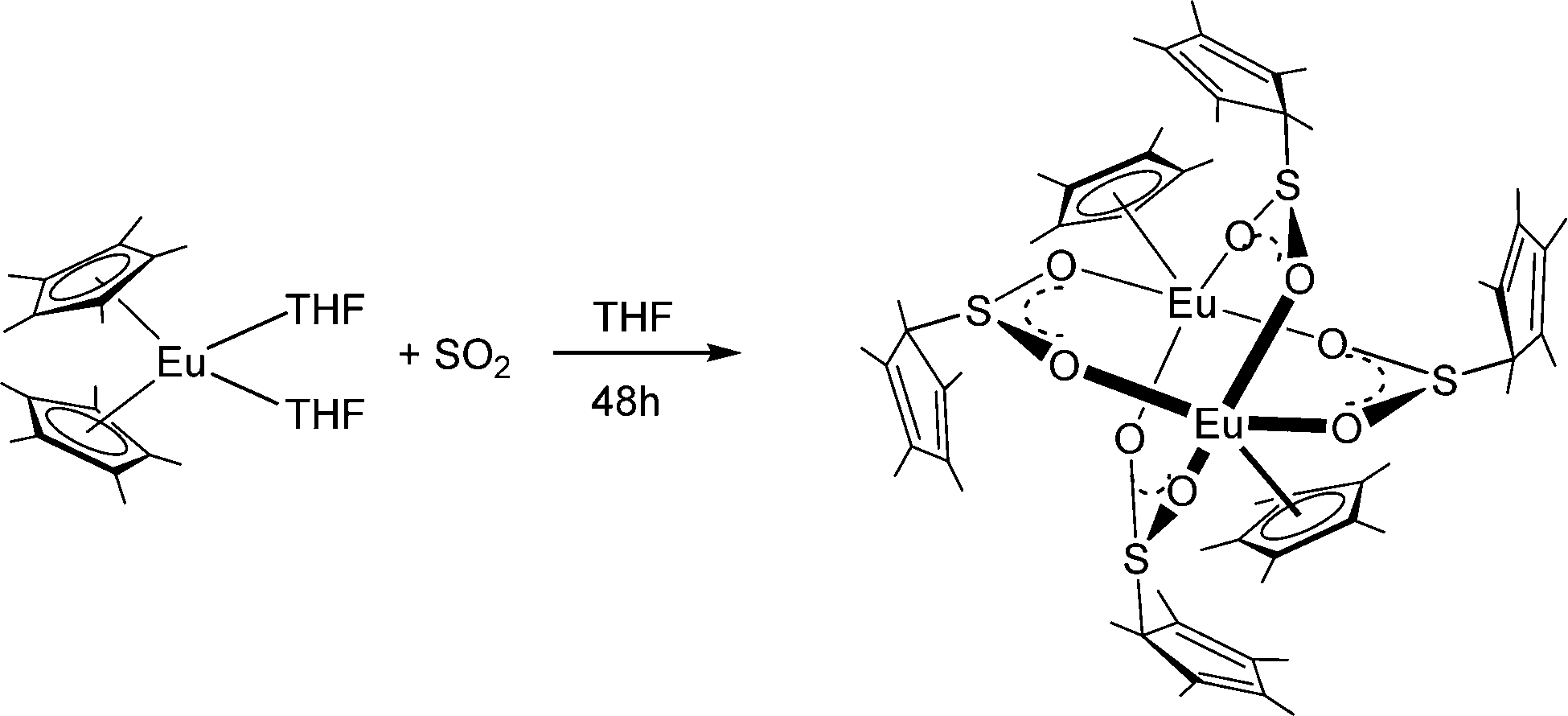 | ||
| Scheme 66 Reaction of [(C5Me5)2Eu(THF)2] with SO2.197 | ||
For [(C5Me5)2Yb(THF)2], three different products were crystallised from the reaction with SO2. By reductive coupling of two SO2 molecules, the dinuclear Yb(III) complex [(C5Me5)2Yb(μ-η2:η2-O2S2O2)Yb(C5Me5)2] was formed as a minor product (Scheme 67).197
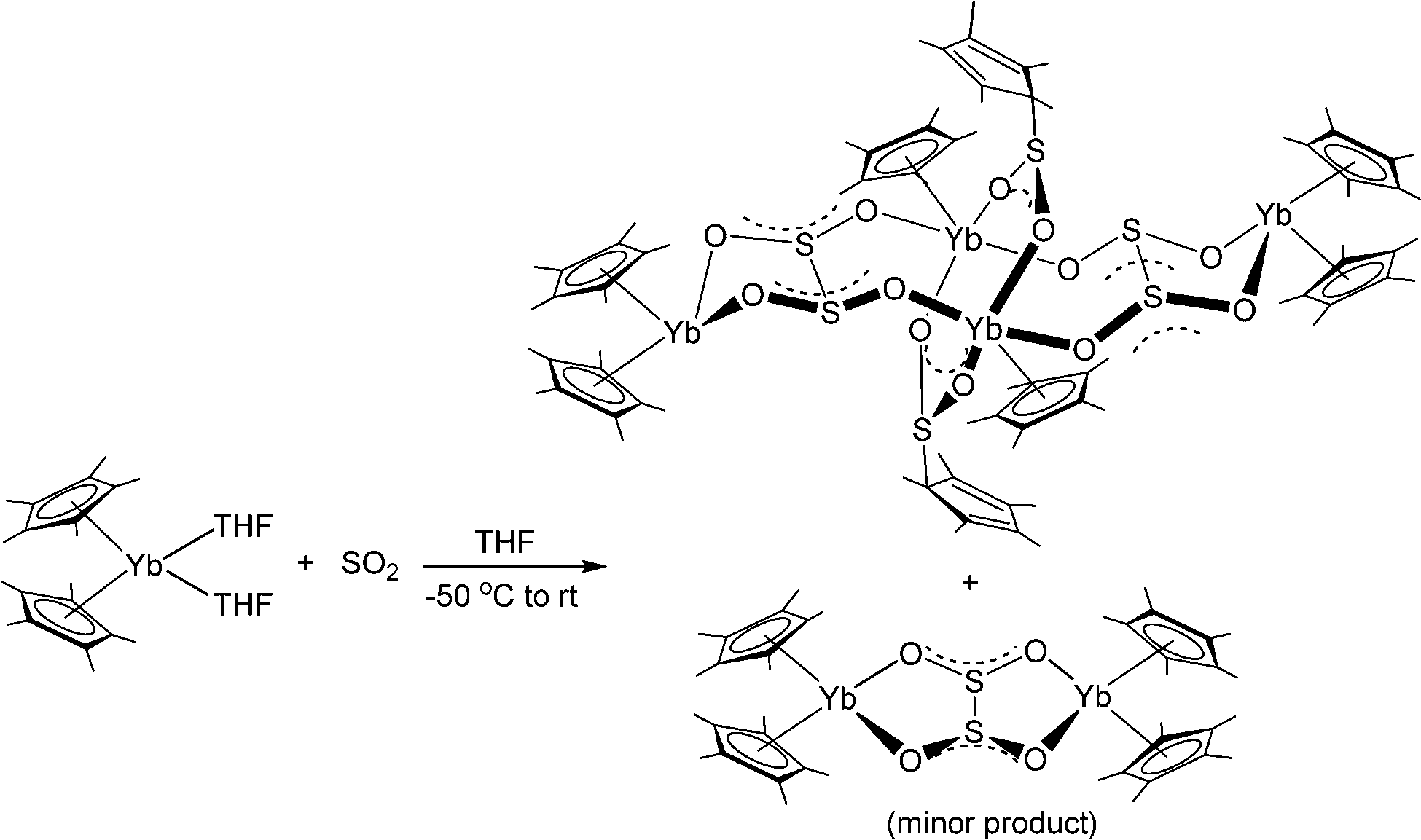 | ||
| Scheme 67 Reaction of [(C5Me5)2Yb(THF)2] with SO2.197 | ||
The coupling results in a bridging dithionite ligand, in which the sulphur atoms adopt a pyramidal structure. The Yb fragments are coordinated by the oxygen atoms. The Yb–O bond lengths range from 2.256(7) to 2.273(8) Å. Since this compound is only a minor product full characterization was not feasible. The main product of the reaction between SO2 and [(C5Me5)2Yb(THF)2] is the tetranuclear, red-violet complex [{(C5Me5)2Yb(S2O4)}2{(C5Me5)Yb(C5Me5SO2)}2], wherein both the reductive coupling of two SO2 molecules and the nucleophilic attack of C5Me5 anion on an SO2 occurred to form the sulfonate ligands C5Me5SO2. Structural investigations also show a cage-like structure, in which two (C5Me5)Yb units are alternatively bridged by sulfonate and dithionite ligands. The two dithionite ligands additionally chelate one more (C5Me5)2Yb fragment each, ultimately resulting in the tetranuclear compound. The (C5Me5)Yb–O bonds to the sulfinate ligands are 2.222(4) and 2.252(4) Å, the (C5Me5)Yb–O dithionite bonds are 2.356(4) and 2.225(4) Å. The outer (C5Me5)2Yb fragments show Yb–O distances of 2.223(4) and 2.255(4) Å. The third product, which was obtained upon heating, is the structural analogue of the dinuclear tetrasulfinate europium complex described above (Scheme 66).
Upon treatment of the more reactive metallocene [(C5Me5)2Sm(THF)2] with SO2, four isolable compounds were obtained (Scheme 68).198 As major products [{(C5Me5)2Sm(S2O4)}2{(C5Me5)Sm(C5Me5SO2)}2], which is an analogue to the corresponding Yb compound, and [{(C5Me5)2Sm(C5Me5SO2)}2] were formed. The latter is a dinuclear sulfinate complex, in which the two C5Me5SO ligands were created by the nucleophilic attack of a C5Me5 anion to SO2. The resulting mono-(C5Me5)Sm units were also found in the other products. The two Sm centres in [{(C5Me5)2Sm(C5Me5SO2)}2] are bridged by the oxygen atoms of the sulfinate, creating an eight-membered ring. The Sm–O distances determined in the structure are 2.339(2) and 2.355(2) Å, respectively. The other two minor products are [(C5Me5)2Sm(μ-η2:η2-O2S2O2)Sm(C5Me5)2], also observed for Yb, and the dinuclear compound [(C5Me5)Sm}2(μ,κO,κO’-C5Me5SO2)4]. All structures were investigated by single crystal X-ray diffraction and revealed no unexpected features when compared with the corresponding Yb and Eu complexes.
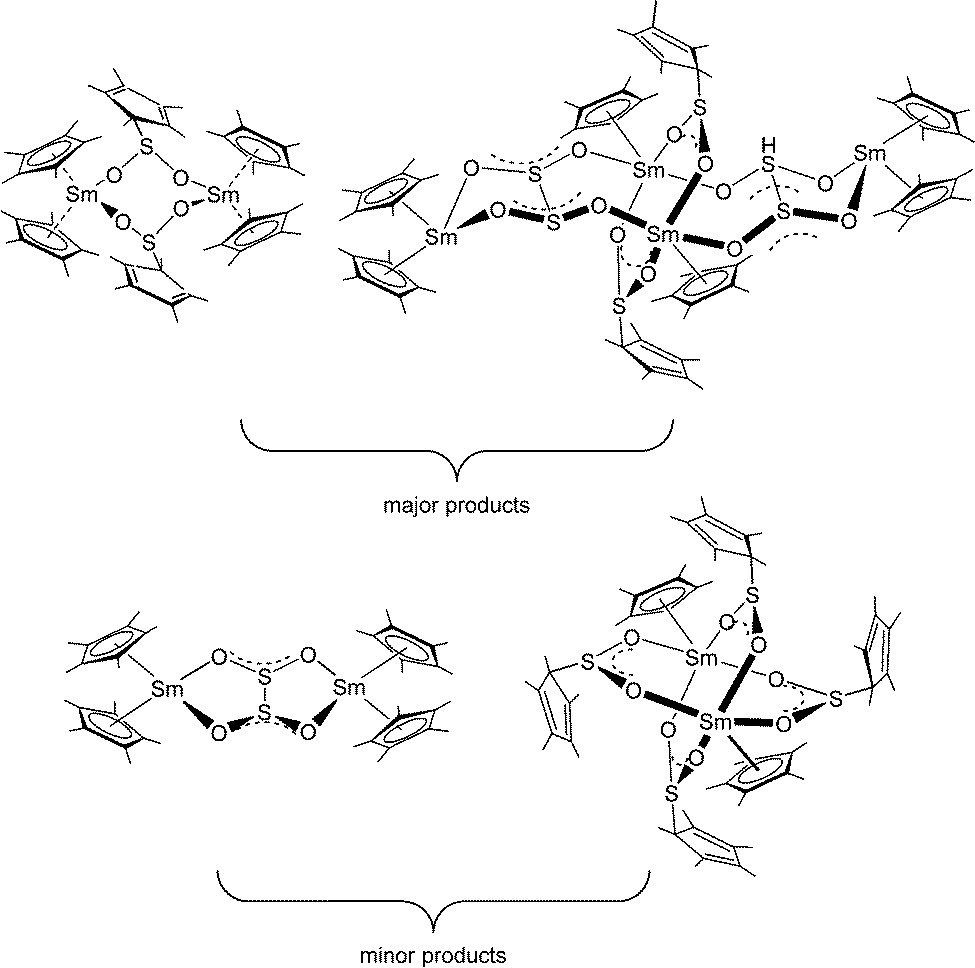 | ||
| Scheme 68 Products from the reaction of [(C5Me5)2Sm(THF)2] with SO2.198 | ||
The influence of the steric demand of the ligands was seen in the reaction of various metallocenes with the mineral realgar (As4S4). The reaction of [(C5Me5)2Ln(THF)2] (Ln = Sm, Yb) with As4S4 resulted the tetrametallic cage complex [{(C5Me5)2Sm}((C5Me5)Sm)3AsS3{(C5Me5)AsS2}2(THF)3] or the trimetallic cage compound [{(C5Me5)Yb}3As2S4((C5Me5)AsS2)(THF)2] (Scheme 69).199 The reduction of realgar leads to the formation of the unprecedented (C5Me5)AsS22− anion in both compounds. Moreover, thioarsenate (AsS33−) is found in the Sm complex, while an As2S44− anion is seen in the Yb compound.
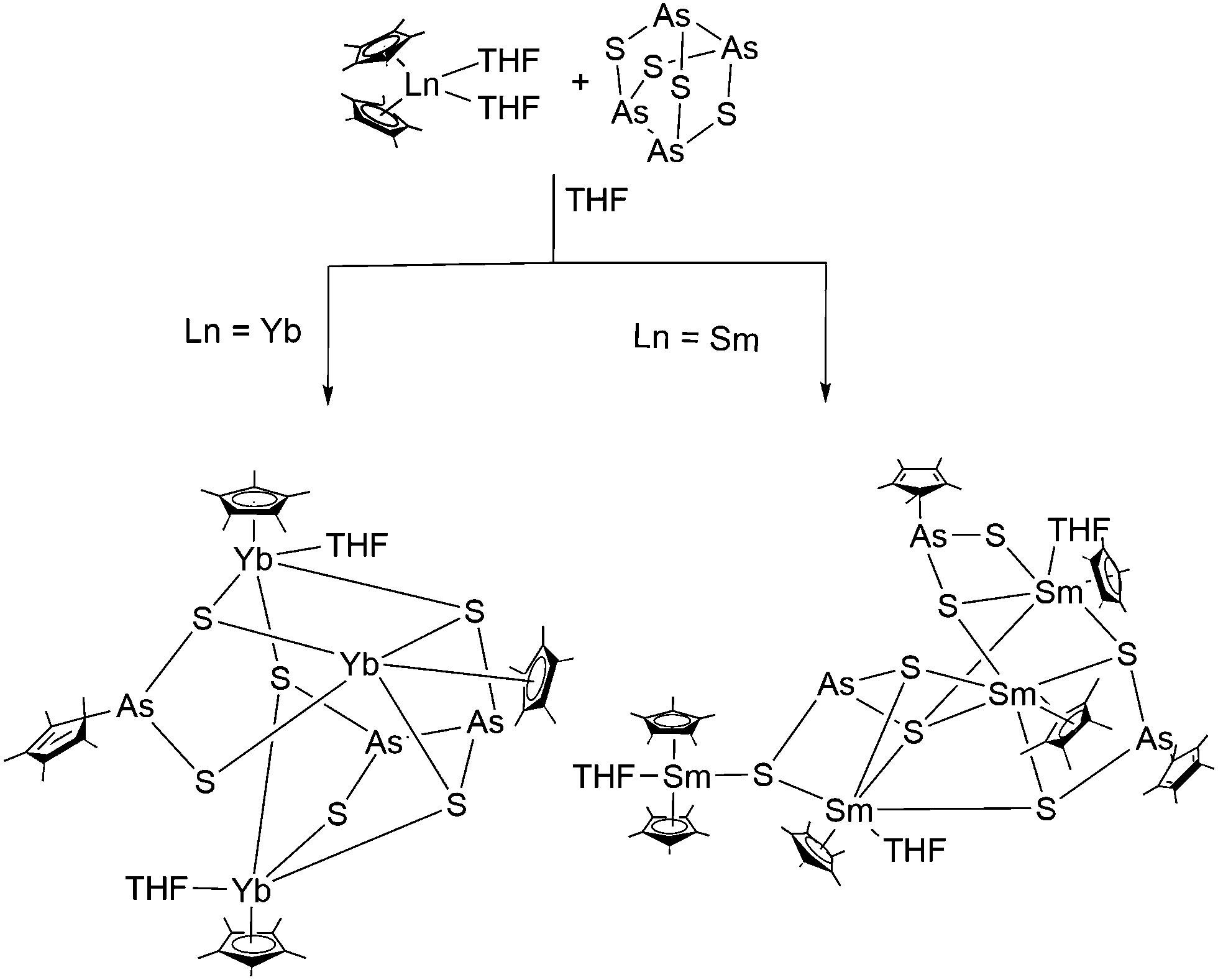 | ||
| Scheme 69 Reaction of [(C5Me5)2Ln(THF)2] with the mineral realgar (As4S4).199 | ||
Closed cage compounds are formed by either using bulkier ligands or a different As/S cage. The reaction of the bulky substituted samarocene [(1,2,4-C5H2tBu3)2Sm] with As4S4 and the reaction of [(C5Me5)2Yb(THF)2] with dimorphite (As4S3) gave the closed eleven-vertex cage clusters [{(1,2,4-C5H2tBu3)Sm}3(AsS3)2] and [{(C5Me5)Yb}3(AsS3)2], which have similar structures (Scheme 70).199 Both cages consist of two AsS33− anions and three lanthanide cations.
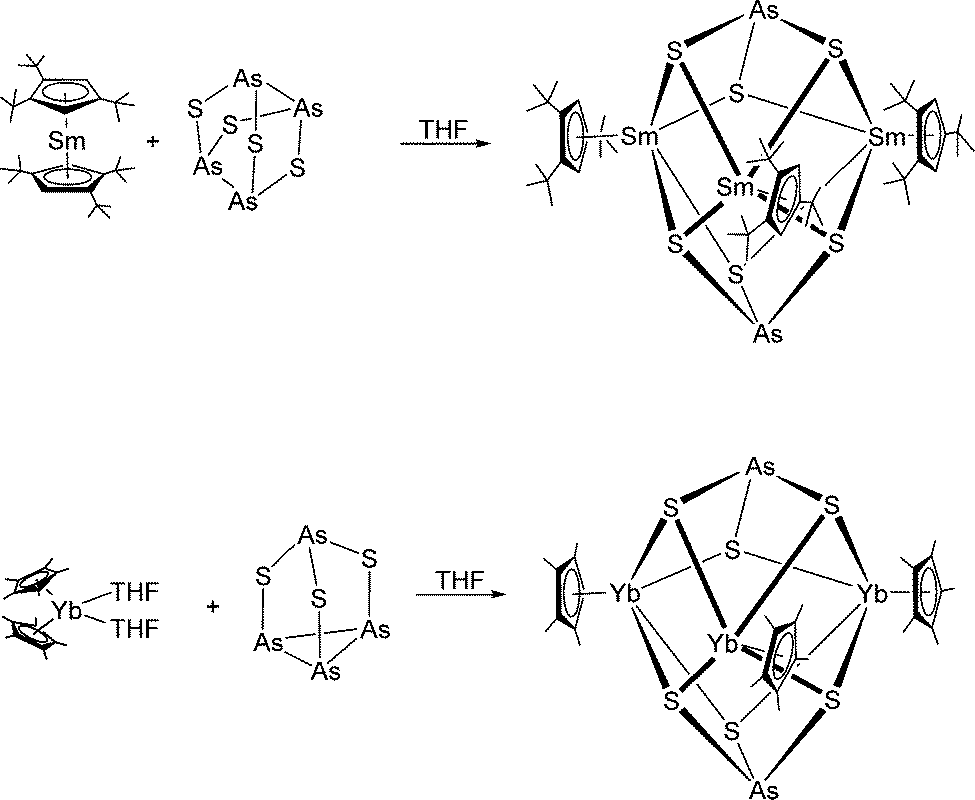 | ||
| Scheme 70 Synthesis of the closed cage compounds [(1,2,4-C5H2tBu3)3(AsS3)2] and [{(C5Me5)Yb}3(AsS3)2].199 | ||
Reaction of [(MeO)2P(S)S]2 with [(C5Me5)2Sm(THF)2] gave the unusual trivalent dithiophosphate mono-pentamethylcyclopentadienyl-complex [(C5Me5)Sm{S2P(OMe)2}2]2.200 In the dinuclear complex, the samarium atom possesses a high coordination number, which is rather atypical for Sm2+ and more often found in Sm3+ ions. This effect would explain this unusual bridging between the Sm atoms.
Heterometallic complexes of samarium and tungsten or molybdenum were prepared from the reaction of [(C5Me5)2Sm(THF)2] and the tetrathiometalates [(PPh4)2MoS4] and [(PPh4)2WS4], respectively (Scheme 71).201 By using the molybdenum compound with samarocene in THF a red compound was obtained, which was proven to be the trimetallic complex [{(C5Me5)2Sm}2Mo(μ-S)4][PPh4]. The central metal atom of the thiometalate was reduced during the reaction. The structure was established by single crystal X-ray diffraction. The central molybdenum atom is surrounded tetrahedrally by four sulphur ions, which are also bridging the Sm ions of the (C5Me5)2Sm units. The Sm–S distances are 2.791(2) and 2.796(2) Å. In contrast to the molybdenum compound, the reaction of [(C5Me5)2Sm(THF)2] with [(PPh4)2WS4] resulted in the bimetallic complex [(C5Me5)2Sm(μ-S)2WS2][PPh4]. In this case, only the reduction of one PPh4 anion occurs. The sulphur atoms are tetrahedrally arranged around the tungsten atom. Only two of them are also binding to the Sm(C5Me5)2 unit, while the remaining two sulphur atoms are arranged terminally. The Sm–S bond distances are 2.817(8) and 2.841(7) Å. The authors explain the different reactivity of group 6 thiometalates with the different redox potentials of Mo(VI) and W(VI).
 | ||
| Scheme 71 Reactions of [(C5Me5)2Sm(THF)2] with [(PPh4)2MoS4] and [(PPh4)2WS4].201 | ||
5.3. Reactivity towards elements
Especially the two stronger reducing metallocenes ytterbocene and samarocene are well known to reduce elements to anionic ligands.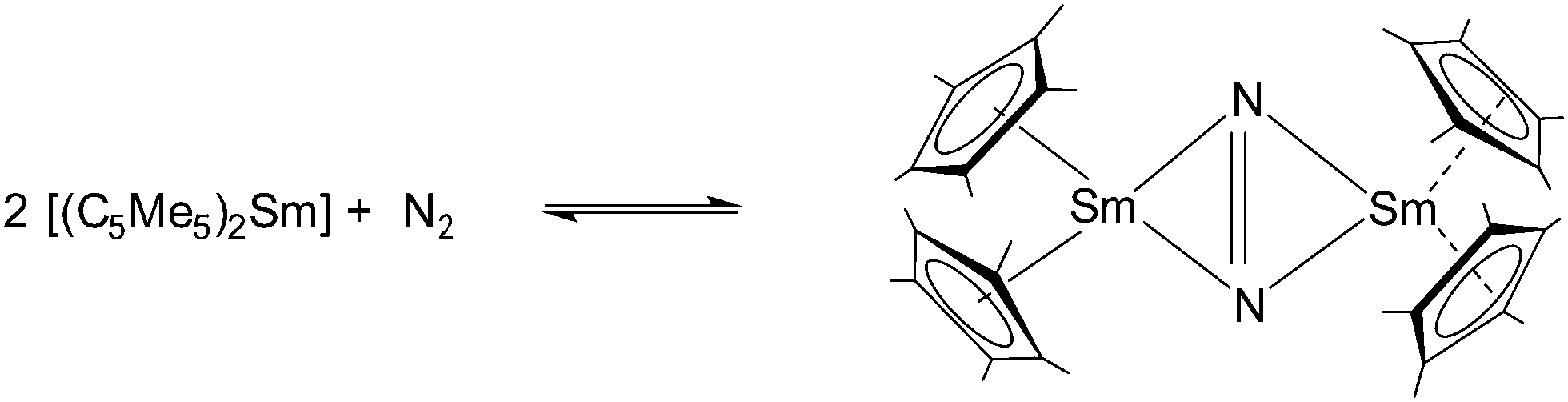 | ||
| Scheme 72 Reactivity of unsolvated decamethylsamarocene towards molecular nitrogen.202 | ||
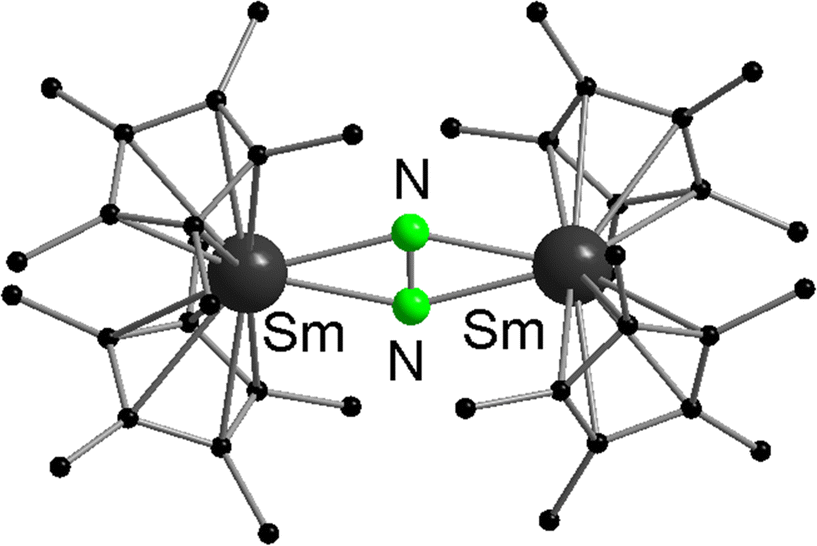 | ||
| Fig. 9 Molecular structure of [(C5Me5)2Sm(μ-η2:η2-N2)Sm(C5Me5)2] in the solid state (Reproduced from the CIF file CCDC: 1181490).202 | ||
NMR studies revealed a temperature dependent equilibrium between samarocene and the corresponding dinitrogen complex in solution.
A similar dinitrogen activation was seen when [(1,2,4-C5H2tBu3)2Nd(μ-I)(K[18]crown-6)] was treated with N2.9 As result the dimer [(1,2,4-C5H2tBu3)2Nd(μ-η2:η2-N2)Nd(1,2,4-C5H2tBu3)2] was isolated.
A cooperative dinitrogen activation was observed by using a mixture of [(C5Me5)2Sm(THF)2] and 9,10-Me2-9,10-diboraanthracene (Scheme 73).203 As result the salt [(C5Me5)2Sm(THF)2][(C5Me5)2Sm(η2-N2B2C14H14)] was obtained, in which the N22− anion is stabilised between a [(C5Me5)2Sm(THF)2]+ cation and the diboraanthracene Lewis acid.
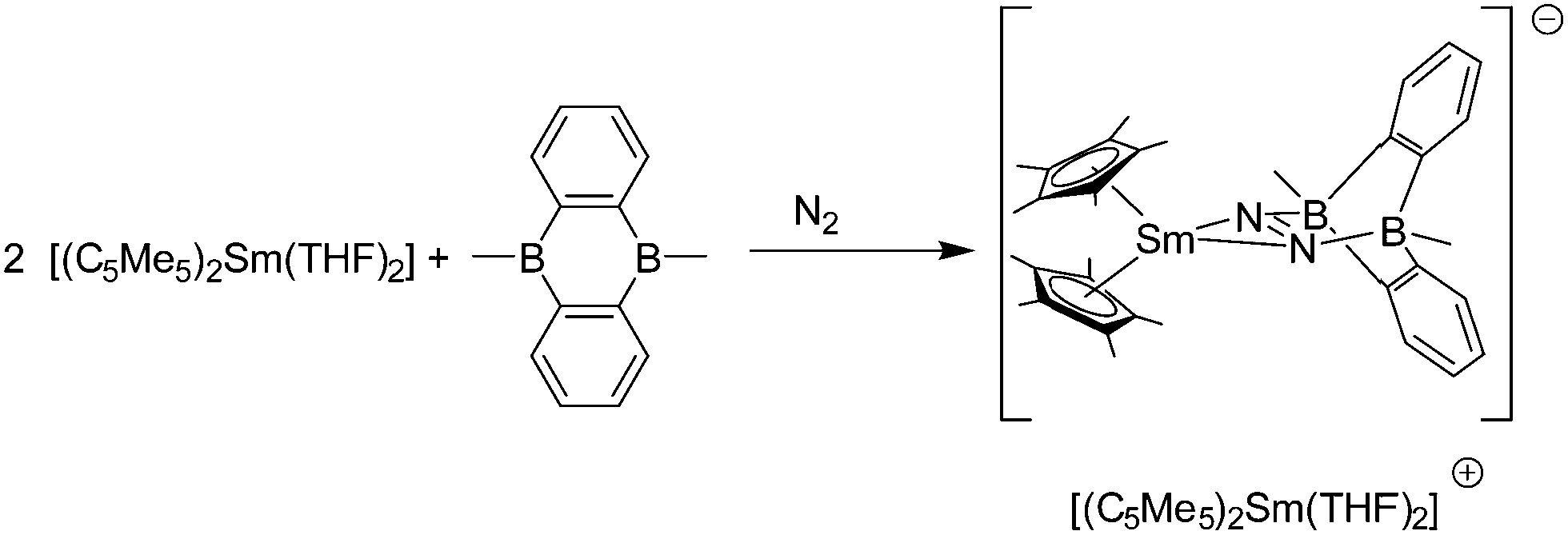 | ||
| Scheme 73 Synthesis of [(C5Me5)2Sm(THF)2][(C5Me5)2Sm(η2-N2B2C14H14)].203 | ||
The reactions of heavier congeners of dinitrogen with the samarocene were investigated by Roesky and co-workers.126,204,205 A toluene solution of desolvated [(C5Me5)2Sm] activates white phosphorus (P4) vapour to yield a molecular polyphosphide [{(C5Me5)2Sm}4P8] (Scheme 74).206 By diffusing P4 vapour in the solution, pyrophoric red crystals were formed. Structural studies show a tetranuclear Sm compound with a realgar-type P8 cage in the centre of the molecule. The four {(C5Me5)2Sm} moieties are residing on the corners of a square, each one binding to two phosphorous atoms (Sm–P 2.997(2)–3.100(2) Å). The authors note the analogy of the central P84− anion to the well-known Zintl anion P73− and theoretical studies were carried out. The formation of the Zintl type P84− anion by samarium pronounces once more the parallels to the reductive strength of the alkaline metals.
 | ||
| Scheme 74 Reactions of [(C5Me5)2Sm] with P4 (Reproduced from the CIF file CCDC 740249).206 | ||
The heavier congeners of P4 are either highly reactive (As) or non-existing under ambient conditions (Sb, Bi). Yellow arsenic, As4, is inconvenient to synthesise, extremely photosensitive and highly prone towards decomposition into its thermodynamically stable modification, grey arsenic. Thus, the reaction of freshly prepared solution of As4 in toluene at room temperature with unsolvated decamethylsamarocene [(C5Me5)2Sm] as divalent lanthanide source resulted besides unidentified and inseparable side products in [{(C5Me5)2Sm}2(μ-η2:η2-As2)].205 [{(C5Me5)2Sm}2(μ-η2:η2-As2)] (Scheme 75) is similar to Evans et al. dinitrogen complex [{(C5Me5)2Sm}2(μ-η2:η2-N2)] (Scheme 72).202 The As–As bond length is 2.278(2) Å, which corresponds to a double bond rather than a single bond, since {AsAs}2− moieties have bond lengths of 2.2 to 2.3 Å in transition metal complexes.207,208
 | ||
| Scheme 75 Reactions of [(C5Me5)2Sm] with As4 and As0nano.205 | ||
A more straightforward approach to polyarsenides starts from elemental As0 nanoparticles (As0nano, d = 7.2 ± 1.8 nm), which were obtained by the reduction of AsI3 with a freshly prepared solution of lithium naphthalenide.205 Treatment of the as-prepared As nanoparticles with [(C5Me5)2Sm] at 60 °C for short reaction times (<1 day) resulted again in [{(C5Me5)2Sm}2(μ-η2:η2-As2)] in low yields. Also here, by-products formed during the reaction accompanied the product.
In contrast, the samarium polyarsenide [{(C5Me5)2Sm}4As8], which is the heavier congener of the realgar-type [{(C5Me5)2Sm}4P8] polyphosphide, was formed after prolonged heating of As0nano and [(C5Me5)2Sm] at 120 °C. This polyarsenide is the most important congener of the realgar type [{(C5Me5)2Sm}4P8]. All Sm–As bond distances are similar and are between 3.0814(10) Å and 3.1734(10) Å. Within the {As8}4− tetraanion, all As–As bonds are similar (2.4044(12) Å and 2.5003(12) Å) and in the range of single bonds (Fig. 10).
 | ||
| Fig. 10 Molecular structure of [{(C5Me5)2Sm}4As8] in the solid (Reproduced from the CIF file CCDC 1880716).205 | ||
As already mentioned, there is no reactive elemental modification for antimony. To increase the reactivity, Sb/Hg alloy or Sb0 nanoparticles were used as an activated antimony source.204 Reaction of [(C5Me5)2Sm] with Sb/Hg stirred at room temperature for 48 hours resulted in the polystibide complexes [{(C5Me5)2Sm}2(μ-η2:η2-Sb2)] and surprisingly, a mercury-containing compound [{{(C5Me5)2Sm}2Sb}2(μ-Hg)] (Scheme 76).204,205 The authors could not separate the complexes on a preparative scale. The compound [{(C5Me5)2Sm}2(μ-η2:η2-Sb2)] is isostructural to other group 15 compounds such as [{(C5Me5)2Sm}2(μ-η2:η2-E2)] (E = N, As, Bi).179,202,205 The Sm–Sb bond lengths of 3.2141(9) Å are in the range of those in [{(C5Me5)2Sm}3(μ-η2:η2:η1-Sb3)(THF)] (Sm–Sb 3.162(1) Å - 3.205(1) Å)185 and the short Sb–Sb bond length of 2.6593(15) Å is similar to other Sb–Sb double bonds (Sb–Sb’ 2.642(1) Å) in the distibene [TbtSbSbTbt] (Tbt = 2,4,6-tris[bis(trimethylsilyl)-methyl]phenyl).209 [{{(C5Me5)2Sm}2Sb}2(μ-Hg)] is the first molecular Sb–Hg bond ever reported with d(Hg–Sb) = 2.6400(6) Å.204
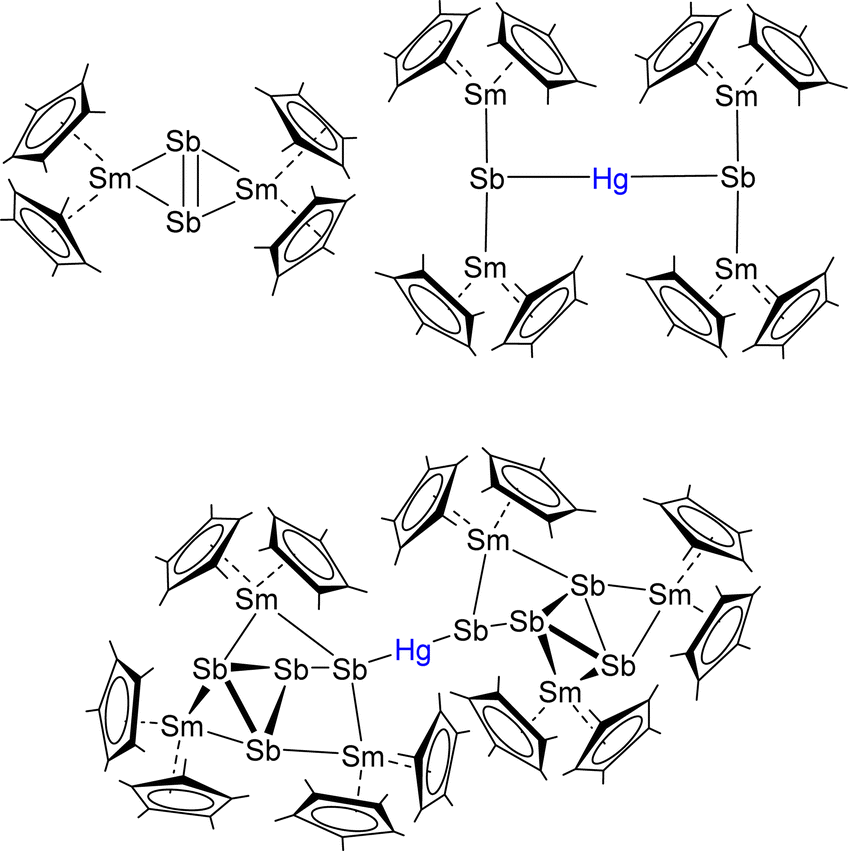 | ||
| Scheme 76 Isolated intermediates from the reaction of [(C5Me5)2Sm] with Sb/Hg alloy.204 | ||
Performing the reaction with an identical reaction mixture at 60–70 °C for two days resulted in [{{(C5Me5)2Sm}3(μ4,η1:2:2:2-Sb4)}2Hg] as minor product. The elevated reaction temperature thus led to compounds with a higher Sb and a lower Hg ratio. As shown by X-ray analysis, two [{(C5Me5)2Sm}3Sb4]−-moieties are linked by Hg2+ (Scheme 76). In [{{(C5Me5)2Sm}3(μ4,η1:2:2:2-Sb4)}2Hg] three samarocene fragments are connected by a [Sb4]4− anion, in which the Sb atoms clearly adopt different formal oxidation states with an average of +1.204
Finally, the authors heated the reaction to 120 °C for two days in order to obtain the f-element realgar-type polystibide complex [{(C5Me5)2Sm}4Sb8] (Scheme 77).204 [{(C5Me5)2Sm}4Sb8] is isostructural to its lighter analogue [{(C5Me5)2Sm}4E8] (E = P, As),205,206 demonstrating the accessibility of also heavier group 15 polyanions in f-element chemistry. Four {(C5Me5)2Sm}+-fragments bridge the corners of a [Sb8]4− Zintl anion.
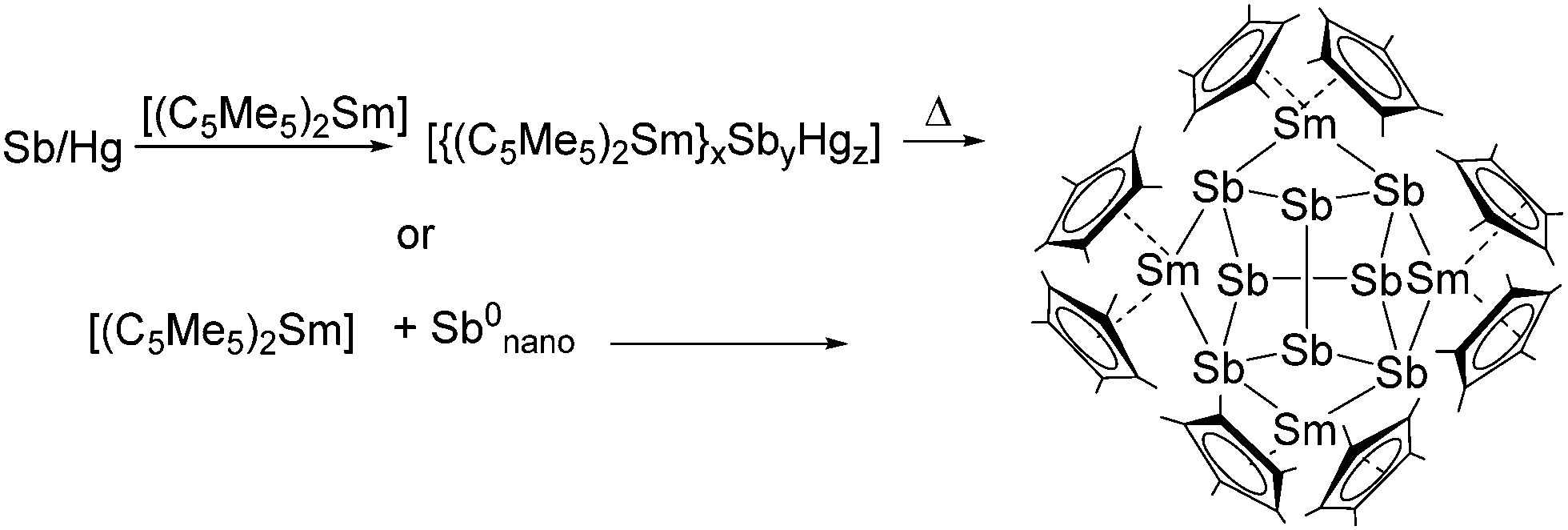 | ||
| Scheme 77 Reaction of Sb/Hg alloy with [(C5Me5)2Sm] to give different {(C5Me5)2Sm}/Sb/Hg species, which further react to give [{(C5Me5)2Sm}4Sb8] (left). Reactions of Sb0 nanoparticles with [(C5Me5)2Sm] yield [{(C5Me5)2Sm}4Sb8] (right).204 | ||
As an alternative approach, Sb0 nanoparticles synthesised under salt-free and inert conditions from SbCl3 and 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-diaza-2,5-cyclo-hexadiene210 were employed as reactive antimony source.204 The nanoparticles were subsequently reacted with [(C5Me5)2Sm] to give the polystibide complexes [{(C5Me5)2Sm}2(μ-η2:η2-Sb2)] again. After prolonged heating of the reaction mixture, the Zintl complex [{(C5Me5)2Sm}4Sb8] was isolated as the sole product demonstrating that the formation of this compound is the thermodynamically favoured pathway (Scheme 77).
:3 ratio to give [(C5Me5)2Sm(THF)(μ-η1:η3-S3)Sm(C5Me5)2] (Scheme 78).190
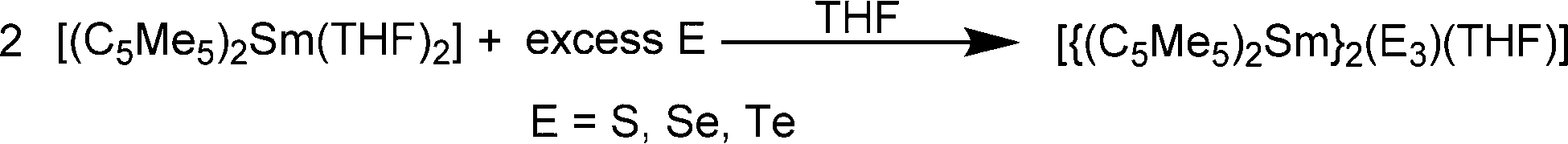 | ||
| Scheme 78 Reactivity of [(C5Me5)2Sm(THF)2] towards sulphur, selenium and tellurium.190 | ||
The reaction is so rapid that the S2− containing complex (see above, Scheme 59) cannot be isolated from this reaction. However, [(C5Me5)2Yb(μ-S)Yb(C5Me5)2] was isolated from the reaction mixture of [(C5Me5)2Yb(THF)2] and [(VMeCp)2(μ-S)(μ,η1:1-S2)(μ,η2:2-S2)] (MeCp = C5H4Me).211
The more reactive thulium reagent [(1,2,4-C5H2tBu3)2Tm] reacts with S8 to give a [(1,2,4-C5H2tBu3)2Tm2(μ-η2:η2-S2)], in which the S22− unit is side-on coordinated with both Tm ions.212
By treating [(C5Me5)2Sm(THF)2] with excess of selenium, the selenium analogue [(C5Me5)2Sm(THF)(μ-η1:η3-Se3)Sm(C5Me5)2] was isolated as dark red crystals. In the case of solid tellurium, the corresponding tellurium complexes are formed; however, the reaction with tellurium is slower, owing to the lower oxidative reactivity of this element. [(C5Me5)2Sm(THF)(μ-η1:η3-Se3)Sm(C5Me5)2] was characterised by single crystal X-ray crystallography. The structure reveals a dinuclear Sm compound, in which the two metal centres are bridged by a Se32− anion. One of the Sm atoms has an additional THF ligand bound to it. The Se32− anion is asymmetrically coordinated, in the sense that one (C5Me5)2Sm unit coordinates to all three selenium atoms in an η3-fashion, while the other (C5Me5)2Sm moiety coordinates only to one Se atom from the opposing side of the triangular anion (Scheme 79). [(C5Me5)2Sm(THF)(μ-η1:η3-Se3)Sm(C5Me5)2] slowly transformed to the interesting hexanuclear Se-Sm cluster [(C5Me5)Sm]6(Se11)].213
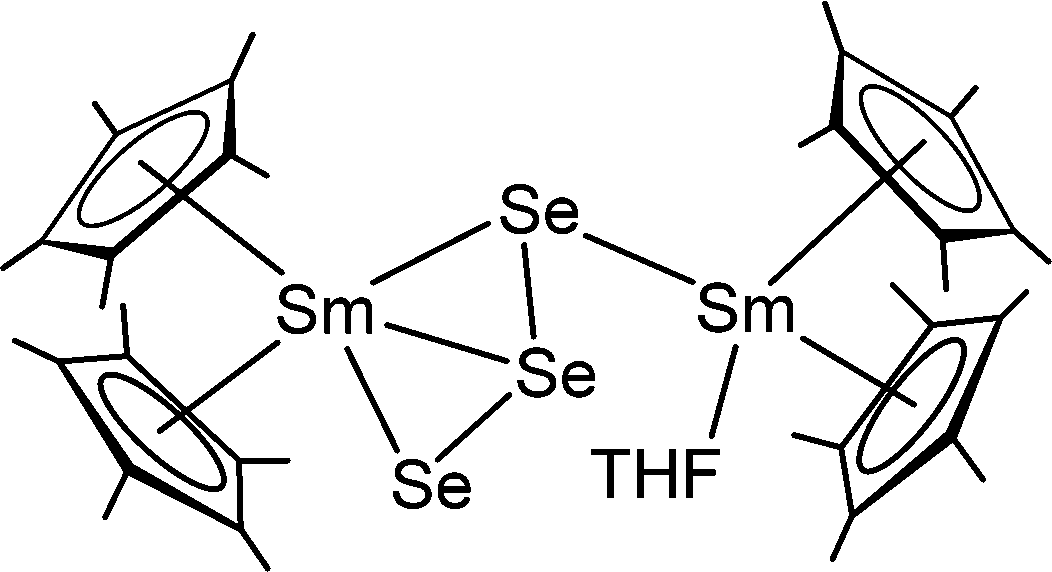 | ||
| Scheme 79 Structure of [(C5Me5)2Sm(THF)(μ-η1:η3-Se3)Sm(C5Me5)2].190 | ||
[(C5Me5)2Yb(OEt2)] reacted with selenium and tellurium to give the anticipated chalcogenide bridged compounds [{(C5Me5)2Yb}2(μ-Se)] and [{(C5Me5)2Yb}2(μ-Te)] (also accessible from the reaction of phosphine chalcogenide compounds, see above, Scheme 59).191 However, when [(C5Me5)2Yb(OEt2)] is stirred with a “large excess” of tellurium in hexane over a longer time, black crystals of [(C5Me5)2Yb(μ-η2:η2-Te2)Yb(C5Me5)2] were isolated (Scheme 80).214 The ditelluride anion is located between the two (C5Me5)2Yb units. The Te–Te bond is perpendicular to the Yb–Yb axis. The Yb–Te bond lengths are 3.153(9) and 3.1598(7) Å.
 | ||
| Scheme 80 Structure of [(C5Me5)2Yb(μ-η2:η2-Te2)Yb(C5Me5)2].214 | ||
Similar compounds can also be obtained by the reaction of polychalcogenide complexes of vanadium with [(C5Me5)2Sm(THF)2] and [(C5Me5)2Yb(THF)2]. Thus, a mixture of the mono-[(C5Me5)2Yb(μ-Se)Yb(C5Me5)2] and the diselenides, [(C5Me5)2Yb(μ-η2:η2-Se2)Yb(C5Me5)2] was isolated from the reaction of [(C5Me5)2Yb(THF)2] and [(V(C5H4Me)2(μ-Se)(μ,η1:1-Se2)(μ,η2:2-Se2)].211 [(C5Me5)2Sm(μ-Te)Sm(C5Me5)2] was obtained from [(C5Me5)2Sm(THF)2] and [{V(nacnac)}2(μ-Te)2] ((nacnac) = (HC(C(Me)NC6H3-iPr2)2)).211
The reaction of the metallocenes with organometallic reagents leads us to the next chapter.
5.4. Reaction with coordination compounds and organometallic reagents
Especially in the early 1980s, Andersen investigated the reactivity of [(C5Me5)2Yb] towards transition metal–carbonyl complexes.215–217 In general, reactions of [(C5Me5)2Ln(OEt2)] (Ln = Sm, Yb) with [Co2(CO)8],215,218 [Fe2(CO)9],216 [Mn2(CO)10]217,219 and [Re2(CO)10]217,219 were studied. In every case reduction of the carbonyl complexes occurs, yielding compounds with bridging carbonyl ligands. Isocarbonyl complexes with oxygen atoms binding to the metal centres were formed in all cases. For [Co2(CO)8], the heterobimetallic complex [(C5Me5)2Yb(OC)Co(CO)3(THF)] was obtained (Scheme 81). | ||
| Scheme 81 Reaction of [(C5Me5)2Yb(OEt2)] and [Co2(CO)8].215 | ||
The stronger reducing agent [(C5Me5)2Sm(THF)2] compared to the above mentioned [(C5Me5)2Yb(THF)2] also reacts with [Co2(CO)8] and forms the structurally related product [(C5Me5)2Sm(OC)Co(CO)3(THF)].218 The infinite chain [{(C5Me5)2Sm(THF)}{(μ-CO)2(CO)3Mn}]∞ was obtained upon reaction of [(C5Me5)2Sm(THF)2] with [Mn2(CO)10]. Here, two CO ligands are coordinating with each samarocene (Scheme 82).219
 | ||
| Scheme 82 Reaction of [(C5Me5)2Sm(THF)2] with [Mn2(CO)10].219 | ||
In contrast, the reaction with [Re2(CO)10] led to the formation of [{(C5Me5)2Sm}3{(O4C4)(μ3-CO)2(μ-CO)(CO)5Re2}Sm(C5Me5)2(THF)] (Scheme 83).219 The central [(O4C4)(μ3-CO)2(μ-CO)(CO)5Re2]4− core has been formed by a four-fold reduction process and the [μ-O4C4] unit can formally be considered as tetra-anionic where each oxygen atom is negatively charged. The Re–Re bond is slightly shorter than the Re–Re single bond in Re2(CO)10220 (2.934(3) vs. 3.041(11) Å, respectively) but theoretical investigations indicate only a weak interaction.
 | ||
| Scheme 83 Reaction of [(C5Me5)2Sm(THF)2] with [Re2(CO)10].219 | ||
The reaction with iron and manganese carbonyls resulted in more sophisticated structures. The pentanuclear Yb–Fe complex [{(C5Me5)2Yb}2{Fe3(CO)7(μ-CO)4}]216 and the polymeric Mn–Yb compound [{(C5Me5)2Yb}Mn(CO)5]n217 consist of polymeric chains with both [(C5Me5)2Yb(μ-CO)3Mn(CO)2] moieties and dimeric (C5Me5)2Yb(μ-CO)2Mn(CO)3 subunits. A similar structure was found for the reaction with [Re2(CO)10].217 No significant influence of bulkier cyclopentadienyl ligands is observed on the reactivity. Thus, reaction of [(CpBz5)2Sm] with [Co2(CO)8] or [Mn2(CO)10] in toluene gave the bridged tetrametallic complexes [{(CpBz5)2Sm}2{(μ-OC)2Co(CO)2}2] or [{(CpBz5)2Sm}2{(μ-OC)2Mn(CO)3}2].48
Besides pure metal carbonyls also organometallic carbonyl derivatives were reacted with [(C5Me5)2Sm(THF)2]. Thus, the reaction with [(C5Me5)Fe(CO)2]2 led to the tetranuclear complex [(C5Me5)2Sm(μ-OC)2Fe(C5Me5)]2, which forms a twelve membered ring.221
The sulphur carbonyl complex [Fe2(μ-S2)(CO)6] was reacted with [(C5Me5)2Ln(THF)2] (Ln = Eu, Sm, Yb) in toluene (Scheme 84).222 While for [(C5Me5)2Eu(THF)2] no reaction was observed, [(C5Me5)2Sm(THF)2] and [(C5Me5)2Yb(THF)] yielded octanuclear complexes of the type [Fe6Ln2(μ3-S)6(μ,η2-CO)4(CO)8(η5-C5Me5)4] as black crystals. The compounds form wheel-type structures, with a dianionic Fe6(μ3-S2)(CO)12 unit in the centre. The (C5Me5)2Ln moieties are bound via a bridging sulphur atom and the oxygen atoms of the carbonyl ligands. The sulphur atoms are connected to the inner iron atoms.
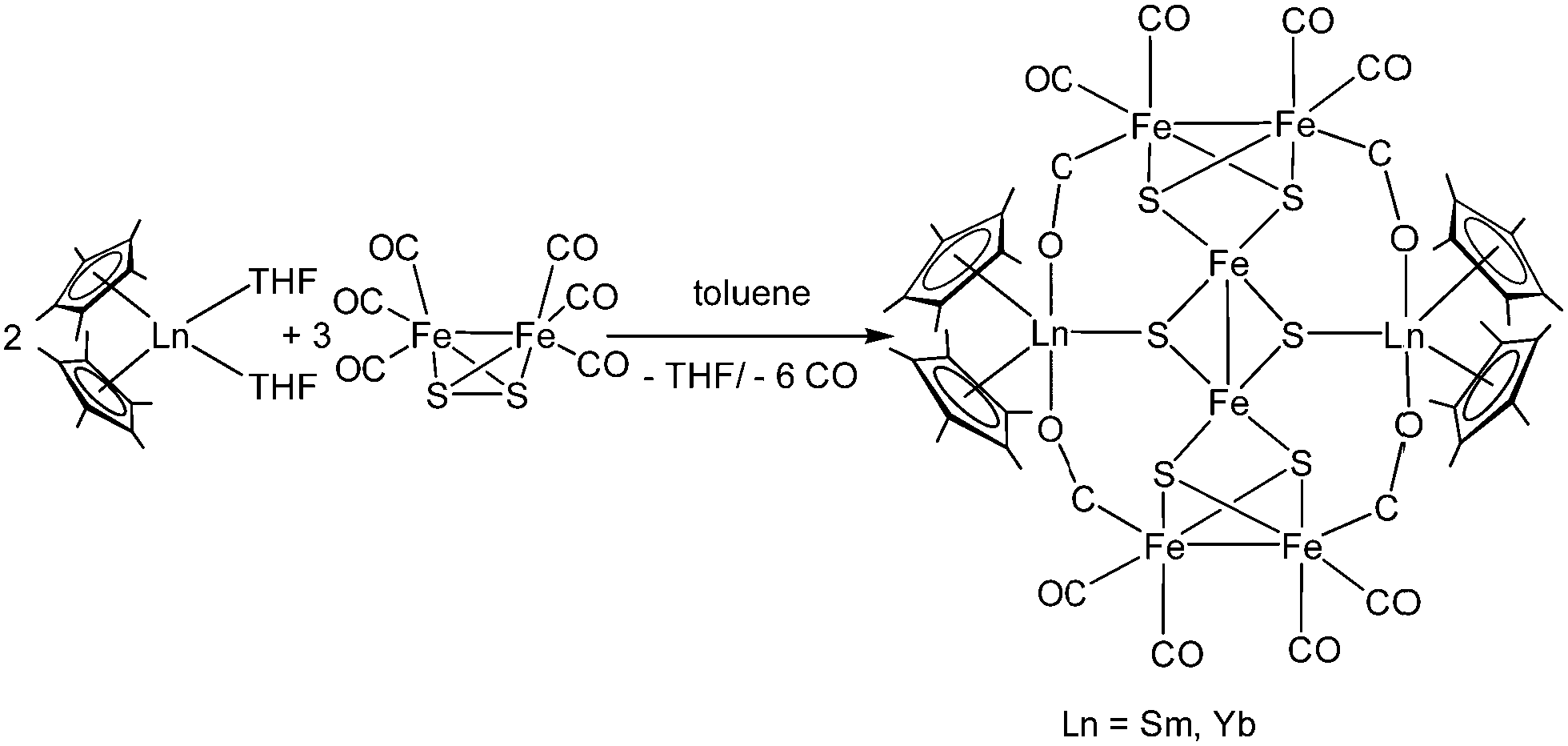 | ||
| Scheme 84 Reactions of [(C5Me5)2Ln(THF)2] (Ln = Sm, Yb) with [Fe2(μ-S2)(CO)6].222 | ||
Solvent-free [(C5Me5)2Sm] is able to reduce the organometallic compound AlEt3. With an excess of AlEt3 in toluene, elemental aluminium was deposited and the red complex [(C5Me5)2Sm(μ-Et)2AlEt2] was formed.223,224 Two carbon atoms attached to the aluminium ion form a bridge to the samarium atom with Sm–C distances of 2.662(4) Å. Here, the difference in the reactivity between [(C5Me5)2Sm] and [(C5Me5)2Yb] was emphasised: R3Al compounds react with ytterbocene with the formation of Lewis acid–base adducts (see above).
Samarocene [(C5Me4R)2Sm(THF)2] (R = H, Me) also reduces AlMe3 in toluene to form [(C5Me4R)2Sm{(μ-Me)AlMe2(μ-Me)}2Sm(C5Me4R)2] (Fig. 11).225,226 Each samarocene unit is linked to two tetrahedral (μ-Me)2AlMe2 moieties with the Sm(μ-Me)-Al linkages being nearly linear with angles in [(C5Me5)2Sm{(μ-Me)AlMe2(μ-Me)}2Sm(C5Me5)2] of 175.2(9)° and 177.8(7)° and a planar arrangement.
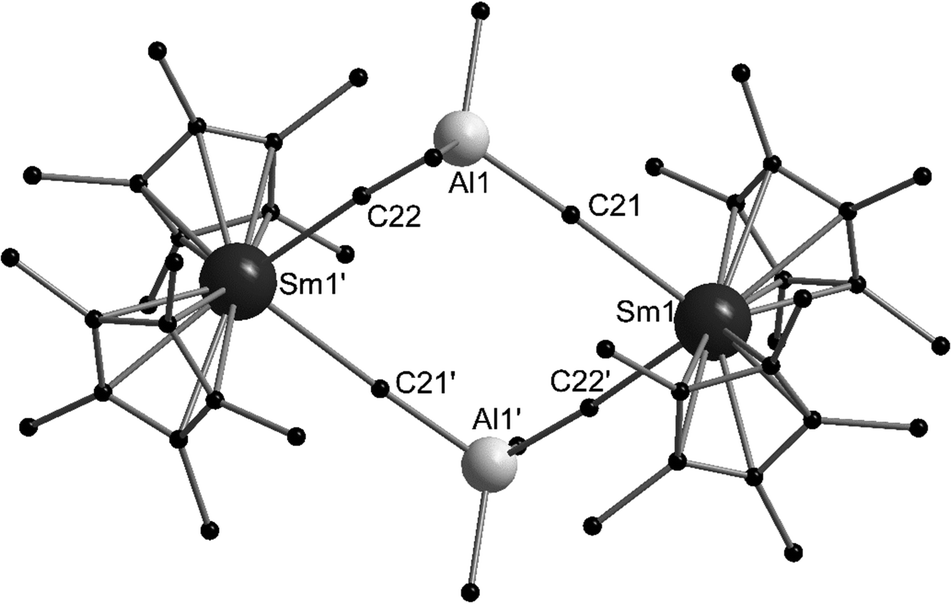 | ||
| Fig. 11 Molecular structure of [(C5Me5)2Sm{(μ-Me)AlMe2(μ-Me)}2Sm(C5Me5)2] in the solid state (hydrogen atoms omitted for clarity). The central [Sm–C–Al–C–Sm–C–Al–C–] unit is almost planar (Reproduced from the CIF file CCDC: 1278704).225 | ||
Upon reaction with dimethylzinc [Me2Zn], [(C5Me5)2Yb] reacts with the methyl-bridged zincate [(C5Me5)2Yb(μ-Me)2ZnMe], which can be readily prepared in 2–3 g scale.227 Upon recrystallization from THF the [ZnMe]-fragment is cleaved and [(C5Me5)2YbMe(THF)] is obtained. When [ZnMe2] is replaced with the more reactive [CuMe], the compounds [(C5Me5)2Yb(μ-Me)Yb(C5Me5)2] and [(C5Me5)2Yb(μ-Me)Yb(C5Me5)2(Me)] were obtained.227 [(C5Me5)2Yb(μ-Me)Yb(C5Me5)2] is a mixed valent compound containing a Yb(II) and a Yb(III) species. When [(C5Me5)2Yb(Et2O)n] (n = 0, 1) is combined with [ZnPh2] in toluene, [(C5Me5)2Yb(μ-Ph)2ZnPh] is yielded (Scheme 85). Its structural motive is similar to the one of [(C5Me5)2Yb(μ-Me)2ZnMe].
 | ||
| Scheme 85 Reaction of [(C5Me5)2Yb(Et2O)n] (n = 0, 1) with [ZnPh2].227 | ||
Since the reaction with diphenyl mercury [HgPh2] did not lead to any results, [Hg(C6F5)2] was investigated. This led to the compound [(C5Me5)2Yb(C6F5)], which is similar to the previously reported reaction of [(C5H5)2Yb] and [Hg(C6F5)2].228 To investigate more into the formation of [(C5Me5)2YbMe] other organometallic methyl transfer reagents were employed. When [(C5Me5)2VMe] is mixed with [(C5Me5)2Yb] in a 2:1 ratio, [(C5Me5)2Yb(μ-Me)Yb(C5Me5)2] is formed and [(C5Me5)2V] can be isolated from the mother liquor. This is also applicable for halides and other anions and [(C5Me5)2Yb(μ-X)Yb(C5Me5)2] (X = F, Cl, Br, I, BH4, H) can be synthesised in the same manner. When the same reaction was attempted with [(C5Me5)2TiX] (X = Cl, Br, H, Me, BH4) the bridged compounds [(C5Me5)2Yb(μ-X)Ti(C5Me5)2] were obtained, in which the key feature is the nearly linear Yb–X–Ti bridge.227
[(C5Me5)2Sm(THF)2] reduces diphenylmercury to clean elemental mercury and the orange complex [(C5Me5)2Sm(Ph)(THF)].229 The structure was established by X-ray crystallography.
In 2001, [{(C5Me5)2SmPh}2] was synthesised again from [(C5Me5)2Sm] with HgPh2 for mechanistic studies of the samarium-catalyzed redistribution of Ph3SiH3 to Ph2SiH2 and SiH4.230 It was assumed that Sm–Ph would play a key role as an intermediate. The first attempts to synthesise [{(C5Me5)2SmPh}2] were conducted in toluene, but the reaction led to [(C5Me5)2Sm(CH2Ph)]. The anticipated product was finally obtained with benzene as solvent. However, [{(C5Me5)2SmPh}2] is thermally unstable and decomposes even at −35 °C under a nitrogen atmosphere. Thus, the complex was only characterised in solution by NMR.
[(C5Me5)2Yb] reacts with [(C5H5)2M] (M = V, Cr, Mn, Co and Ni) in an exchange reaction to yield [(C5Me5)2Yb(C5H5)(THF)n] (n = 0 or 2, depending on the conditions). It is noteworthy that the reaction proceeds on different timescales. The Ni and Mn compounds react within 24 hours, whereas the other metallocenes need timespans ranging from days to weeks.231
In recent years, the reactivity towards polyphosphide complexes was investigated.232–234 Pentaphosphaferrocene [(C5Me5)Fe(P5)] and [(C5Me4R)2Sm(THF)2] (R = Me, nPr) reacted under reductive coupling to [((C5Me5)Fe)2P10{Sm(η5-C5Me4R)2}2] (R = Me, nPr) (Scheme 86).232 A phosphorous atom of each P5 ring was coupled, thus forming the bicyclic (P10)4− ion. The iron atoms bind to four phosphorous atoms each, while the fifth is connected to the other ring. Each samarocene unit connects to two phosphorus atoms, each on opposing sides of the polyphosphide ion.
 | ||
| Scheme 86 Synthesis of [((C5Me5)Fe)2P10{Sm(η5-C5Me4R)2}2] (R = Me, nPr).232 | ||
In contrast to pentaphosphaferrocene [(C5Me5)Fe(P5)], the corresponding arsenide species [(C5Me5)Fe(As5)] is less stable due to a weaker As–As bond. Reduction of [(C5Me5)Fe(As5)] with [(1,3-C5H3tBu2)2Sm(THF)] led to the arsenic poor [{(1,3-C5H3tBu2)2Sm}(μ,η4:η4-As4){Fe(C5Me5)}] and the arsenic-rich species [{(1,3-C5H3tBu2)2Sm}2(As7){Fe(C5Me5)}] (Scheme 87).235 These compounds were the first polyarsenides of rare earth metals. Both compounds form parallel. They were separated by crystallization from different solvents. [{(1,3-C5H3tBu2)2Sm}(μ,η4:η4-As4)[Fe(C5Me5)] is a nice example of a d/f-triple decker sandwich complex with a purely inorganic planar middle deck. As supported by DFT calculations the As42− unit is a 6π-aromatic system, which is related to the cyclobutadiene dianion (CH)42−. The bond distances within the As42− dianion are equal with an average As–As bond length of 2.410 Å. The arsenic-rich species [{(1,3-C5H3tBu2)2Sm}2(As7){Fe(C5Me5)}] features an As73− cage, which has a norbornadiene-like structure with two short As–As bonds in the scaffold.
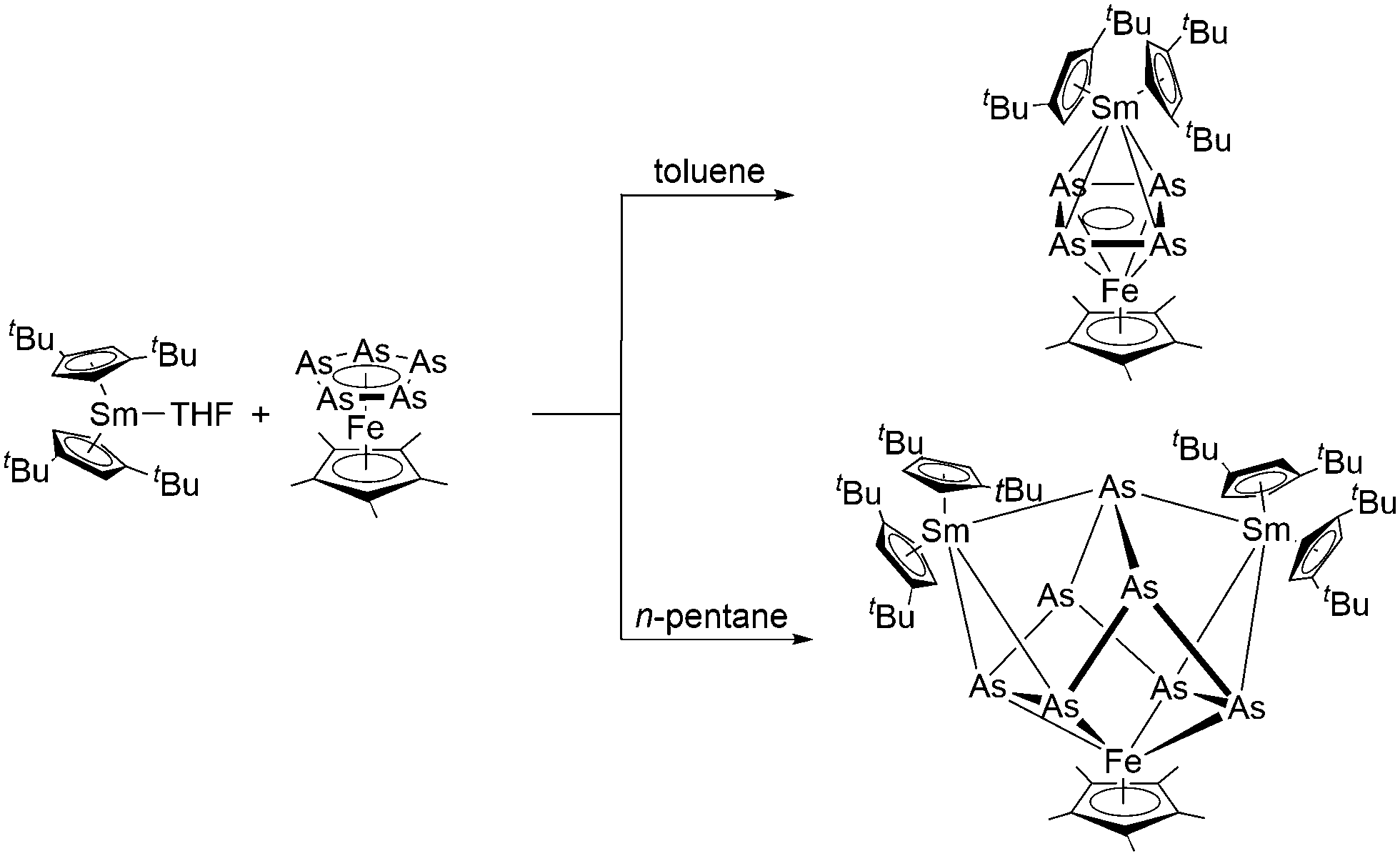 | ||
| Scheme 87 Synthesis of the first polyarsenides of rare earth metals: the arsenic poor species [{(1,3-C5H3tBu2)2Sm}(μ,η4:η4-As4){Fe(C5Me5)}] and the arsenic-rich species [{(1,3-C5H3tBu2)2Sm}2(As7){Fe(C5Me5)}].235 | ||
The molecule [(C5H2(tBu)3Co)2(μ,η2:2-P2)2] containing two P2 units was reacted with [(C5Me4R)2Sm(THF)2] (R = Me, nPr) in heptane.236 Single crystal X-ray diffraction studies revealed the structure of the product as [(C5H2(tBu)3Co)2P4Sm(C5Me4R)2] (R = Me, nPr) (Scheme 88). Upon reduction, a bond between the two P2 ligands was formed to establish a P4 chain ligand. The samarocene unit is located between the C5H2(tBu)3Co units, bonding to two phosphorus atoms. The Sm–P bond lengths are 2.874(3) and 2.921(3) Å. Quantum chemical calculations were carried out. They showed that the P–P bond formation was not a direct consequence of reduction by samarium, as all the spin density resides on the Co atoms.
 | ||
| Scheme 88 Synthesis of [(C5H2(tBu)3Co)2E4Sm(C5Me5)2] (E = P, As).236,237 | ||
In a similar reaction, the arsenic compound [(C5H2(tBu)3Co)2(μ,η2:2-As2)2] featuring two As2 units was reacted with [(C5Me4R)2Sm(THF)2] (R = Me, nPr) to give [(C5H2(tBu)3Co)2As4Sm(C5Me4R)2] (R = Me, nPr) (Scheme 88), which is the first structural representative of open chain-like polyarsenides as ligands in the coordination sphere of the lanthanides.237 The central As4Co2 scaffold forms a (non-crystallographic) C2-symmetric distorted trigonal prism, which is chiral. However, both enantiomers crystallised as racemates. The observed As–As bonds within the chain are in the range of those observed in yellow arsenic (2.44 Å) and thus can be considered single bonds.238
Treatment of [(C5Me5)2Ln(THF)2] (Ln = Sm and Yb) with the phosphide complex [{(C5H5)Mo(CO)2}2(μ,η2:2-P2)] resulted in a mixture of phosphide containing complexes, which were separated by fractional crystallization. The major product is the sixteen-membered, bicyclic complexes [{(C5Me5)2Ln}2P2{(C5H5)Mo(CO)2}4] (Scheme 89).234 In the case of samarocene, two further complexes were isolated as side products, namely the P4 complex [{(C5Me5)2Sm}2P4{(C5H5)Mo(CO)2}2] and the P5 complex [{(C5Me5)2Sm}3P5{(C5H5)Mo(CO)2}3] (Scheme 89). Both complexes consist of cyclo-phosphide ligands resulting from reduction by the lanthanide. All compounds were crystallographically characterised. DFT calculations elucidate the electronic structure of the complexes.
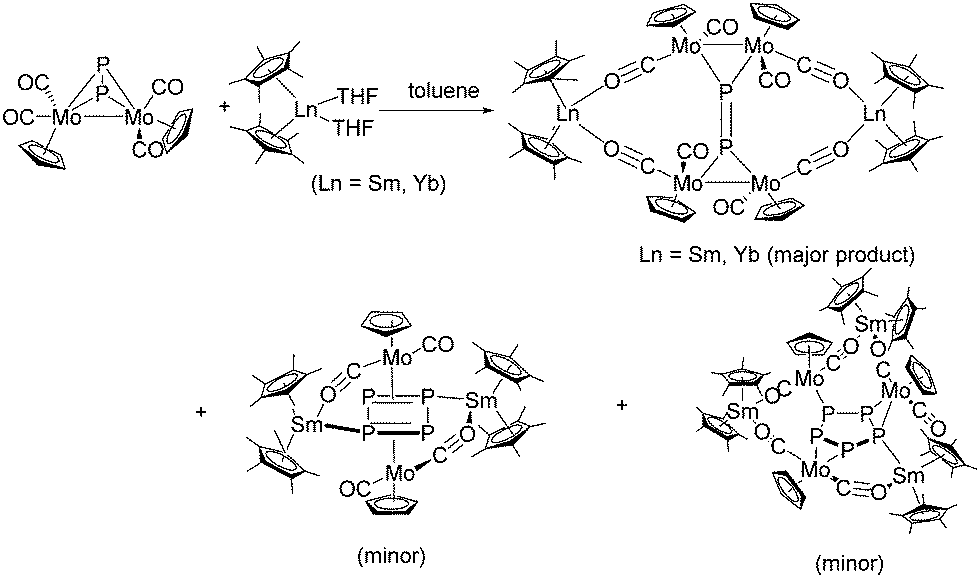 | ||
| Scheme 89 Reactions of [(C5Me5)2Ln(THF)2] (Ln = Sm, Yb) with [{(C5H5)Mo(CO)2}2(μ,η2:2-P2)].234 | ||
By reacting the heavier group 15 congener [{(C5H4tBu)Mo(CO)2}2(μ,η2:2-As2)] with solvent-free [(C5Me5)2Sm], it resulted in a straightforward formation of the mixed d/f-metal species [{(C5Me5)2Sm}2As2{(C5H4tBu)Mo(CO)2}2] (Scheme 90).239 Due to the two-electron reduction of [{(C5H4tBu)Mo(CO)2}2(μ,η2:2-As2)] the Mo–Mo bond is cleaved and the [{(C5H4tBu)Mo(CO)2}2(μ,η2:2-As2)]2− dianion is formed. The central As22− unit is side-on coordinated with the Mo atoms. This is in contrast to [{(C5Me5)2Ln}2P2{(C5H5)Mo(CO)2}4] (Scheme 89)234 in which the P22− unit is end-on bound. The As–As-bond length of 2.238(2) Å is in the range of As–As double bonds known in the literature and only slightly shortened in comparison to [{(C5H5)Mo(CO)2}2(μ,η2:2-As2)].240,241 On the other hand, quantum chemical calculations suggest a weakened double bond.
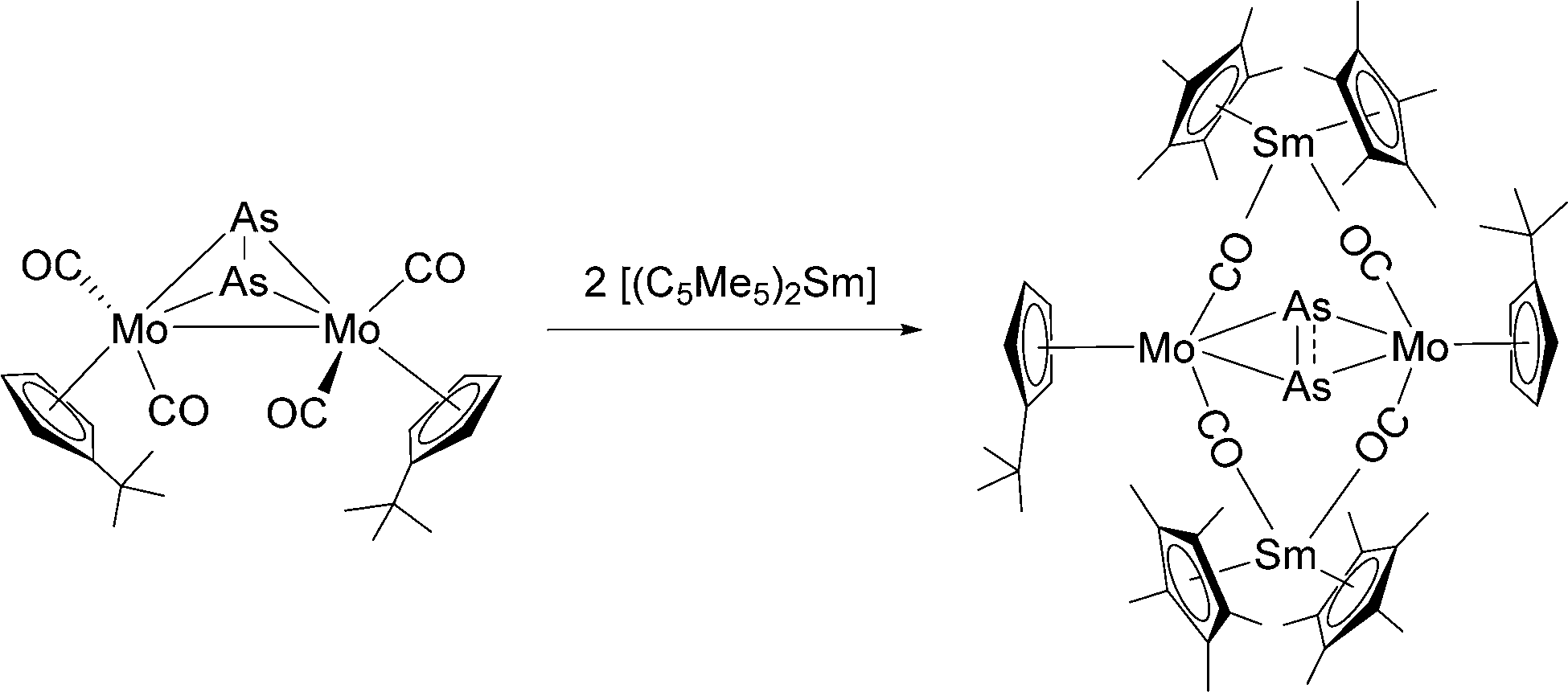 | ||
| Scheme 90 Reduction of [{(C5H4tBu)Mo(CO)2}2(μ,η2:2-As2)] with [(C5Me5)2Sm(THF)2] resulting in [{(C5Me5)2Sm}2As2{(C5H4tBu)Mo(CO)2}2].239 | ||
When the analogue reaction of [(C5Me5)2Sm] with [{(C5H4tBu)Mo(CO)2}2(μ,η2:2-Sb2)] was performed the polystibide [{(C5Me5)2Sm}2Sb4{(C5H4tBu)Mo(CO)2}2], featuring a planar Sb4 ring was isolated (Scheme 91).239 The planar Sb4 ring, which consists of two short (Sb1–Sb2 2.7313(8) Å) and two longer (Sb1–Sb2′ 2.8608(7) Å) Sb–Sb bonds, is as a unprecedented ligand for an organometallic or coordination compound. This scaffold can be considered a metal-coordinated tetrastibacyclobutadiene unit.
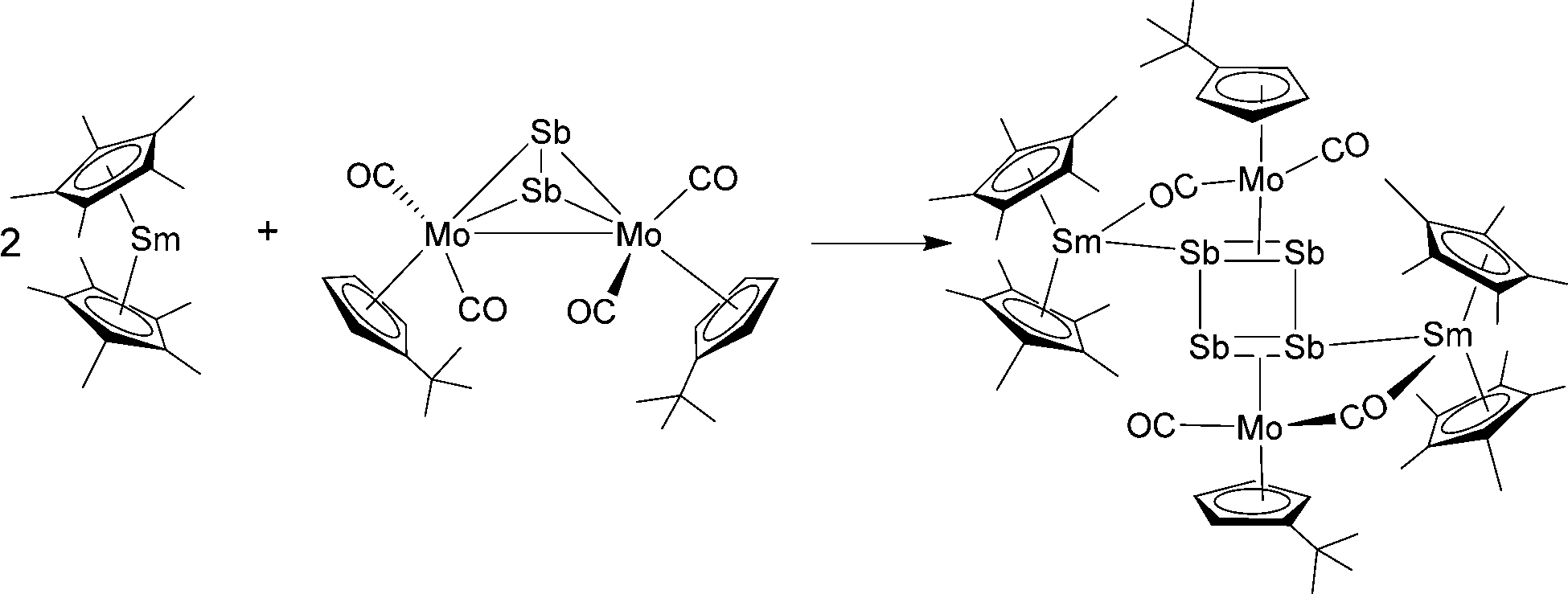 | ||
| Scheme 91 Synthesis of [{(C5Me5)2Sm}2Sb4{(C5H4tBu)Mo(CO)2}2].239 | ||
When [(C5Me5)2Ln(THF)2] (Ln = Sm, Yb) was treated with the molybdenum-polyphosphide sandwich compound [(C5Me5)Mo(CO)2(η3-P3)], reductive coupling of the triangular phosphide ions occurs.234 This resulted in a new P–P bond formation via a reductive dimerization. As a result, the 4d/4f hexaphosphide complexes [{(C5Me5)2Ln}2P6{(C5Me5)Mo(CO)2}2] featuring an unusual P6 ligand in the centre were formed (Scheme 92). The P–P bond length of the newly formed bond is in the range of a P–P single bond. Each of the two molybdenum-containing units and each of the two lanthanocene fragments coordinates with one of the triangular P3 units.234
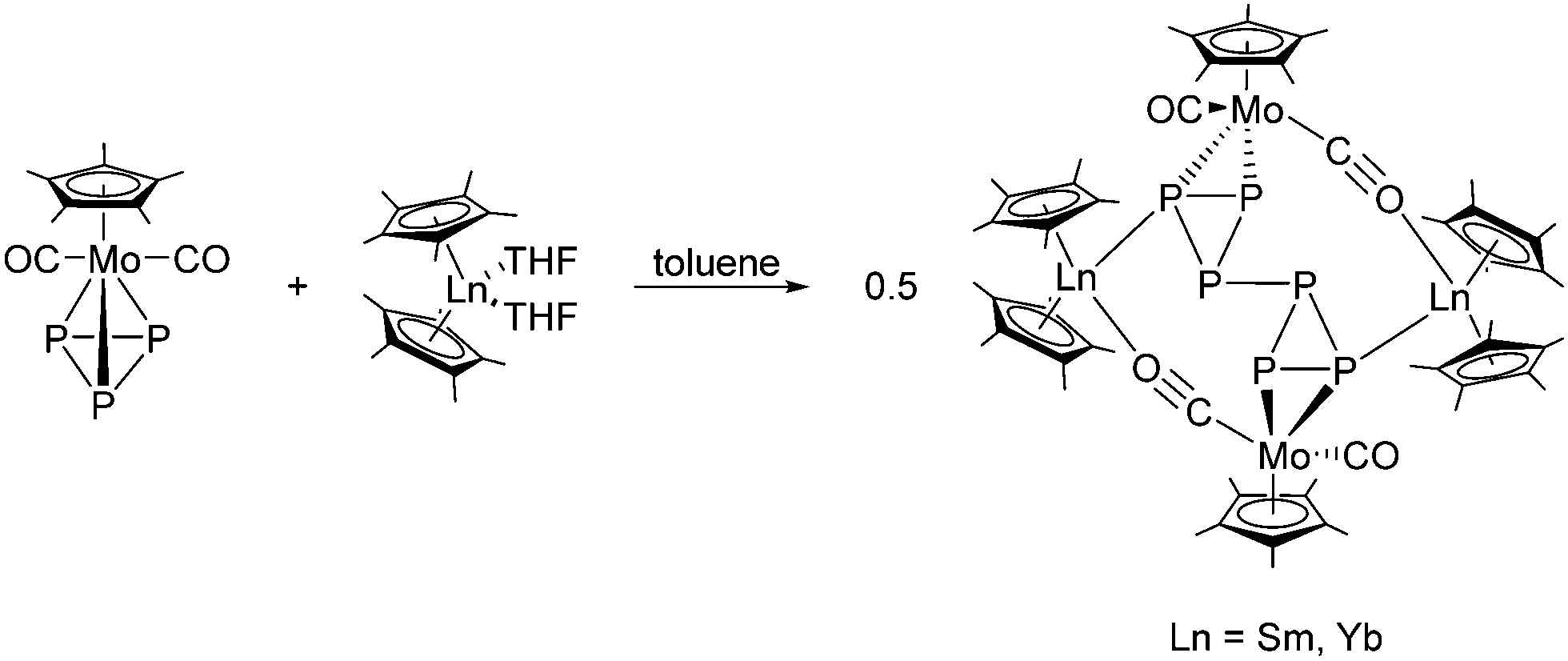 | ||
| Scheme 92 Synthesis of [{(C5Me5)2Ln}2P6{(C5Me5)Mo(CO)2}2] (Ln = Sm, Yb).234 | ||
6. Conclusions
In this article, we provided a comprehensive overview of the syntheses, properties and reactivities of divalent metallocenes of the lanthanides. Two main reaction pathways of these compounds have become clear: Lewis acid–base complex formation and redox reactions in which the lanthanide atom is oxidised in a one-electron reaction. These interesting reagents are characterised by the rich and diverse structures of the resulting products and their high reactivity. The observed reduction chemistry of the divalent lanthanocenes shows some similarities with the reduction chemistry of alkali metals. In addition to the similarities in reactivity, some products also show structural correlations. However, in contrast to the alkaline metals, the reactivity can be fine-tuned via the steric bulk of the substituents of the cyclopentadienyl rings. Moreover, the electron transfer is in some cases reversible. The discovery of the so-called non-classical divalent lanthanides (divalent lanthanides beyond Sm, Eu, Yb) opens another almost unexplored pathway towards unprecedented reactivity. However, even with the established classical compounds, a large number of new reactions have been published in recent years. Thus, the journey is far from being over. We hope that we have been able to show that divalent metallocenes of the lanthanides have an enriching chemistry, which even after almost 40 years of the first reports is far from being fully investigated.Author contributions
All authors did the literature research and wrote parts of the manuscript. PWR originated the idea and supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the Collaborative Research Centre “4f for Future” (CRC 1573 project number 471424360, project C1).Notes and references
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- G. B. Kauffman, J. Chem. Educ., 1983, 60, 185 CrossRef CAS.
- E. O. Fischer and W. Pfab, Z Naturforsch B, 1952, 7, 377–379 CrossRef.
- H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052–6058 CrossRef CAS PubMed.
- G. W. Watt and E. W. Gillow, J. Am. Chem. Soc., 1969, 91, 775–776 CrossRef CAS.
- G. B. Deacon, G. D. Fallon, P. I. MacKinnon, R. H. Newnham, G. N. Pain, T. D. Tuong and D. L. Wilkinson, J. Organomet. Chem., 1984, 277, C21–C24 CrossRef CAS.
- F. Jaroschik, F. Nief and L. Ricard, Chem. Commun., 2006, 426–428 RSC.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 1123–1125 CrossRef CAS.
- F. Jaroschik, A. Momin, F. Nief, X.-F. Le Goff, G. B. Deacon and P. C. Junk, Angew. Chem., Int. Ed., 2009, 48, 1117–1121 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed.
- K. R. McClain, C. A. Gould, D. A. Marchiori, H. Kwon, T. T. Nguyen, K. E. Rosenkoetter, D. Kuzmina, F. Tuna, R. D. Britt, J. R. Long and B. G. Harvey, J. Am. Chem. Soc., 2022, 144, 22193–22201 CrossRef CAS PubMed.
- P. L. Watson, J Chem Soc Chem Comm, 1980, 14, 652–653 RSC.
- T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Inorg. Chem., 1980, 19, 2999–3003 CrossRef CAS.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1981, 103, 6507–6508 CrossRef CAS.
- W. J. Evans, J. W. Grate, H. W. Choi, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 941–946 CrossRef CAS.
- M. Schultz, C. D. Sofield, M. D. Walter and R. A. Andersen, New J. Chem, 2005, 29, 919–927 RSC.
- C. A. P. Goodwin, D. Reta, F. Ortu, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2017, 139, 18714–18724 CrossRef CAS PubMed.
- W. J. Evans, T. S. Gummersheimer and J. W. Ziller, Appl. Organomet. Chem., 1995, 9, 437–447 CrossRef CAS.
- M. Schultz, C. J. Burns, D. J. Schwartz and R. A. Andersen, Organometallics, 2000, 19, 781–789 CrossRef CAS.
- P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS.
- W. J. Evans, T. S. Gummersheimer, T. J. Boyle and J. W. Ziller, Organometallics, 1994, 13, 1281–1284 CrossRef CAS.
- W. J. Evans, K. J. Forrestal and J. W. Ziller, Polyhedron, 1998, 17, 4015–4021 CrossRef CAS.
- W. J. Evans, J. M. Perotti, J. C. Brady and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 5204–5212 CrossRef CAS PubMed.
- H. Nakamura, Y. Nakayama, H. Yasuda, T. Maruo, N. Kanehisa and Y. Kai, Organometallics, 2000, 19, 5392–5399 CrossRef CAS.
- C. Qian, G. Zou, W. Jiang, Y. Chen, J. Sun and N. Li, Organometallics, 2004, 23, 4980–4986 CrossRef CAS.
- M. Visseaux, D. Barbier-Baudry, O. Blacque, A. Hafid, P. Richard and F. Weber, New J. Chem, 2000, 24, 939–942 RSC.
- V. K. Bel'sky, Y. K. Gunko, B. M. Bulychev, A. I. Sizov and G. L. Soloveichik, J. Organomet. Chem., 1990, 390, 35–44 CrossRef.
- S. K. Adas and G. J. Balaich, J. Organomet. Chem., 2018, 857, 200–206 CrossRef CAS.
- F. Weber, M. Schultz, C. D. Sofield and R. A. Andersen, Organometallics, 2002, 21, 3139–3146 CrossRef CAS.
- S. Zhou, Z. Wu, L. Zhou, S. Wang, L. Zhang, X. Zhu, Y. Wei, J. Zhai and J. Wu, Inorg. Chem., 2013, 52, 6417–6426 CrossRef CAS PubMed.
- M. Xémard, V. Goudy, A. Braun, M. Tricoire, M. Cordier, L. Ricard, L. Castro, E. Louyriac, C. E. Kefalidis, C. Clavaguéra, L. Maron and G. Nocton, Organometallics, 2017, 36, 4660–4668 CrossRef.
- E. Ihara, M. Nodono, K. Katsura, Y. Adachi, H. Yasuda, M. Yamagashira, H. Hashimoto, N. Kanehisa and Y. Kai, Organometallics, 1998, 17, 3945–3956 CrossRef CAS.
- C. A. P. Goodwin, J. Su, L. M. Stevens, F. D. White, N. H. Anderson, J. D. Auxier, T. E. Albrecht-Schönzart, E. R. Batista, S. F. Briscoe, J. N. Cross, W. J. Evans, A. N. Gaiser, A. J. Gaunt, M. R. James, M. T. Janicke, T. F. Jenkins, Z. R. Jones, S. A. Kozimor, B. L. Scott, J. M. Sperling, J. C. Wedal, C. J. Windorff, P. Yang and J. W. Ziller, Nature, 2021, 599, 421–424 CrossRef CAS PubMed.
- H. Sitzmann, T. Dezember, O. Schmitt, F. Weber, G. Wolmershäuser and M. Ruck, Z. Anorg. Allg. Chem., 2000, 626, 2241–2244 CrossRef CAS.
- W. J. Evans, G. Kociok-Köhn, V. S. Leong and J. W. Ziller, Inorg. Chem., 1992, 31, 3592–3600 CrossRef CAS.
- A. N. Selikhov, T. V. Mahrova, A. V. Cherkasov, G. K. Fukin, J. Larionova, J. Long and A. A. Trifonov, Organometallics, 2015, 34, 1991–1999 CrossRef CAS.
- M. N. Bochkarev, I. L. Fedushkin, A. A. Fagin, T. V. Petrovskaya, J. W. Ziller, R. N. R. Broomhall-Dillard and W. J. Evans, Angew. Chem., Int. Ed. Engl., 1997, 36, 133–135 CrossRef CAS.
- W. J. Evans, N. T. Allen and J. W. Ziller, Angew. Chem., Int. Ed., 2002, 41, 359–361 CrossRef CAS PubMed.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 3552–3558 CrossRef CAS.
- F. Nief, B. T. de Borms, L. Ricard and D. Carmichael, Eur. J. Inorg. Chem., 2005, 637–643 CrossRef CAS.
- T. Simler, K. N. McCabe, L. Maron and G. Nocton, Chem. Sci, 2022, 13, 7449–7461 RSC.
- W. J. Evans, L. A. Hughes and T. P. Hanusa, J. Am. Chem. Soc., 1984, 106, 4270–4272 CrossRef CAS.
- W. J. Evans, G. Kociok-Köhn, S. E. Foster, J. W. Ziller and R. J. Doedens, J. Organomet. Chem., 1993, 444, 61–66 CrossRef CAS.
- W. J. Evans, L. A. Hughes and T. P. Hanusa, Organometallics, 1986, 5, 1285–1291 CrossRef CAS.
- P. B. Hitchcock, J. A. K. Howard, M. F. Lappert and S. Prashar, J. Organomet. Chem., 1992, 437, 177–189 CrossRef CAS.
- G. Nocton and L. Ricard, Chem. Commun., 2015, 51, 3578–3581 RSC.
- G. B. Deacon, C. M. Forsyth, F. Jaroschik, P. C. Junk, D. L. Kay, T. Maschmeyer, A. F. Masters, J. Wang and L. D. Field, Organometallics, 2008, 27, 4772–4778 CrossRef CAS.
- N. Reinfandt and P. W. Roesky, Inorganics, 2022, 10, 25 CrossRef CAS.
- M. H. Kuiper and H. Lueken, Z. Anorg. Allg. Chem., 2007, 633, 1407–1409 CrossRef CAS.
- A. L. Wayda, J. L. Dye and R. D. Rogers, Organometallics, 1984, 3, 1605–1610 CrossRef CAS.
- B. M. Wolf, C. Stuhl, C. Maichle-Mössmer and R. Anwander, Chem. – Eur. J., 2018, 24, 15921–15929 CrossRef CAS PubMed.
- R. P. Kelly, T. D. M. Bell, R. P. Cox, D. P. Daniels, G. B. Deacon, F. Jaroschik, P. C. Junk, X. F. Le Goff, G. Lemercier, A. Martinez, J. Wang and D. Werner, Organometallics, 2015, 34, 5624–5636 CrossRef CAS.
- G. Lin and W.-T. Wong, J. Organomet. Chem., 1996, 523, 93–98 CrossRef CAS.
- A. C. G. Shephard, D. P. Daniels, G. B. Deacon, Z. Guo, F. Jaroschik and P. C. Junk, Chem. Commun., 2022, 58, 4344–4347 RSC.
- I. L. Fedushkin, S. Dechert and H. Schumann, Angew. Chem., Int. Ed., 2001, 40, 561–563 CrossRef CAS PubMed.
- J. Tao, S. Qi, L. Yonghua and J. Songchun, J. Organomet. Chem., 1993, 450, 121–124 CrossRef.
- M. F. Lappert, P. I. W. Yarrow, J. L. Atwood, R. Shakir and J. Holton, J. Chem. Soc., Chem. Commun., 1980, 987–988 RSC.
- E. Sheng, S. Zhou, S. Wang, G. Yang, Y. Wu, Y. Feng, L. Mao and Z. Huang, Eur. J. Inorg. Chem., 2004, 2923–2932 CrossRef CAS.
- K. Zhang, W. Zhang, S. Wang, E. Sheng, G. Yang, M. Xie, S. Zhou, Y. Feng, L. Mao and Z. Huang, Dalton Trans, 2004, 1029–1037 RSC.
- S. Zhou, S. Wang, E. Sheng, L. Zhang, Z. Yu, X. Xi, G. Chen, W. Luo and Y. Li, Eur. J. Inorg. Chem., 2007, 1519–1528 CrossRef CAS.
- S. Wang, S. Wang, S. Zhou, G. Yang, W. Luo, N. Hu, Z. Zhou and H.-B. Song, J. Organomet. Chem., 2007, 692, 2099–2106 CrossRef CAS.
- C. Ruspic, J. R. Moss, M. Schürmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 2121–2126 CrossRef CAS PubMed.
- N. J. C. van Velzen and S. Harder, Organometallics, 2018, 37, 2263–2271 CrossRef CAS.
- S. Harder, D. Naglav, C. Ruspic, C. Wickleder, M. Adlung, W. Hermes, M. Eul, R. Pöttgen, D. B. Rego, F. Poineau, K. R. Czerwinski, R. H. Herber and I. Nowik, Chem. – Eur. J., 2013, 19, 12272–12280 CrossRef CAS PubMed.
- R. A. Andersen, J. M. Boncella, C. J. Burns, R. Blom, A. Haaland and H. V. Volden, J. Organomet. Chem., 1986, 312, C49–C52 CrossRef CAS.
- R. A. Andersen, R. Blom, C. J. Burns and H. V. Volden, J Chem Soc Chem Comm, 1987, 10, 768–769 RSC.
- J. Marçalo, A. Pires de Matos and W. J. Evans, Organometallics, 1996, 15, 345–349 CrossRef.
- T. P. Hanusa, Chem. Rev., 1993, 93, 1023–1036 CrossRef CAS.
- C. Elschenbroich, Organometallchemie, Teubner, Wiesbaden, 2008 Search PubMed.
- R. L. De Kock, M. A. Peterson, L. K. Timmer, E. J. Baerends and P. Vernooijs, Polyhedron, 1990, 9, 1919–1934 CrossRef CAS.
- T. V. Timofeeva, J. H. Lii and N. L. Allinger, J. Am. Chem. Soc., 1995, 117, 7452–7459 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- N. Kaltsoyannis and M. R. Russo, J. Nucl. Sci. Technol., 2014, 39, 393–399 CrossRef.
- J. C. Green, D. Hohl and N. Roesch, Organometallics, 1987, 6, 712–720 CrossRef CAS.
- S. Labouille, C. Clavaguera and F. Nief, Organometallics, 2013, 32, 1265–1271 CrossRef CAS.
- A. F. Williams, F. Grandjean, G. J. Long, T. A. Ulibarri and W. J. Evans, Inorg. Chem., 1989, 28, 4584–4588 CrossRef CAS.
- A. G. Avent, M. A. Edelman, M. F. Lappert and G. A. Lawless, J. Am. Chem. Soc., 1989, 111, 3423–3425 CrossRef CAS.
- S. P. Nolan, D. Stern and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 7844–7853 CrossRef CAS.
- S. J. Swamy, J. Loebel, J. Pickardt and H. Schumann, J. Organomet. Chem., 1988, 353, 27–34 CrossRef CAS.
- W. J. Evans and T. A. Ulibarri, Polyhedron, 1989, 8, 1007–1014 CrossRef CAS.
- W. J. Evans, J. W. Grate, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 405–409 CrossRef CAS.
- D. J. Schwartz and R. A. Andersen, Organometallics, 1995, 14, 4308–4318 CrossRef CAS.
- Z. Hou, Y. Zhang, H. Tezuka, P. Xie, O. Tardif, T.-A. Koizumi, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 2000, 122, 10533–10543 CrossRef CAS.
- Z. M. Hou, Y. G. Zhang, T. Yoshimura and Y. Wakatsuki, Organometallics, 1997, 16, 2963–2970 CrossRef CAS.
- M. Nishiura, Z. Hou and Y. Wakatsuki, Organometallics, 2004, 23, 1359–1368 CrossRef CAS.
- W. J. Evans, G. W. Rabe and J. W. Ziller, J. Organomet. Chem., 1994, 483, 39–45 CrossRef CAS.
- W. J. Evans, E. Montalvo, S. E. Foster, K. A. Harada and J. W. Ziller, Organometallics, 2007, 26, 2904–2910 CrossRef CAS.
- T. D. Tilley, R. A. Andersen, B. Spencer and A. Zalkin, Inorg. Chem., 1982, 21, 2647–2649 CrossRef CAS.
- G. Nocton and L. Ricard, Dalton Trans, 2014, 43, 4380–4387 RSC.
- M. Schultz, J. M. Boncella, D. J. Berg, T. D. Tilley and R. A. Andersen, Organometallics, 2002, 21, 460–472 CrossRef CAS.
- W. J. Evans and D. K. Drummond, J. Am. Chem. Soc., 1989, 111, 3329–3335 CrossRef CAS.
- C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y. J. Hu, L. Maron, O. Eisenstein, M. Head-Gordon and R. A. Andersen, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS PubMed.
- G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed.
- J. M. Veauthier, E. J. Schelter, C. N. Carlson, B. L. Scott, R. E. Da Re, J. D. Thompson, J. L. Kiplinger, D. E. Morris and K. D. John, Inorg. Chem., 2008, 47, 5841–5849 Search PubMed.
- C. N. Carlson, C. J. Kuehl, R. E. Da Re, J. M. Veauthier, E. J. Schelter, A. E. Milligan, B. L. Scott, E. D. Bauer, J. D. Thompson, D. E. Morris and K. D. John, J. Am. Chem. Soc., 2006, 128, 7230–7241 Search PubMed.
- F. Reichart, M. Kischel and K. Zeckert, Chem. – Eur. J., 2009, 15, 10018–10020 CrossRef CAS PubMed.
- K. Zeckert, Organometallics, 2013, 32, 1387–1393 CrossRef CAS.
- K. Zeckert, S. Zahn and B. Kirchner, Chem. Commun., 2010, 46, 2638–2640 RSC.
- K. Zeckert, Dalton Trans, 2012, 41, 14101–14106 RSC.
- V. Goudy, A. Jaoul, M. Cordier, C. Clavaguéra and G. Nocton, J. Am. Chem. Soc., 2017, 139, 10633–10636 CrossRef CAS PubMed.
- D. Wang, J. Moutet, M. Tricoire, M. Cordier and G. Nocton, Inorganics, 2019, 7, 58 CrossRef CAS PubMed.
- M. Tricoire, D. Wang, T. Rajeshkumar, L. Maron, G. Danoun and G. Nocton, JACS Au, 2022, 2, 1881–1888 CrossRef CAS PubMed.
- D. Wang, M. Tricoire, V. Cemortan, J. Moutet and G. Nocton, Inorg. Chem. Front, 2021, 8, 647–657 RSC.
- S. A. J. Boyce, J. Moutet, L. Niederegger, T. Simler, G. Nocton and C. R. Hess, Inorg. Chem., 2021, 60, 403–411 CrossRef CAS PubMed.
- E. J. Schelter, J. M. Veauthier, J. D. Thompson, B. L. Scott, K. D. John, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2006, 128, 2198–2199 CrossRef CAS PubMed.
- E. J. Schelter, R. Wu, J. M. Veauthier, E. D. Bauer, C. H. Booth, R. K. Thomson, C. R. Graves, K. D. John, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2010, 49, 1995–2007 CrossRef CAS PubMed.
- M. Schultz, C. J. Burns, D. J. Schwartz and R. A. Andersen, Organometallics, 2001, 20, 5690–5699 CrossRef CAS.
- P. Selg, H. H. Brintzinger, M. Schultz and R. A. Andersen, Organometallics, 2002, 21, 3100–3107 CrossRef CAS.
- L. Maron, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2002, 124, 5614–5615 CrossRef CAS PubMed.
- F. Weber, H. Sitzmann, M. Schultz, C. D. Sofield and R. A. Andersen, Organometallics, 2002, 21, 3139–3146 CrossRef CAS.
- A. J. Arduengo, M. Tamm, S. J. Mclain, J. C. Calabrese, F. Davidson and W. J. Marshall, J. Am. Chem. Soc., 1994, 116, 7927–7928 CrossRef CAS.
- M. Glanz, S. Dechert, H. Schumann, D. Wolff and J. Springer, Z. Anorg. Allg. Chem., 2000, 626, 2467–2477 CrossRef CAS.
- H. Schumann, M. Glanz, J. Winterfeld, H. Hemling, N. Kuhn and T. Kratz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1733–1734 CrossRef.
- C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 941–942 CrossRef CAS.
- C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 915–917 CrossRef CAS.
- Z. M. Hou, Y. Zhang, M. Nishiura and Y. Wakatsuki, Organometallics, 2003, 22, 129–135 CrossRef CAS.
- C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 5853–5855 CrossRef CAS.
- H. Yamamoto, H. Yasuda, K. Yokota, A. Nakamura, Y. Kai and N. Kasai, Chem. Lett., 1988, 1963–1966 CrossRef CAS.
- W. J. Evans, T. M. Champagne and J. W. Ziller, Organometallics, 2007, 26, 1204–1211 CrossRef CAS.
- W. J. Evans, J. R. Walensky, F. Furche, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2009, 28, 6073–6078 CrossRef CAS.
- C. T. Palumbo, J. W. Ziller and W. J. Evans, J. Organomet. Chem., 2018, 867, 142–148 CrossRef CAS.
- W. J. Evans, J. M. Perotti, J. W. Ziller, D. F. Moser and R. West, Organometallics, 2003, 22, 1160–1163 CrossRef CAS.
- R. Zitz, H. Arp, J. Hlina, M. Walewska, C. Marschner, T. Szilvási, B. Blom and J. Baumgartner, Inorg. Chem., 2015, 54, 3306–3315 CrossRef CAS PubMed.
- T. D. Tilley, R. A. Andersen and A. Zalkin, Inorg. Chem., 1983, 22, 856–859 CrossRef CAS.
- A. Jaoul, C. Clavaguéra and G. Nocton, New J. Chem, 2016, 40, 6643–6649 RSC.
- M. T. Gamer, P. W. Roesky, S. N. Konchenko, P. Nava and R. Ahlrichs, Angew. Chem., Int. Ed., 2006, 45, 4447–4451 CrossRef CAS PubMed.
- M. Wiecko and P. W. Roesky, Organometallics, 2007, 26, 4846–4848 CrossRef CAS.
- L. R. Morss, Chem. Rev., 1976, 76, 827–841 CrossRef CAS.
- R. G. Finke, S. R. Keenan and P. L. Watson, Organometallics, 1989, 8, 263–277 CrossRef CAS.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1983, 105, 1401–1403 CrossRef CAS.
- W. J. Evans, D. G. Giarikos, C. B. Robledo, V. S. Leong and J. W. Ziller, Organometallics, 2001, 20, 5648–5652 CrossRef CAS.
- W. J. Evans, R. A. Keyer and J. W. Ziller, Organometallics, 1993, 12, 2618–2633 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri, L. R. Chamberlain, J. W. Ziller and D. Alvarez, Organometallics, 1990, 9, 2124–2130 CrossRef CAS.
- V. Goudy, M. Xémard, S. Karleskind, M. Cordier, C. Alvarez Lamsfus, L. Maron and G. Nocton, Inorganics, 2018, 6, 82 CrossRef.
- W. J. Evans, R. A. Keyer, H. Zhang and J. L. Atwood, J. Chem. Soc., Chem. Commun., 1987, 837–838 RSC.
- W. J. Evans, R. A. Keyer and J. W. Ziller, Organometallics, 1990, 9, 2628–2631 CrossRef CAS.
- J. M. Boncella, T. D. Tilley and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1984, 710–712 RSC.
- W. J. Evans, G. W. Rabe and J. W. Ziller, J. Organomet. Chem., 1994, 483, 21–25 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1990, 112, 2314–2324 CrossRef CAS.
- W. J. Evans and T. A. Ulibarri, J. Am. Chem. Soc., 1987, 109, 4292–4297 CrossRef CAS.
- M. P. Plesniak, X. Just-Baringo, F. Ortu, D. P. Mills and D. J. Procter, Chem. Commun., 2016, 52, 13503–13506 RSC.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1994, 116, 2600–2608 CrossRef CAS.
- A. Recknagel, M. Noltemeyer and F. T. Edelmann, J. Organomet. Chem., 1991, 410, 53–61 CrossRef CAS.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 7423–7424 CrossRef CAS.
- W. J. Evans, K. J. Forrestal, J. T. Leman and J. W. Ziller, Organometallics, 1996, 15, 527–531 CrossRef CAS.
- R. G. Finke, S. R. Keenan, D. A. Schiraldi and P. L. Watson, Organometallics, 1987, 6, 1356–1358 CrossRef CAS.
- R. G. Finke, S. R. Keenan, D. A. Schiraldi and P. L. Watson, Organometallics, 1986, 5, 598–601 CrossRef CAS.
- W. J. Evans, J. W. Grate, K. R. Levan, I. Bloom, T. T. Peterson, R. J. Doedens, H. Zhang and J. L. Atwood, Inorg. Chem., 1986, 25, 3614–3619 CrossRef CAS.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999–2009 CrossRef CAS.
- C. J. Burns and R. A. Anderson, J. Chem. Soc., Chem. Commun., 1989, 2, 136–137 RSC.
- H. Schumann, M. R. Keitsch, J. Winterfeld and J. Demtschuk, J. Organomet. Chem., 1996, 525, 279–281 CrossRef CAS.
- W. J. Evans and D. K. Drummond, Organometallics, 1988, 7, 797–802 CrossRef CAS.
- A. A. Trifonov, I. D. Gudilenkov, G. K. Fukin, A. V. Cherkasov and J. Larionova, Organometallics, 2009, 28, 3421–3425 CrossRef CAS.
- W. J. Evans, D. K. Drummond, S. G. Bott and J. L. Atwood, Organometallics, 1986, 5, 2389–2391 CrossRef CAS.
- W. J. Evans, D. K. Drummond, L. R. Chamberlain, R. J. Doedens, S. G. Bott, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 4983–4994 CrossRef CAS.
- K. G. Wang, E. D. Stevens and S. P. Nolan, Organometallics, 1992, 11, 1011–1013 CrossRef CAS.
- D. Jin, X. Sun, A. Hinz and P. W. Roesky, Dalton Trans, 2022, 51, 5218–5226 RSC.
- Z. Hou, C. Yoda, T.-A. Koizumi, M. Nishiura, Y. Wakatsuki, S.-I. Fukuzawa and J. Takats, Organometallics, 2003, 22, 3586–3592 CrossRef CAS.
- W. J. Evans, R. A. Keyer, G. W. Rabe, D. K. Drummond and J. W. Ziller, Organometallics, 1993, 12, 4664–4667 CrossRef CAS.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2012, 32, 1150–1158 CrossRef.
- A. A. Trifonov, Russ. Chem. Bull., 2003, 52, 601–606 CrossRef CAS.
- M. D. Walter, D. J. Berg and R. A. Andersen, Organometallics, 2007, 26, 2296–2307 CrossRef CAS.
- A. A. Trifonov, B. G. Shestakov, K. A. Lyssenko, J. Larionova, G. K. Fukin and A. V. Cherkasov, Organometallics, 2011, 30, 4882–4889 CrossRef CAS.
- A. A. Trifonov, I. A. Borovkov, E. A. Fedorova, G. K. Fukin, J. Larionova, N. O. Druzhkov and V. K. Cherkasov, Chem. – Eur. J., 2007, 13, 4981–4987 CrossRef CAS PubMed.
- J. A. Moore, A. H. Cowley and J. C. Gordon, Organometallics, 2006, 25, 5207–5209 CrossRef CAS.
- K. Vasudevan and A. H. Cowley, Chem. Commun., 2007, 3464–3466 RSC.
- G. B. Deacon, E. E. Delbridge, G. D. Fallon, C. Jones, D. E. Hibbs, M. B. Hursthouse, B. W. Skelton and A. H. White, Organometallics, 2000, 19, 1713–1721 CrossRef CAS.
- C. Schoo, S. V. Klementyeva, M. T. Gamer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2016, 52, 6654–6657 RSC.
- K. Takaki, T. Kusudo, S. Uebori, Y. Makioka, Y. Taniguchi and Y. Fujiwara, Tetrahedron Lett, 1995, 36, 1505–1508 CrossRef CAS.
- K. Takaki, T. Kusudo, S. Uebori, T. Nishiyama, T. Kamata, M. Yokoyama, K. Takehira, Y. Makioka and Y. Fujiwara, J. Org. Chem., 1998, 63, 4299–4304 CrossRef CAS.
- K. Takaki, M. Maruo, T. Kamata, Y. Makioka and Y. Fujiwara, J. Org. Chem., 1996, 61, 8332–8334 CrossRef CAS PubMed.
- W. J. Evans, T. P. Hanusa and K. R. Levan, Inorg. Chim. Acta, 1985, 110, 191–195 CrossRef CAS.
- D. J. Berg, R. A. Andersen and A. Zalkin, Organometallics, 1988, 7, 1858–1863 CrossRef CAS.
- G. B. Deacon, C. C. Quitmann, K. Müller-Buschbaum and G. Meyer, Z. Anorg. Allg. Chem., 2001, 627, 1431–1432 CrossRef CAS.
- N. A. Pushkarevsky, M. A. Ogienko, A. I. Smolentsev, I. N. Novozhilov, A. Witt, M. M. Khusniyarov, V. K. Cherkasov and S. N. Konchenko, Dalton Trans, 2016, 45, 1269–1278 RSC.
- Z. M. Hou, A. Fujita, Y. G. Zhang, T. Miyano, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1998, 120, 754–766 CrossRef CAS.
- W. J. Evans, D. K. Drummond, L. A. Hughes, H. M. Zhang and J. L. Atwood, Polyhedron, 1988, 7, 1693–1703 CrossRef CAS.
- W. J. Evans, J. T. Leman, J. W. Ziller and S. I. Khan, Inorg. Chem., 1996, 35, 4283–4291 CrossRef CAS PubMed.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 9880–9882 CrossRef CAS.
- F. Calderazzo, A. Morvillo, G. Pelizzi and R. Poli, J. Chem. Soc., Chem. Commun., 1983, 507–508 RSC.
- E. V. Dikarev and B. Li, Inorg. Chem., 2004, 43, 3461–3466 CrossRef CAS PubMed.
- K. Y. Monakhov, T. Zessin and G. Linti, Eur. J. Inorg. Chem., 2010, 322–332 CrossRef CAS.
- R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2016, 55, 14798–14801 CrossRef CAS PubMed.
- M. G. Zhao, T. T. Hao, X. Zhang, J. P. Ma, J. H. Su and W. Zheng, Inorg. Chem., 2017, 56, 12678–12681 CrossRef CAS PubMed.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J Chem Soc Chem Comm, 1992, 16, 1138 RSC.
- N. A. Pushkarevsky, I. Y. Ilyin, P. A. Petrov, D. G. Samsonenko, M. R. Ryzhikov, P. W. Roesky and S. N. Konchenko, Organometallics, 2017, 36, 1287–1295 CrossRef CAS.
- F. Nief and L. Ricard, Organometallics, 2001, 20, 3884–3890 CrossRef CAS.
- A. Recknagel, D. Stalke, H. W. Roesky and F. T. Edelmann, Angew. Chem., Int. Ed. Engl., 1989, 28, 445–446 CrossRef.
- A. Recknagel, M. Noltemeyer, D. Stalke, U. Pieper, H.-G. Schmidt and F. T. Edelmann, J. Organomet. Chem., 1991, 411, 347–356 CrossRef CAS.
- W. J. Evans, G. W. Rabe, J. W. Ziller and R. J. Doedens, Inorg. Chem., 1994, 33, 2719–2726 CrossRef CAS.
- D. J. Berg, C. J. Burns, R. A. Andersen and A. Zalkin, Organometallics, 1989, 8, 1865–1870 CrossRef CAS.
- S. V. Klementyeva, N. P. Gritsan, M. M. Khusniyarov, A. Witt, A. A. Dmitriev, E. A. Suturina, N. D. Hill, T. L. Roemmele, M. T. Gamer, R. T. Boeré, P. W. Roesky, A. V. Zibarev and S. N. Konchenko, Chem. – Eur. J., 2017, 23, 1278–1290 CrossRef CAS PubMed.
- W. J. Evans, N. T. Allen, M. A. Greci and J. W. Ziller, Organometallics, 2001, 20, 2936–2937 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and S. Prashar, J. Organomet. Chem., 1991, 413, 79–90 CrossRef CAS.
- W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef CAS.
- W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770–776 CrossRef CAS.
- S. V. Klementyeva, M. T. Gamer, A. C. Schmidt, K. Meyer, S. N. Konchenko and P. W. Roesky, Chem. – Eur. J., 2014, 20, 13497–13500 CrossRef CAS PubMed.
- S. V. Klementyeva, N. Arleth, K. Meyer, S. N. Konchenko and P. W. Roesky, New J. Chem, 2015, 39, 7589–7594 RSC.
- N. Arleth, S. Bestgen, M. T. Gamer and P. W. Roesky, J. Am. Chem. Soc., 2014, 136, 14023–14026 CrossRef CAS PubMed.
- M. Rieckhoff, M. Noltemeyer, F. T. Edelmann, I. Haiduc and I. Silaghi-Dumitrescu, J. Organomet. Chem., 1994, 469, C19–C21 CrossRef.
- W. J. Evans, M. A. Ansari, J. W. Ziller and S. I. Khan, Organometallics, 1995, 14, 3–4 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877–6879 CrossRef CAS.
- S. Xu, L. A. Essex, J. Q. Nguyen, P. Farias, J. W. Ziller, W. H. Harman and W. J. Evans, Dalton Trans, 2021, 50, 15000–15002 RSC.
- C. Schoo, S. Bestgen, A. Egeberg, S. Klementyeva, C. Feldmann, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 5912–5916 CrossRef CAS PubMed.
- C. Schoo, S. Bestgen, A. Egeberg, J. Seibert, S. N. Konchenko, C. Feldmann and P. W. Roesky, Angew. Chem., Int. Ed., 2019, 58, 4386–4389 CrossRef CAS PubMed.
- S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnockel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740–5741 CrossRef CAS PubMed.
- D. Fenske, H. Fleischer and C. Persau, Angew. Chem., Int. Ed. Engl., 1989, 28, 1665–1667 CrossRef.
- H. A. Spinney, N. A. Piro and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 16233–16243 CrossRef CAS PubMed.
- N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa and Y. S. Ohashi, J. Am. Chem. Soc., 1998, 120, 433–434 CrossRef CAS.
- T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161–5170 CrossRef CAS PubMed.
- A. Y. Konokhova, M. Y. Afonin, T. S. Sukhikh and S. N. Konchenko, J. Struct. Chem., 2020, 61, 1244–1252 CrossRef CAS.
- A. G. Demkin, B. Y. Savkov, T. S. Sukhikh and S. N. Konchenko, J. Struct. Chem., 2021, 62, 957–965 CrossRef CAS.
- W. J. Evans, G. W. Rabe, M. A. Ansari and J. W. Ziller, Angew. Chem., Int. Ed. Engl., 1994, 33, 2110–2111 CrossRef.
- A. Zalkin and D. J. Berg, Acta Crystallogr C, 1988, 44, 1488–1489 CrossRef.
- T. D. Tilley and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1981, 19, 985–986 RSC.
- T. D. Tilley and R. A. Andersen, J. Am. Chem. Soc., 1982, 104, 1772–1774 CrossRef CAS.
- J. M. Boncella and R. A. Andersen, Inorg. Chem., 1984, 23, 432–437 CrossRef CAS.
- W. J. Evans, I. Bloom, J. W. Grate, L. A. Hughes, W. E. Hunter and J. L. Atwood, Inorg. Chem., 1985, 24, 4620–4623 CrossRef CAS.
- R. Yadav, T. Simler, M. T. Gamer, R. Köppe and P. W. Roesky, J. Chem. Soc., Chem. Commun., 2019, 55, 5765–5768 RSC.
- M. R. Churchill, K. N. Amoh and H. J. Wasserman, Inorg. Chem., 1981, 20, 1609–1611 CrossRef CAS.
- A. Recknagel, A. Steiner, S. Brooker, D. Stalke and F. T. Edelmann, Chem. Ber, 1991, 124, 1373–1375 CrossRef CAS.
- S. N. Konchenko, T. Sanden, N. A. Pushkarevsky, R. Köppe and P. W. Roesky, Chem. – Eur. J., 2010, 16, 14278–14280 CrossRef CAS PubMed.
- W. J. Evans, L. R. Chamberlain and J. W. Ziller, J. Am. Chem. Soc., 1987, 109, 7209–7211 CrossRef CAS.
- W. J. Evans, J. T. Leman, R. D. Clark and J. W. Ziller, Main Group Met. Chem., 2000, 23, 163–168 CAS.
- W. J. Evans, L. R. Chamberlain, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6423–6432 CrossRef CAS.
- M. Glanz, S. Dechert, H. Schumann, D. Wolff, D. Wolff and J. Springer, Z. Anorg. Allg. Chem., 2000, 626, 2467–2477 CrossRef CAS.
- M. D. Walter, P. T. Matsunaga, C. J. Burns, L. Maron and R. A. Andersen, Organometallics, 2017, 36, 4564–4578 CrossRef CAS.
- G. B. Deacon and D. L. Wilkinson, Inorg. Chim. Acta, 1988, 142, 155–159 CrossRef CAS.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, Organometallics, 1985, 4, 112–119 CrossRef CAS.
- I. Castillo and T. D. Tilley, J. Am. Chem. Soc., 2001, 123, 10526–10534 CrossRef CAS PubMed.
- M. D. Walter, C. J. Burns, P. T. Matsunaga, M. E. Smith and R. A. Andersen, Organometallics, 2016, 35, 3488–3497 CrossRef CAS.
- T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183–2185 RSC.
- T. Li, N. Arleth, M. T. Gamer, R. Köppe, T. Augenstein, F. Dielmann, M. Scheer, S. N. Konchenko and P. W. Roesky, Inorg. Chem., 2013, 52, 14231–14236 CrossRef CAS PubMed.
- N. Arleth, M. T. Gamer, R. Köppe, N. A. Pushkarevsky, S. N. Konchenko, M. Fleischmann, M. Bodensteiner, M. Scheer and P. W. Roesky, Chem. Sci, 2015, 6, 7179–7184 RSC.
- N. Arleth, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Fleischmann, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2016, 55, 1557–1560 CrossRef CAS PubMed.
- T. Li, N. Arleth, M. T. Gamer, R. Köppe, T. Augenstein, F. Dielmann, M. Scheer, S. N. Konchenko and P. W. Roesky, Inorg. Chem., 2013, 52, 14231–14236 CrossRef CAS PubMed.
- C. Schoo, R. Köppe, M. Piesch, M. T. Gamer, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem. – Eur. J., 2018, 24, 7890–7895 CrossRef CAS PubMed.
- L. R. Maxwell, S. B. Hendricks and V. M. Mosley, J. Chem. Phys., 1935, 3, 699–709 CrossRef CAS.
- N. Reinfandt, C. Schoo, L. Dütsch, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem. – Eur. J., 2021, 27, 3974–3978 CrossRef CAS PubMed.
- L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem. – Eur. J., 2018, 24, 3241–3250 CrossRef CAS PubMed.
- P. J. Sullivan and A. L. Rheingold, Organometallics, 1982, 1, 1547–1549 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |