
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Niobium carbide nanofibers as a versatile precursor for high power supercapacitor and high energy battery electrodes†
Aura
Tolosa
ab,
Benjamin
Krüner
ab,
Simon
Fleischmann
b,
Nicolas
Jäckel
ab,
Marco
Zeiger
ab,
Mesut
Aslan
a,
Ingrid
Grobelsek
a and
Volker
Presser
*ab
aINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, 66123 Saarbrücken, Germany
First published on 15th September 2016
Abstract
This study presents electrospun niobium carbide/carbon (NbC/C) hybrid nanofibers, with an average diameter of 69 ± 30 nm, as a facile precursor to derive either highly nanoporous niobium carbide-derived carbon (NbC–CDC) fibers for supercapacitor applications or niobium pentoxide/carbon (Nb2O5/C) hybrid fibers for battery-like energy storage. In all cases, the electrodes consist of binder-free and free-standing nanofiber mats that can be used without further conductive additives. Chlorine gas treatment conformally transforms NbC nanofiber mats into NbC–CDC fibers with a specific surface area of 1508 m2 g−1. These nanofibers show a maximum specific energy of 19.5 W h kg−1 at low power and 7.6 W h kg−1 at a high specific power of 30 kW kg−1 in an organic electrolyte. CO2 treatment transforms NbC into T-Nb2O5/C hybrid nanofiber mats that provide a maximum capacity of 156 mA h g−1. The presence of graphitic carbon in the hybrid nanofibers enabled high power handling, maintaining 50% of the initial energy storage capacity at a high rate of 10 A g−1 (64 C-rate). When benchmarked for an asymmetric full-cell, a maximum specific energy of 86 W h kg−1 was obtained. The high specific power for both systems, NbC–CDC and T-Nb2O5/C, resulted from the excellent charge propagation in the continuous nanofiber network and the high graphitization of the carbon structure.
Introduction
Energy storage systems with high power and high energy ratings are in high demand for a variety of applications; ranging from mobile devices to power leveling for wind and solar energy installations.1 In particular, electrochemical energy storage (EES) has emerged as a highly promising and efficient technology.2 With EES, energy can be stored by several mechanisms, ranging from physical ion electrosorption (electrical double-layer capacitors, EDLCs, also known as supercapacitors) and fast surface redox processes to ion intercalation (batteries).3,4 The process of electrical double-layer formation is a non-faradaic interface mechanism, enabling high specific power and long cycling lifetime, but rather low specific energy.5 Ion intercalation batteries accomplish charge storage and recovery by faradaic charge transfer between the electrolyte ions and the electrode materials.6 By this way, high specific energy can be obtained, while the cycling lifetime and power handling are limited by mechanical/structural degradation and diffusion processes through bulk materials.6 The ultimate goal is to achieve a combination of high power and high energy ratings. This is sought to be achieved by the development of hybrid technologies, such as lithium ion capacitors,7 and employment of rapid pseudocapacitive materials8 or redox-active electrolytes.9,10A particularly attractive group of electrode materials are binder-free and free standing fiber mats.11,12 The open-mesh percolated network and the possibility of creating a continuous active material are highly attractive to achieve an improved power performance. Commonly, different precursor materials and electrode architectures are employed when exploring supercapacitor or battery-like systems. The challenge is to satisfy the very different requirements for these two applications: (1) nanoporous carbon with a highly tailorable and controllable pore size distribution and (2) a redox-active material enabling, for example, rapid and reversible Li-ion intercalation.
Nanoporous carbon fibers have been extensively explored for supercapacitor applications, especially those derived from high carbon yield polymers such as polyacrylonitrile (PAN) and polymer blends such as polyvinylpyrrolidone and methylacrylate as pore formers.13 To achieve a large surface area, physical or chemical activation of such PAN fibers can be employed.13,14 An alternative approach, which allows more control over the size and size distribution of nanopores, is the selective removal of metal atoms from metal carbide fibers.15 This can be done via thermal treatment with chlorine gas, yielding highly nanoporous carbon (carbide-derived carbon, CDC).16 So far, this approach has only been employed with electrospun fibers of TiC–CDC12,17 and SiC–CDC.18,19 More detailed work has been carried out on the synthesis of CDC powders, including systems such as NbC,20 TiC,21,22 or B4C,23 with surface areas between 1000 and 3000 m2 g−1 and pore volumes between 0.5 and 2 cm3 g−1.
Carbon hybrid fibers containing redox-active materials have been proposed for pseudocapacitors, redox-hybrids, and battery-like energy storage.24,25 Different approaches have been reported, including functionalization of carbon fibers with quinones,26 coating with pseudocapacitive oxides (mainly MnO2),27,28 and synthesis of composite fibers with RuO2 or NiO.29,30 The hybridization of carbon and redox-active materials combines two synergetic aspects: the high electrical conductivity of carbon with the high faradaic charge storage capacity of redox-active materials. The latter often lack a good electron propagation ability, which makes the presence of a conductive phase mandatory in order to achieve a high power performance. A very promising metal oxide that has not been explored so far for carbon-hybrid fibers is niobium pentoxide (Nb2O5). Niobium pentoxide presents a high specific energy and power capability related to fast lithium ion intercalation within (110) or (001) planes, depending on the crystal phase.8,31,32 There are several Nb2O5 crystal phases based on octahedrally coordinated niobium atoms and the most common phases are TT-Nb2O5 (pseudohexagonal), T-Nb2O5 (orthorhombic), M-Nb2O5 (tetragonal), and H-Nb2O5 (monoclinic).33 The monoclinic phase is the most thermodynamically stable (formed above 1000 °C), while the TT-phase is the least thermodynamically stable and is formed at lower temperatures (400–500 °C).33
First studies showed that Nb2O5 presents Li-ion intercalation at a potential below 2.0 V vs. Li/Li+,34 and it was initially explored as a cathode material in secondary lithium batteries using a Li–metal anode.35 Different polymorphs have later been investigated as electrode materials not just for Li-ion batteries,36 but also for carbon/Nb2O5 hybrid systems.8,37 It has been observed that the energy storage capacity of Nb2O5 for lithium-ion batteries highly depends on its crystalline structure; H-Nb2O5 shows the highest specific energy, delivering a second cycle discharge capacity of 242 mA h g−1, compared to 152 mA h g−1 for TT-Nb2O5.35,36 For battery-like systems, TT-, T- and M-Nb2O5 have been widely studied. T-Nb2O5 has shown superior power handling due to fast two dimensional Li+ transport within the crystal structure that causes no phase transition during the electrochemical reaction.8,37 So far, polymer-bound electrodes have been mainly produced by mixing Nb2O5 fibers,36 nanosheets,38 and nanoparticles8,31 with conductive additives. Thin electrodes without conductive additives have also been explored by direct drop casting of Nb2O5 nanoparticles on the current collector32 or from thin mesoporous films.39 However, this approach remains limited with regards to scalability.
Our work presents, for the first time, electrospun NbC/C hybrid nanofibers as a versatile precursor to derive either (1) NbC–CDC nanofibers for high power, capacitive energy storage or (2) Nb2O5/C hybrid nanofibers for high energy, battery-like energy storage. We will first establish the synthesis process and optimum processing parameters, and then provide comprehensive material characterization. Supercapacitor performance was benchmarked in an organic electrolyte (1 M TEA-BF4 in acetonitrile), and the battery-like system is investigated in 1 M LiClO4 in ethylene carbonate/dimethyl carbonate electrolyte. The choice of different electrolytes was motivated by enabling high ion mobility for the supercapacitor system, while a large voltage stability window and lithium intercalation were needed for the battery-like system. A key feature of our system is the ability to directly use free-standing, binder-free nanofiber mats; hence, the need for a binder (i.e., dead mass) is eliminated, the possible blocking of pores by the polymer binder is avoided, and an improved electrical conductivity of the electrode can be obtained, as shown in our recent study.18
Experimental description
Materials synthesis
Niobium carbide/carbon hybrid nanofibers were synthesized following a sol–gel synthesis, as described in our previous work.40 Niobium n-butoxide (NbBO) (99% metal basis) was purchased from Alfa Aesar, while acetic anhydride (Ac2O) (≥99% purity), anhydrous N,N-dimethylformamide (DMF) (99.8% purity), and polyvinylpyrrolidone (PVP) (Mw ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 g mol−1) were purchased from Sigma Aldrich. The spinning dope was prepared by mixing 9 mass% NbBO and 2 mass% Ac2O for 2 min. Then, 79 mass% of DMF (solvent) and 10 mass% of PVP (carrier polymer) were added. The spinning dope was stirred for 48 h prior to electrospinning, until a translucent solution was obtained. The fiber mats were produced by electrospinning in a MECC nanofiber system. The solution was pumped at 0.5 mL h−1 through a spinneret (inner diameter: 400 μm), inductively charged at 23 kV. The fibers were collected at a distance of 165 mm from the spinneret. The humidity and the temperature were maintained between 10 and 20% and 25 and 30 °C.
300000 g mol−1) were purchased from Sigma Aldrich. The spinning dope was prepared by mixing 9 mass% NbBO and 2 mass% Ac2O for 2 min. Then, 79 mass% of DMF (solvent) and 10 mass% of PVP (carrier polymer) were added. The spinning dope was stirred for 48 h prior to electrospinning, until a translucent solution was obtained. The fiber mats were produced by electrospinning in a MECC nanofiber system. The solution was pumped at 0.5 mL h−1 through a spinneret (inner diameter: 400 μm), inductively charged at 23 kV. The fibers were collected at a distance of 165 mm from the spinneret. The humidity and the temperature were maintained between 10 and 20% and 25 and 30 °C.
Post spinning, the as-spun fibers were exposed at room humidity for 72 h, to promote further hydrolysis and condensation reactions. Annealing was performed under 99.5% argon in a graphite heated furnace, Thermal Technology. The fibers were heated at 5 °C min−1 to 400 °C for 3 h, and then heated to 1500 or 1700 °C for 4 h for carbothermal reduction.
Materials characterization
The composition of the fibers was determined by energy dispersive X-ray spectroscopy (EDX) using an X-Max-150 detector from Oxford Instruments attached to the SEM chamber. The spectra of 10 fibers were measured using an accelerating voltage of 10 kV, in uncoated fibers attached to a sticky carbon tape. A silicon standard was used for standardization.
XRD diffractograms were recorded with a Bruker D8 Discover diffractometer using CuKα radiation (0.154 nm; without a monochromator) with a step size of 0.02° and a measurement time of 1–3 s per step. The system was calibrated with a corundum alumina standard. The samples were pestled and placed on a sapphire single crystal for the measurement. The full width at half maximum (FWHM) was measured by using EVA software from Bruker. The values of the average coherence length (roughly corresponding with the domain size) were obtained using the Scherrer equation.41
Raman spectra were recorded with an InVia Raman system, from Renishaw, using a laser with 532 nm excitation wavelength and 0.5 mW power on the sample with a spectral resolution of ca. 1.2 cm−1, using a 50× objective (numeric aperture: 0.9). Peak analysis was performed by baseline correction and assuming four Voigt peak fittings between 500 and 2000 cm−1. In situ Raman spectra were recorded during thermal treatment under synthetic air (heating rate 5 °C min−1) by using a temperature controlled Linkam T95-HT heating stage. A laser with 532 nm excitation wavelength was used, which exposed the sample to a power of 0.5 mW at the focal point.
Thermogravimetric analysis (TGA) was carried out in synthetic air or CO2 with a heating rate of 5 °C min−1 with a TG 209 F1 Libra system (Netzsch). Nitrogen gas sorption measurements were carried out at −196 °C with an Autosorb iQ system (Quantachrome), after outgassing at 300 °C for 10 h under vacuum conditions (about 102 Pa) to remove volatile surface functionalities present in the sample. The specific surface area (SSA) was calculated with the Quantachrome ASiQwin software using the Brunauer–Emmett–Teller (BET) equation42 in the linear relative pressure range of 0.05–0.2. Using the quenched-solid density functional theory (QSDFT),43 the SSA and pore size distribution (PSD) were calculated assuming slit-shaped pores and pore sizes between 0.56 and 37.5 nm. The values of the total pore volume correspond to p/p0 = 0.95. The values of the mean pore size are volume-weighted. Thermodynamic calculations were carried out with FactSage 7.0 (GTT-technologies).
For electrochemical characterization of electrical double-layer capacitor cells, the performance of NbC–CDC electrodes was characterized in half- and symmetric full-cell setups employing 1 M tetraethylammonium tetrafluoroborate (TEA-BF4) in electrochemical grade acetonitrile (ACN) from BASF as the electrolyte. Half-cell tests were performed with an oversized activated carbon (AC) counter electrode (10 mm diameter, 15 mg mass; YP-80F from Kuraray Chemicals with 5 mass% PTFE). Symmetric full-cell tests used two NbC–CDC fiber electrodes of the same mass. CVs were recorded at 10 mV s−1 scan rate in the potential range from −1.0 V to +1.0 V vs. carbon in half-cells and from 0 to +2.7 V cell voltage for full-cells. Galvanostatic cycling for half-cells was carried out from −1.0 V to +1.0 V vs. carbon at 2 A g−1 with a holding time of 10 min. The specific capacitance (Csp) was calculated by integrating the discharge current between t0, the discharge starting time, and t, the end time of discharge, divided by U (voltage vs. carbon or applied cell voltage, for half- and full-cells, respectively) viaeqn (1).45m corresponds to the mass of the CDC electrode for the half-cells, and for the full-cells it corresponds to the mass of both electrodes divided by four.
 | (1) |
Rate handling was quantified with the IR drop corrected voltage measured after 5 s resting time. Electrical impedance spectroscopy (EIS) was performed in the range of 100 kHz to 100 mHz at 0 V with 10 points per decade and averaged over 5 measurements. Performance stability tests were carried out in full-cells by galvanostatic cycling over 10000 cycles to 2.5 V at 1 A g−1.
For electrochemical characterization of battery-like cells, Nb2O5/C electrodes were characterized by using half- and asymmetric full-cell setups in 1 M lithium perchlorate (LiClO4) in electrochemical grade ethylene carbonate (EC)/dimethyl carbonate (DMC) from Sigma-Aldrich (volume ratio 1:1). Half-cell tests were performed with an oversized AC counter electrode (10 mm diameter, 15 mg mass; YP-80F from Kuraray Chemicals with 5 mass% PTFE) and asymmetric full-cell tests by combining a Nb2O5/C hybrid electrode with an AC electrode (YP-80F with 5 mass% PTFE). Charge balancing for asymmetric full-cells was achieved by mass balancing, according to the capacity values obtained by half-cell measurements at 0.1 A g−1. The lower gravimetric capacity of the AC electrode compared to the Nb2O5/C electrode was compensated by using electrodes with different masses. The mass of the AC electrode was between 1.5 and 2.5 times higher than the mass of the Nb2O5/C electrode. The cell voltage was chosen in order to achieve a maximum potential of −2.0 V vs. carbon at the negative electrode (Nb2O5/C). For Nb2O5/C fiber electrodes, CVs were recorded at 1 mV s−1 scan rate in the potential range from 0 to −2.0 V vs. carbon in half-cells and from 0 to +3.6 V cell voltage in full-cells. Rate handling for half- and asymmetric full-cells was carried out by GCPL, using the same potential ranges, from 0.1 to 40 A g−1 with a holding time of 10 min. Capacity (Qsp) was calculated by integrating the discharge current between t0, the discharge starting time, and t, the end time of discharge, and normalized to the electrode mass, m, viaeqn (2):
 | (2) |
Performance stability tests were carried out by galvanostatic cycling over 5000 cycles from 0 to −2.0 V vs. carbon and 3.0 V cell voltage at 1 A g−1 for half- and asymmetric full-cells, respectively.
Data from the electrodes with the best electrochemical performance for electrical double-layer capacitors or battery-like systems are presented in a Ragone plot. Using GCPL data of full-cells (asymmetric and symmetric), specific energy (Esp) and specific power (Psp) were calculated, using eqn (3) and (4), respectively, with U and I as the voltage and current applied to the full-cell, and M corresponds to the mass of both electrodes (positive and negative electrodes).
 | (3) |
 | (4) |
Results and discussion
Niobium carbide/carbon hybrid nanofibers
We briefly outline the important aspects of NbC/C hybrid nanofiber synthesis, which can be found in more detail in our previous work.40 Using NbBO as the sol–gel precursor and PVP as the carrier polymer, we have established a facile synthesis route for hybrid nanofibers with an average fiber diameter of 69 ± 30 nm. This approach employs electrospinning to obtain free-standing fiber mats, and thermal treatment yielding NbC/C hybrid nanofibers via carbothermal reduction. The fibers synthesized at 1700 °C (NbC-1700) presented a higher crystallinity when compared to the fibers synthesized at 1500 °C (NbC-1500), with an NbC average domain size (i.e., XRD coherence length) of 65 nm compared to 38 nm, respectively.40 Also, at a higher pyrolysis temperature, carbon presented a higher degree of graphitization (see ESI, Fig. S1†). NbC/C fibers were later evaluated as a versatile precursor to derive (1) NbC–CDC nanofibers and (2) Nb2O5/C hybrid nanofibers. To promote the carbide-to-carbon conversion, a lower degree of carbon ordering is preferred.22 The graphitic carbon engulfing the metal carbide crystals affects the reaction kinetics between Cl2 and the NbC; therefore, longer Cl2 treatment durations or higher synthesis temperatures will be needed for complete etching. To promote the carbide-to-oxide conversion without a complete loss of carbon, a higher degree of carbon ordering is desirable due to the higher stability in oxidizing atmospheres.22 Therefore, NbC-1500 was chosen as the precursor for NbC–CDC, and NbC-1700 was preferable as the precursor for Nb2O5/C hybrid nanofibers.Niobium carbide-derived carbon nanofibers
No influence on the morphology is observed as a function of the heat treatment parameters. The SEM micrographs of fibers treated for 3 h (Fig. 1) and 1 h (see ESI, Fig. S3†) show a mean fiber diameter of around 61 ± 22 nm (see ESI, Fig. S2B†). The conformal etching process during the CDC transformation is not expected to modify the macroscopic structure of the material, but rather the nanoporosity of the material (Fig. 1, inset).16 The TEM micrograph of the sample after 3 h holding time at 400 °C shows mesopores, which at higher temperatures become more organized, leading to a more densified fiber structure.
 | ||
| Fig. 1 SEM and TEM micrographs (inset) of NbC–CDC fibers synthesized by chlorine gas treatment for 3 h at (A) 400 °C, (B) 600 °C, and (C) 900 °C. | ||
A better understanding of the influence of the treatment conditions on the carbon structure is achieved by the analysis of the Raman spectra and XRD patterns (Fig. 2). The data of samples treated for 1 h are presented in the ESI (see Fig. S4†). To understand these data, we have to consider the presence of two different carbon species: (1) carbon formed during pyrolysis at 1500 °C and present in NbC/C hybrid nanofibers and (2) carbide-derived carbon formed between 400 and 900 °C via chlorine gas treatment of NbC nanocrystals. The latter co-exists in NbC–CDC nanofibers in addition to the carbon formed at 1500 °C, because the CDC synthesis process does not remove initially formed carbon.16 All Raman spectra (Fig. 2A) present two characteristic peaks for sp2-hybridized carbon, the D- and G-modes at around 1340 and 1600 cm−1 related to sp2-hybridized carbon atoms in rings and the presence of defects in this structure.46 Additionally, we see the corresponding combination and overtone modes between 2500 and 3500 cm−1. The position of the G-mode is shifted from 1603 to 1597 cm−1 when increasing the synthesis temperature from 400 °C to 900 °C, due to the successive transformation from nanocrystalline into larger graphitic structures.46 This graphitization correlates well with the decrease in the intensity of the amorphous carbon phase (1500–1550 cm−1) with increasing temperature.46,47
 | ||
| Fig. 2 Structural characterization of NbC-1500 fibers before and after chlorine treatment for 3 h. (A) Raman spectra, (B) XRD diffractograms and literature values of the diffraction peak positions of NbC (PDF: 38-1364) and graphite (PDF: 89-8487), (C) isotherms from nitrogen gas sorption at −196 °C (STP: standard temperature pressure), and (D) cumulative pore volume as a function of the pore width from QSDFT deconvolution. | ||
For the analysis of the different carbon phases, a four Voigt peak deconvolution was applied to the Raman data (see ESI, Fig. S5†). At 400 °C the ID/IG ratio (i.e., the intensity ratio of the D- and G-mode peaks) is similar to that of the precursor (see ESI, Table S2†). At this temperature graphitic carbon, preserved from NbC-1500, co-exists with new partially amorphous/nanocrystalline CDC. At 600 °C, the grain size of nanocrystalline CDC increases proportionally to the defect concentration in hexagonal carbon rings (higher ID/IG ratio).46 By treatment at 900 °C, the domains exceed a specific size (around 2 nm),46 leading to graphitic carbon with less defects (lower ID/IG ratio). The successive transformation from amorphous into graphitic carbon is further supported by the sharpening of the D-mode FWHM from 96.2 ± 0.8 cm−1 to 82.8 ± 3.6 cm−1 when increasing the synthesis temperature from 400 to 900 °C.
XRD data (Fig. 2B) corroborate the transformation of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m niobium carbide after chlorine gas treatment into amorphous carbon at 400 °C. While all crystalline niobium carbide peaks vanish, we observed a sharp (002)-graphite reflection (P63mc) at around 26.5° 2θ at 400 °C corresponding to the graphitic carbon present in the precursor. When increasing the temperature, two broad peaks are observed at around 26.5 and 44° 2θ related to the (002) and (110)-graphite reflections,48 related to the transformation of the formed amorphous or nanocrystalline phase into graphitic carbon.
m niobium carbide after chlorine gas treatment into amorphous carbon at 400 °C. While all crystalline niobium carbide peaks vanish, we observed a sharp (002)-graphite reflection (P63mc) at around 26.5° 2θ at 400 °C corresponding to the graphitic carbon present in the precursor. When increasing the temperature, two broad peaks are observed at around 26.5 and 44° 2θ related to the (002) and (110)-graphite reflections,48 related to the transformation of the formed amorphous or nanocrystalline phase into graphitic carbon.
The pore structure of the material was analyzed before and after Cl2 treatment by nitrogen gas sorption analysis (Fig. 2C and D). NbC-1500 presents a type I(b) isotherm,49 with mainly micropores with an average size of 1.2 nm. With non-porous NbC nanocrystals, this porosity is completely accomplished by microporous carbon in the hybrid nanofibers. After Cl2 treatment, the surface area is drastically increased from 120 m2 g−1 to maximum 1370 m2 g−1 (DFT SSA) and the pore volume from 0.06 cm3 g−1 to 1.0 cm3 g−1 (Table 1). The removal of Nb from the NbC crystal lattice at 400 °C for 1 h led to the formation of micro- and meso-pores (below 3 nm; see ESI, Fig. S4D†), with a mean pore size of 1.8 nm. At 600 °C for 1 h, more micropores are formed (below 2 nm), related to a higher amount of Nb etching compared to that at 400 °C, and a mean pore size of 1.1 nm was obtained. At a higher temperature (900 °C for 1 h) the redistribution of carbon slightly increases the mean pore size (1.2 nm). The isotherms after treatment for 1 h correspond to a type I(b) isotherm, with mainly micropores (see ESI, Fig. S4C†).49 When increasing the time to 3 h, all the isotherms become type II because of pore growth, while maintaining a type I(b) shape at low pressures. The change to the type II isotherm comes with an H4 hysteresis loop related to capillary condensation49 and a broader distribution of mesopores. The increase in the pore size when increasing the synthesis time leads to a decrease in the surface area and an increase in the pore volume (Table 1).
| Sample | DFT SSA (m2 g−1) | BET SSA (m2 g−1) | Pore volume (cm3 g−1) | Mean pore size (nm) |
|---|---|---|---|---|
| NbC-1500 | 122 | 121 | 0.06 | 1.2 |
| CDC-400-1 | 1065 | 1141 | 0.78 | 1.8 |
| CDC-400-3 | 744 | 750 | 1.00 | 3.5 |
| CDC-600-1 | 1369 | 1508 | 0.80 | 1.1 |
| CDC-600-3 | 1207 | 1326 | 0.96 | 2.4 |
| CDC-900-1 | 1092 | 1123 | 0.63 | 1.2 |
| CDC-900-3 | 980 | 1122 | 0.87 | 2.6 |
 | ||
| Fig. 3 Electrochemical analysis of CDC nanofiber mats in 1 M TEA-BF4 in acetonitrile. Half-cell measurements with (A) cyclic voltammetry at 10 mV s−1 and (B) GCPL profiles at 2 A g−1. Full-cell measurements using (C) cyclic voltammetry at 10 mV s−1, (D) GCPL power handling between 0.02 and 40 A g−1 (d), (E) electrochemical impedance spectra, and (F) galvanostatic cycling stability up to 2.5 V cell voltage at 1 A g−1. | ||
Full-cell performance was evaluated by cyclic voltammetry and galvanostatic cycling. The CVs (Fig. 3C) present a typical rectangular shape, with a typical increase in the capacitance when increasing the applied voltage. At 2.7 V, maximum values are achieved between 90 and 125 F g−1. Rate handling for full-cells was characterized by galvanostatic cycling to 2.5 V cell voltage. At a specific current of 0.02 A g−1, capacitance values between 64 and 83 F g−1 were obtained for the samples with the lowest SSA (CDC-400-3 and CDC-900-3) (Table 2). The highest capacitance was achieved for the samples synthesized at 600 °C (101–108 F g−1 for 1–3 h of chlorine gas treatment; see Fig. 3D and Table 2). When increasing the specific current to 0.1 A g−1, most of the samples present a decrease in capacitance between 10 and 20%. CDC-900-3, which presents a larger mean pore size (2.6 nm), maintained at 0.1 A g−1 97% of the capacitance at 0.02 A g−1. At a very high specific current of 40 A g−1, all samples retained still between 44 and 56% of the initial capacitance. Compared to other CDC materials, the electrochemical performance and energy storage capacity of NbC–CDC-600 in an organic electrolyte are good, as seen from Table 3. Compared to commercial electrodes (AC + 5% PTFE), NbC–CDC-600 presents similar capacitance values (104 F g−1), but a superior capacitance retention above 10 A g−1 related to the excellent charge propagation in the continuous fiber network.
| Sample | ESR (Ω cm2) | EDR (Ω cm2) | CPE exponent | C (F g−1) at 0.02 A g−1 | C loss (%) at 40 A g−1 |
C/C0 (%) after 10000 cycles |
|---|---|---|---|---|---|---|
| CDC-400-1 | 0.89 | 0.16 | 0.95 | 97 | 68 | 85 |
| CDC-400-3 | 0.73 | 0.28 | 0.89 | 64 | 60 | 76 |
| CDC-600-1 | 0.78 | 0.18 | 0.93 | 101 | 80 | 91 |
| CDC-600-3 | 0.53 | 0.45 | 0.94 | 108 | 44 | 85 |
| CDC-900-1 | 0.47 | 0.21 | 0.96 | 85 | 56 | 92 |
| CDC-900-3 | 0.43 | 0.49 | 0.97 | 83 | 50 | 90 |
| Sample | BET-SSA (m2 g−1) | Pore volume (cm3 g−1) | C (F g−1) | Capacitance retention | Reference |
|---|---|---|---|---|---|
| NbC–CDC fibers | 1508 | 0.80 | 101 | 20% at 40 A g−1 | This work, 1 M TEA-BF4 in ACN |
| NbC–CDC fibers | 1326 | 0.96 | 108 | 56% at 40 A g−1 | This work, 1 M TEA-BF4 in ACN |
| SiC–CDC particles | 2430 | 1.50 | 125 | 85% at 20 A g−1 | Ref. 19 1 M TEA-BF4 in ACN |
| SiC–CDC particles | 2250 | 1.40 | 160 | 81% at 20 A g−1 | Ref. 19, 1 M TEA-BF4 in ACN |
| SiOC–CDC fibers | 3089 | 1.78 | 135 | 63% at 50 A g−1 | Ref. 18, 1 M TEA-BF4 in ACN |
| SiOC–CDC particles | 2480 | 1.29 | 110 | 83% at 30 A g−1 | Ref. 61, 1 M TEA-BF4 in ACN |
| TiC–CDC fibers | 1390 | 1.50 | 102 | 50% at 5 V s−1 | Ref. 12, 1.5 M TEA-BF4 in ACN |
| AC + 5% PTFE | 1481 | 0.8 | 104 | 68% at 10 A g−1 | Ref. 62, 1.0 M TEA-BF4 in ACN |
To further characterize the electrochemical behavior, impedance spectroscopy (EIS) was carried out for full-cells (Fig. 3E). As seen from the inset in Fig. 3E, the evolving semi-circle correlates with a high contact resistance between the fibers and the current collector. Compared to free-standing polymer-bound carbon electrodes, fiber electrodes are less smooth at the interface with the current collector, which is why an additional contact impedance arises.18,53 At very low frequencies, the constant phase element (CPE) is close to 1 with values from 0.89 to 0.97 indicative of a near-ideal capacitive behavior (Table 2).53 Furthermore a direct correlation between the degree of carbon ordering, the pore structure, and the rate handling performance (Table 2) is observed. The samples with the highest degree of carbon ordering lead to the lowest equivalent serial resistance (ESR), namely the samples Cl2 treated at higher temperatures and times, CDC-600-3, CDC-900-1 and CDC-900-3 (43–53 Ω cm2).
The stability of the system was benchmarked by galvanostatic cycling (Table 2). Most of the samples retain more than 80% of the initial capacitance after 10000 charge/discharge cycles. The only exception is CDC-400-3 with a capacitance loss of 24%. The highest capacitance retention was found for CDC-600-1 (91%) and CDC-900-1 or CDC-900-3 (90–92%). This enhanced stability may be related to the lower content of oxygen containing functional groups (Table S1†).
Niobium pentoxide–carbon hybrid nanofibers
To identify the most suitable processing parameters, we employed thermogravimetric analysis. In CO2 at 5 °C min−1, we see an onset of mass increase at 720 °C, reaching a maximum at 950 °C (+12%). Further temperature increase resulted in a progressing mass loss associated with the burn-off of carbon. In this range of temperatures, T-Nb2O5 formation is expected36,55 as the only crystalline phase in co-existence with carbon. Therefore, 850 °C was selected as the preferred temperature for treatment under CO2 to produce the hybrid material.
Using synthetic air, the volatilization of carbon occurs at lower temperatures, at around 500 °C (see ESI, Fig. S7A†), and at similar temperatures to that of the carbide oxidation, at around 450 °C. At 410 °C, the mass of the system increases related to oxidation of the NbC, reaching a maximum at 460 °C (+20%). At higher temperatures, carbon burning takes place (TGA derivate peak, 590 °C) leading to mass decrease. In alignment with these data and the literature, the only expected crystalline phase in this temperature range is TT-Nb2O5.36,55 For a better identification of an optimum temperature for the treatment in air, we carried out in situ Raman measurements (see ESI, Fig. S7B†). In situ Raman spectra show the emergence of niobia-related signals (646 to 670 cm−1)56 for temperatures above 600 °C and the decrease in carbon signal intensities for the D- and G-mode peaks at 1323 and 1575 cm−1, respectively. To avoid the enhanced loss of carbon, lower synthesis temperatures in air were chosen, namely 450 °C and 500 °C.
After treatment under CO2 (850 °C) or air (450 and 500 °C), the sample mass increased between 7 and 38% (Table 4). Yet, the nanofiber morphology was fully maintained, as seen from the SEM micrographs in Fig. 4. Image analysis documents an increase in the average fiber diameter by 55% (87 ± 25 nm; see ESI, Fig. S8†), related to the difference in density between NbC and Nb2O5 (7.8 and 4.6 g cm−3, respectively).57 In the TEM micrographs (Fig. 4A, inset), only highly amorphous material was identified for the samples synthesized in air at 450 °C. The corresponding SAED pattern confirms the highly disordered nature of the material at 450 °C. Nanocrystalline TT-Nb2O5 domains emerge when using a higher synthesis temperature (500 °C) while the majority of the diffraction signal is still indicative of a highly amorphous material. Higher temperatures are required to obtain a well-developed TT-Nb2O5 crystal structure. However, at 500 °C, already a strong oxidation of carbon takes place as observed from the change in the carbon content according to the EDX data (Table 4). Therefore, a less oxidative environment was chosen in alignment with theoretical calculation, namely thermal treatment in CO2 (see ESI, Fig. S7A†). At 850 °C, TEM micrographs show Nb2O5 nanocrystals engulfed in graphitic carbon (Fig. 4C). The corresponding SAED pattern is characteristic of a highly crystalline material, with diffraction patterns falling into almost continuous rings related to T-Nb2O5.
| Sample | Mass change | C (mass%) | O (mass%) | Nb (mass%) |
|---|---|---|---|---|
| NbC-1700 | — | 27.6 ± 0.5 | 2.0 ± 0.1 | 70.3 ± 1.1 |
| Nb2O5-450 | +16% | 11.8 ± 0.7 | 27.8 ± 1.3 | 60.4 ± 1.2 |
| Nb2O5-500 | +7% | 8.9 ± 0.6 | 28.8 ± 1.9 | 62.4 ± 1.9 |
| Nb2O5-850 | +38% | 18.6 ± 0.8 | 25.0 ± 1.2 | 56.4 ± 1.0 |
 | ||
| Fig. 4 SEM micrographs, TEM micrographs (inset, lower left corner), and selected area electron diffraction pattern (inset, upper right corner) of Nb2O5/C hybrid nanofibers synthetized at (A) 450 °C in air, (B) 500 °C in air, and (C) 850 °C in CO2. | ||
For a more detailed characterization of the crystal structure of the synthesized materials, XRD patterns and Raman spectra were recorded (Fig. 5). All Raman spectra indicate the presence of partially graphitic carbon in all samples. Yet, the carbon signal was very high for NbC-1700 and almost identical to that of Nb2O5-850. The apparent absence of clear signals from crystalline niobia in Nb2O5-850 is related to the very high sensitivity of Raman spectroscopy to detect sp2-hybridized carbon species.58 In contrast, the TT-Nb2O5 signal and the fluorescence-related background were stronger after oxidation in air. Yet, there are differences also related to the carbon signal as seen after peak deconvolution (see ESI, Table S3†). For Nb2O5-450, we found broader D- and G-peaks compared to the precursor and an overall increase in the amount of amorphous carbon (see ESI, Fig. S9†) as a result of on-setting carbon oxidation. The high fluorescence background for Nb2O5-500 made peak deconvolution unreliable, but there was still a redshift of the G-mode peak from 1599 to 1584 cm−1 due to the co-existing amorphous phase. According to the mass change during the process and EDX results (Table 4), the carbon content drastically decreased for this sample.
 | ||
| Fig. 5 (A) Raman spectra of Nb2O5/C hybrid nanofibers and the precursor (NbC-1700). (B) XRD diffractograms of Nb2O5/C hybrid nanofibers and literature values of diffraction peak positions of T-Nb2O5 (PDF: 27-1003) and TT-Nb2O5 (PDF: 07-0061). | ||
According to the XRD pattern (Fig. 5B), NbC is transformed after treatment in an oxidative atmosphere into a highly amorphous material for Nb2O5-450 and into nanocrystalline niobium pentoxide for Nb2O5-500 and Nb2O5-850. By applying the Scherrer equation,41 in air at 500 °C, TT-Nb2O5 is formed with an average domain size of ca. 20 nm and lattice parameters, a = 0.36 nm, b = 0.31 nm, and c = 0.39 nm. By treatment in CO2 at 850 °C, orthorhombic T-Nb2O5 is formed with an average domain size of ca. 35 nm and lattice parameters, a = 0.62 nm, b = 2.93 nm, and c = 0.39 nm.
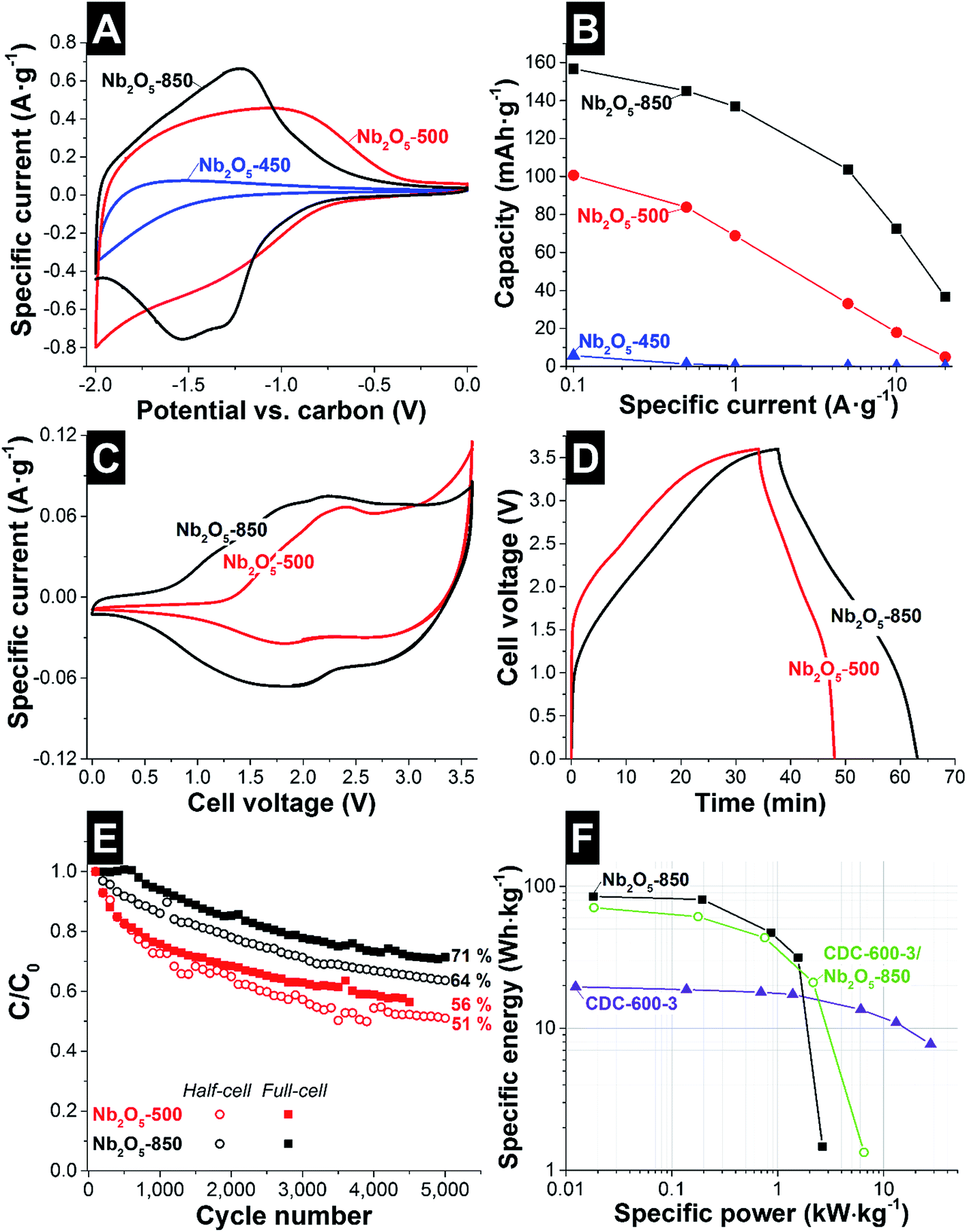 | ||
| Fig. 6 Electrochemical characterization in the half-cell configuration of Nb2O5/C hybrid nanofibers in 1 M LiClO4 in EC/DMC using (A) cyclic voltammetry at 1 mV s−1 and (B) galvanostatic charge/discharge rate handling to −2 V vs. carbon. Electrochemical characterization in the full-cell configuration using (C) cyclic voltammetry up to 3.6 V cell voltage at 1 mV s−1 and (D) GCPL charge/discharge profile at 0.1 A g−1. (E) Capacity retention during galvanostatic charge/discharge cycling to 3.0 V cell voltage for full-cells or from 0 to −2 V vs. carbon for the half-cell configuration. (F) Ragone plot of full-cells. | ||
The observed peaks are related to the electron transfer resulting from lithium intercalation according to the reaction in eqn (5), for redox couples Nb5+/4+ and Nb4+/3+.36
| Nb2O5 + xLi+ + xe− ↔ LixNb2O5 (x = 1, 2) | (5) |
By GCPL to −2.0 V vs. carbon, the rate handling was evaluated for half-cells (Fig. 6B). The specific capacities of the third discharge cycle at 0.1 A g−1 for Nb2O5-450, Nb2O5-500 and Nb2O5-850 (1 C-rate), were 6, 101, and 156 mA h g−1, respectively. When normalizing the results to the mass of niobium pentoxide, as commonly done for battery systems, we obtained capacities of 111 mA h g−1 and 191 mA h g−1 for Nb2O5-500 and Nb2O5-850, respectively. At 0.1 A g−1 the coulombic efficiency of Nb2O5-500 and Nb2O5-850 corresponds to 91 and 92%, respectively. As presented in Table 5, these values are comparable to those reported in the literature for TT-Nb2O5 (150 mA h g−1) and T-Nb2O5 (between 142 and 190 mA h g−1). At 20 A g−1, 5% and 23% of the capacity were retained for Nb2O5-500 and Nb2O5-850, respectively. The higher rate handling performance of Nb2O5-850 is related not just to the graphitic carbon engulfing the T-Nb2O5 crystals, but also to the short diffusion length in the nanofibers and the faster ionic transport characteristic of T-Nb2O5. The latter was explained by Augustyn et al. by the almost empty octahedral sites between (001) planes presenting low energy barriers for Li+-ion transport throughout the a–b plane.8
| Sample | Electrode | Capacity (mA h g−1) | Capacity retention | Reference |
|---|---|---|---|---|
| Nb2O5/C electrospun fibers | Free standing | TT-Nb2O5: 101 (am: 111) | TT-Nb2O5: 18% at 10 A g−1 (100 C-rate) | This work, 1 M LiClO4 in EC/DMC |
| T-Nb2O5: 156 (am: 191) | T-Nb2O5: 46% at 10 A g−1 (64 C-rate); 23% at 20 A g−1 (128 C-rate) | |||
| Nb2O5 electrospun fibers | Mixture with CA and PTFE | TT-Nb2O5: 99 (am: 152) | — | Ref. 36 |
| T-Nb2O5: 123 (am: 189) | ||||
| Nb2O5 nano-particles | Drop cast mixture with CA | T-Nb2O5: 71 (am: 142) | T-Nb2O5: 31% at 1000 C-rate | Ref. 8, 1 M LiClO4 in PC |
| Nb2O5 nanosheets | Mixture with CA and PVdF | T-Nb2O5: 117 (am: 147) | T-Nb2O5: 77% at 1 A g−1 (5 C-rate) | Ref. 38, 1 M LiPF6 in EC/DMC/DEC |
| Nb2O5 mesoporous films | Free-standing thin film | T-Nb2O5: 190 | — | Ref. 39, 1 M LiClO4 in PC |
In order to benchmark Nb2O5/C hybrid nanofibers as a cathode (electrode at negative potentials) in a device, asymmetric full-cell tests were performed (Fig. 6C and D). As an anode (electrode at positive potentials), a charge balanced AC electrode was used. This set-up has demonstrated to improve the performance of the full-cell, by limiting the polarization resistance.31,59 For the CVs at 1 mV s−1, shown in Fig. 6C, a maximum applied cell voltage of 3.6 V was used, which was expected to lead to an appropriate operating potential range for positive and negative electrodes. For asymmetric full-cells, the different charge-voltage-behavior of the two electrodes led to asymmetric voltage development at the anode and cathode. Therefore, the voltage in each electrode was recorded by using an activated carbon spectator reference electrode (see ESI, Fig. S10B and C†). According to the CV (Fig. 6C) of Nb2O5-500, a cathodic peak can be observed at 2.5 V cell voltage and broad anodic peaks are detected at 1.8 and 2.8 V cell voltages. A strong increase in current close to 3.6 V cell voltage is related to fast voltage development at the positive electrode, above the potential limit for the stability of the electrolyte, and therefore the system is not stable. The voltage at the negative electrode reaches −2.2 V vs. carbon, while for the positive electrode a voltage maximum of 1.4 V vs. carbon was obtained (see ESI, Fig. S10B†). For Nb2O5-850, at low voltage, a capacitive behavior was observed, related to the higher mass of AC present as the positive electrode, compared to Nb2O5-500. When increasing the cell voltage, Li+ intercalation/deintercalation takes place in the Nb2O5 electrode, as indicated by broad anodic peaks between 2.2 and 1.8 V cell voltages, and cathodic peaks at 1.9 and 2.6 V cell voltages. At high cell voltage, the system is more stable than Nb2O5-500, and no strong increase in current is observed (see ESI, Fig. S10B†).
The performance of full-cells was further characterized by GCPL to 3.6 V at 0.1 A g−1 (Fig. 6D). No significant charge storage in the cell is observed below 1.2 V and 1.6 V for Nb2O5-850 and Nb2O5-500, respectively, indicating no significant faradic charge transfer to the Nb2O5 electrode. Above this potential, a voltage plateau is observed for both samples, indicating a transfer of charge to the electrode when Li+ ion intercalation takes place. From the voltage plateau during discharging and the difference in potential development for each electrode (see ESI, Fig. S10B†), it can be concluded that both samples present a high difference in charge transfer efficiency. The coulombic efficiency of Nb2O5-500 and Nb2O5-850 corresponds to 40 and 80% respectively, for asymmetric full-cells.
Cycling stability tests were performed in half- and full-cell configurations (Fig. 6E). After 5000 cycles, Nb2O5-850 retained 64% and 71% of the capacity, in half- and full-cell setups, respectively. For Nb2O5-850, the lower stability of half-cells is related to the higher voltage (−2.0 V for half-cells; 3.0 V for full-cells) and the high stability of the positive material, AC. For Nb2O5-500, 51% of the capacity was retained for the half-cell, while for the full-cell, 56% was maintained. The lower stability is related to the low order of crystallinity of the TT-Nb2O5.
The materials with the best electrochemical performance for electrical double-layer capacitors and battery-like systems are plotted in the Ragone plot (Fig. 6F). CDC-600-3 presents high power handling, maintaining a specific energy of 7.6 W h kg−1 at 30 kW kg−1 and a maximum specific energy of 19.5 W h kg−1 at 0.01 kW kg−1. For Nb2O5-850 asymmetric full-cells, a specific energy of 86 W h kg−1 at 0.02 kW kg−1 was exhibited. At 1.5 kW kg−1, the specific energy corresponds to 32 W h kg−1. For comparison, a hybrid full-cell was also evaluated using as positive and negative electrodes CDC-600-3 and Nb2O5-850 respectively. Due to the lower capacitance of the CDC fibers compared to the AC (YP80 + 5% PTFE; 112 F g−1),60 a maximum specific energy of 71 W h kg−1 was obtained. Above 1 kW kg−1, the hybrid electrode system presents a better electrochemical performance maintaining 21 W h kg−1 at 2.2 kW kg−1. Cyclic voltammetry and GCPL of this full-cell are presented in the ESI (see Fig. S11†).
Conclusions
We demonstrated the suitability of NbC/C hybrid nanofibers as a versatile precursor for synthesizing electrode materials for both EDLCs and battery-like systems. Optimized EDLC electrodes were fabricated by chlorine treatment of NbC/C nanofibers at 600 °C in order to obtain a maximum pore volume of 1 cm3 g−1 and a DFT-SSA of 1369 m2 g−1. According to the electrochemical performance, due to the highly accessible pore structure, an efficient use of the surface leads to a maximum specific energy of 19.5 W h kg−1 and 7.6 W h kg−1 for a maximum specific power of 30 kW kg−1 in a full-cell configuration. The high specific power was accomplished due to the continuous path for electron propagation and the high graphitization of the carbon structure.By the treatment of NbC/C nanofibers in oxidative atmospheres, TT-Nb2O5 and T-Nb2O5/C hybrid nanofibers were synthesized at 500 °C and 850 °C, respectively. The synthesis in air (500 °C) does not allow for obtaining a highly crystalline TT-Nb2O5 phase without oxidizing the carbon phase. By employing CO2 (850 °C), highly crystalline T-Nb2O5 engulfed in graphitic carbon was obtained. The higher faradic transfer of charge led to a maximum capacity of 156 mA h g−1 with respect to the full electrode mass. This value is higher than the values reported in the literature for electrode capacity, since no polymer binder or conductive additive was added to the system. The rapid ion transport characteristic of the T-Nb2O5 and the co-existence of a continuous graphitic carbon phase led to an outstanding capacity retention of 50% at a high rate of 10 A g−1 (64 C-rate) in the battery-like system. The electrochemical performance is further highlighted by the superior stability over 5000 discharge cycles (64% retention). The combination of Nb2O5/C and AC in an asymmetric full-cell setup led to a specific energy of 86 W h kg−1, which is among the best values reported for a hybrid full-cell.
Further optimization steps of this promising system will involve enhanced engineering of the synthesis setup. For example, by use of the same furnace for pyrolysis and either chlorine gas treatment or CO2 annealing, the synthesis process can be simplified. Further adaptations of our work may also involve the use of niobium carbide nanopowder instead of electrospun mats. While this may come at the expense of lowered power handling due to the loss of the free-standing and binder-free feature of our electrospun mats, we see possibilities for cost reduction by the use of powder materials.
Acknowledgements
We acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. This work was supported by the CREATe-Network Project, Horizon 2020 of the European Commission (RISE Project No. 644013). We thank Prof. Eduard Arzt (INM) for his continuing support, and Juhan Lee and Anna Schreiber (all at INM) for useful discussions and technical support.References
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- P. R. Bandaru, H. Yamada, R. Narayanan and M. Hoefer, Mater. Sci. Eng., R, 2015, 96, 1–69 CrossRef.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nature Energy, 2016, 1, 16070 CrossRef.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- M. Schroeder, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2013, 238, 388–394 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- J. Lee, B. Kruner, A. Tolosa, S. Sathyamoorthi, D. Kim, S. Choudhury, K.-H. Seo and V. Presser, Energy Environ. Sci., 2016 10.1039/c6ee00712k.
- B. Krüner, J. Lee, N. Jäckel, A. Tolosa and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 9104–9115 Search PubMed.
- B. Zhang, F. Kang, J.-M. Tarascon and J.-K. Kim, Prog. Mater. Sci., 2016, 76, 319–380 CrossRef CAS.
- V. Presser, L. F. Zhang, J. J. Niu, J. McDonough, C. Perez, H. Fong and Y. Gogotsi, Adv. Energy Mater., 2011, 1, 423–430 CrossRef CAS.
- L. F. Zhang, A. Aboagye, A. Kelkar, C. L. Lai and H. Fong, J. Mater. Sci., 2014, 49, 463–480 CrossRef CAS.
- N. C. Abeykoon, J. S. Bonso and J. P. Ferraris, RSC Adv., 2015, 5, 19865–19873 RSC.
- M. Rose, E. Kockrick, I. Senkovska and S. Kaskel, Carbon, 2010, 48, 403–407 CrossRef CAS.
- V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810–833 CrossRef CAS.
- J. R. Martin, L. Borchardt, M. Oschatz, G. Mondin and S. Kaskel, Chem. Ing. Tech., 2013, 85, 1742–1748 CrossRef CAS.
- A. Tolosa, B. Krüner, N. Jäckel, M. Aslan, C. Vakifahmetoglu and V. Presser, J. Power Sources, 2016, 313, 178–188 CrossRef CAS.
- Y. Korenblit, M. Rose, E. Kockrick, L. Borchardt, A. Kvit, S. Kaskel and G. Yushin, ACS Nano, 2010, 4, 1337–1344 CrossRef CAS PubMed.
- J. Xu, R. Zhang, P. Chen, D. Shen, X. Ye and S. Ge, Carbon, 2013, 64, 444–455 CrossRef CAS.
- R. Dash, J. Chmiola, G. Yushin, Y. Gogotsi, G. Laudisio, J. Singer, J. Fischer and S. Kucheyev, Carbon, 2006, 44, 2489–2497 CrossRef CAS.
- C. R. Pérez, S.-H. Yeon, J. Ségalini, V. Presser, P.-L. Taberna, P. Simon and Y. Gogotsi, Adv. Funct. Mater., 2013, 23, 1081–1089 CrossRef.
- R. K. Dash, A. Nikitin and Y. Gogotsi, Microporous Mesoporous Mater., 2004, 72, 203–208 CrossRef CAS.
- M. Xianwen, T. A. Hatton and C. R. Gregory, Curr. Org. Chem., 2013, 17, 1390–1401 CrossRef.
- S. Cavaliere, S. Subianto, I. Savych, D. J. Jones and J. Roziere, Energy Environ. Sci., 2011, 4, 4761–4785 CAS.
- M. Zeiger, D. Weingarth and V. Presser, ChemElectroChem, 2015, 2, 1117–1127 CrossRef CAS.
- M. Zhi, A. Manivannan, F. Meng and N. Wu, J. Power Sources, 2012, 208, 345–353 CrossRef CAS.
- J.-G. Wang, Y. Yang, Z.-H. Huang and F. Kang, Electrochim. Acta, 2011, 56, 9240–9247 CrossRef CAS.
- Y.-W. Ju, G.-R. Choi, H.-R. Jung, C. Kim, K.-S. Yang and W.-J. Lee, J. Electrochem. Soc., 2007, 154, A192–A197 CrossRef CAS.
- Y. Wu, R. Balakrishna, M. V. Reddy, A. S. Nair, B. V. R. Chowdari and S. Ramakrishna, J. Alloys Compd., 2012, 517, 69–74 CrossRef CAS.
- J. Come, V. Augustyn, J. W. Kim, P. Rozier, P.-L. Taberna, P. Gogotsi, J. W. Long, B. Dunn and P. Simon, J. Electrochem. Soc., 2014, 161, A718–A725 CrossRef CAS.
- J. W. Kim, V. Augustyn and B. Dunn, Adv. Energy Mater., 2012, 2, 141–148 CrossRef CAS.
- R. A. Rani, A. S. Zoolfakar, A. P. O'Mullane, M. W. Austin and K. Kalantar-Zadeh, J. Mater. Chem. A, 2014, 2, 15683–15703 CAS.
- B. Reichman and A. J. Bard, J. Electrochem. Soc., 1980, 127, 241–242 CrossRef CAS.
- R. Kodama, Y. Terada, I. Nakai, S. Komaba and N. Kumagai, J. Electrochem. Soc., 2006, 153, A583–A588 CrossRef CAS.
- A. L. Viet, M. V. Reddy, R. Jose, B. V. R. Chowdari and S. Ramakrishna, J. Phys. Chem. C, 2010, 114, 664–671 Search PubMed.
- C. Zhang, R. Maloney, M. R. Lukatskaya, M. Beidaghi, B. Dyatkin, E. Perre, D. Long, W. Qiao, B. Dunn and Y. Gogotsi, J. Power Sources, 2015, 274, 121–129 CrossRef CAS.
- M. Liu, C. Yan and Y. Zhang, Sci. Rep., 2015, 5, 8326 CrossRef PubMed.
- K. Brezesinski, J. Wang, J. Haetge, C. Reitz, S. O. Steinmueller, S. H. Tolbert, B. M. Smarsly, B. Dunn and T. Brezesinski, J. Am. Chem. Soc., 2010, 132, 6982–6990 CrossRef CAS PubMed.
- J. S. Atchison, M. Zeiger, A. Tolosa, L. M. Funke, N. Jackel and V. Presser, RSC Adv., 2015, 5, 35683–35692 RSC.
- P. Scherrer, Nachr. Ges. Wiss. Goettingen, Math.-Phys. Kl., 1918, 98–100 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583–1590 CrossRef CAS.
- P. W. Ruch, D. Cericola, M. Hahn, R. Kötz and A. Wokaun, J. Electroanal. Chem., 2009, 636, 128–131 CrossRef CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- K. Faber, F. Badaczewski, M. Oschatz, G. Mondin, W. Nickel, S. Kaskel and B. M. Smarsly, J. Phys. Chem. C, 2014, 118, 15705–15715 CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report), 2015 Search PubMed.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- J. Segalini, E. Iwama, P.-L. Taberna, Y. Gogotsi and P. Simon, Electrochem. Commun., 2012, 15, 63–65 CrossRef CAS.
- R. Kötz, M. Hahn and R. Gallay, J. Power Sources, 2006, 154, 550–555 CrossRef.
- L. Shi, Y. Gu, L. Chen, Z. Yang, J. Ma and Y. Qian, Solid State Ionics, 2005, 176, 841–843 CrossRef CAS.
- M. V. Reddy, R. Jose, A. Le Viet, K. I. Ozoemena, B. V. R. Chowdari and S. Ramakrishna, Electrochim. Acta, 2014, 128, 198–202 CrossRef CAS.
- T. Ikeya and M. Senna, J. Non-Cryst. Solids, 1988, 105, 243–250 CrossRef CAS.
- V. L. S. Teixeira da Silva, M. Schmal and S. T. Oyama, J. Solid State Chem., 1996, 123, 168–182 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- S. Fleischmann, N. Jäckel, M. Zeiger, B. Krüner, I. Grobelsek, P. Formanek, S. Choudhury, D. Weingarth and V. Presser, Chem. Mater., 2016, 28, 2802–2813 CrossRef CAS.
- N. Jäckel, D. Weingarth, A. Schreiber, B. Krüner, M. Zeiger, A. Tolosa, M. Aslan and V. Presser, Electrochim. Acta, 2016, 191, 284–298 CrossRef.
- A. Meier, M. Weinberger, K. Pinkert, M. Oschatz, S. Paasch, L. Giebeler, H. Althues, E. Brunner, J. Eckert and S. Kaskel, Microporous Mesoporous Mater., 2014, 188, 140–148 CrossRef CAS.
- N. Jäckel, D. Weingarth, A. Schreiber, B. Krüner, M. Zeiger, A. Tolosa, M. Aslan and V. Presser, Electrochim. Acta, 2016, 191, 284–298 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive Raman data (including peak deconvolution analysis), additional high resolution transmission and scanning electron micrographs, additional fiber diameter distribution analysis, additional gas sorption data, comprehensive data analysis (SEM, Raman, XRD, gas sorption analysis, and electrochemistry) for CDC materials obtained for 1 h of chlorine gas treatment (compared to 3 h in the manuscript), complementary thermogravimetric analysis and thermodynamic calculations with FACTSAGE, and additional electrochemical galvanostatic benchmarking of a half-cell and full-cell setup for the battery-system. See DOI: 10.1039/c6ta06224e |
| This journal is © The Royal Society of Chemistry 2016 |