The role of apoptosis in response to photodynamic therapy: what, where, why, and how
Nancy L. Oleinick*, Rachel L. Morris and Irina Belichenko
Department of Radiation Oncology and the CWRU/UHC Ireland Comprehensive Cancer Center, School of Medicine, Case Western Reserve University, Cleveland, Ohio, USA. E-mail: nlo@po.cwru.edu
First published on 2nd January 2002
Abstract
Photodynamic therapy (PDT), a treatment for cancer and for certain benign conditions, utilizes a photosensitizer and light to produce reactive oxygen in cells. PDT is primarily employed to kill tumor and other abnormal cells, so it is important to ask how this occurs. Many of the photosensitizers currently in clinical or pre-clinical studies of PDT localize in or have a major influence on mitochondria, and PDT is a strong inducer of apoptosis in many situations. The purpose of this review is to critically evaluate all of the recently published research on PDT-induced apoptosis, with a focus on studies providing mechanistic insights. Components of the mechanism whereby PDT causes cells to undergo apoptosis are becoming understood, as are the influences of several signal transduction pathways on the response. Future research should be directed to elucidating the role(s) of the multiple steps in apoptosis in directing damaged cells to an apoptotic vs. necrotic pathway and for producing tumor ablation in conjunction with tissue-level mechanisms operating in vivo.
 Nancy L. Oleinick Nancy L. Oleinick | Nancy L. Oleinick, PhD, is Professor of Radiation Oncology, Biochemistry, Oncology, and Environmental Health Sciences, at Case Western Reserve University School of Medicine, Cleveland, OH, USA, and director of the Radiation Biology Program of the CWRU/Ireland Comprehensive Cancer Center. She earned her PhD in biochemistry from the University of Pittsburgh. Dr Oleinick's research specialties are in radiobiology and photobiology. In recent years, her research has focused on the cellular and molecular effects of photodynamic therapy, with a special interest in mechanisms of apoptosis. Dr Oleinick has served on the editorial boards of Radiation Research, International Journal of Radiation Biology, and Photochemistry and Photobiology, on two grant review panels of the US National Institutes of Health, and on numerous advisory boards reviewing policies and programs in radiobiology and photobiology. She is a past-president of the American Society for Photobiology and served as scientific program chair for the 13th International Congress on Photobiology in 2000. |
 Rachel L. Morris Rachel L. Morris | Rachel L. Morris received her BA in Psychology and Sociology from York University in Toronto, Canada, then did post-baccalaureate work in Biomedical Sciences at John Carroll University and Case Western Reserve University in Cleveland, Ohio. She gained extensive laboratory experience working under the direction of Dr Bruce Trapp at the Cleveland Clinic Foundation. Since 1999, she has been working on photosensitizer binding sites and control of PDT-induced apoptosis in the Oleinick laboratory. |
 Irina Belichenko Irina Belichenko | Irina Belichenko, MD PhD, received her PhD in biophysics in 1996 from the Russian State Medical University in Moscow. She was a visiting scientist in the Photodynamic Therapy Unit of Alexis Vautrin Cancer Center, Nancy, France, where she collaborated with Professor F. Guillemin on the development of photodynamic therapy including clinical, instrumental and biological aspects. She then joined the laboratory of Dr Peter Traub at the Max Planck Institute in Ladenburg, Germany, to work on the biochemistry of cytoskeletal intermediate filaments and mitochondria. She carried out postdoctoral research under the guidance of Drs D. Separovic and N. Oleinick at Case Western Reserve University, Cleveland, where she worked on signal transduction mechanisms activated in response to photodynamic therapy. Dr Belichenko's current research concerns the role of intermediate filaments in tumor cell response to photodynamic treatment and signal transduction pathways mediated by small ubiquitin-related modifiers. |
1 Introduction to photodynamic therapy and apoptosis
Photodynamic therapy (PDT) is a novel treatment for cancer and certain non-cancerous conditions that are generally characterized by overgrowth of unwanted or abnormal cells.1–10 The procedure requires exposure of cells or tissues to a photosensitizing drug followed by irradiation with visible light of the appropriate wavelength, usually in the red or near-infrared region and compatible with the absorption spectrum of the drug. Upon absorption of a photon, the photosensitizer undergoes one or more energy transitions and usually emerges in its excited triplet state. The triplet can participate in a one-electron oxidation–reduction reaction (Type I photochemistry) with a neighboring molecule, producing free radical intermediates that can react with oxygen to produce peroxy radicals and various reactive oxygen species (ROS). Alternatively, the triplet-state photosensitizer can transfer energy to ground state oxygen (Type II photochemistry), generating singlet molecular oxygen, a highly reactive form of oxygen that reacts with many biological molecules, including lipids, proteins, and nucleic acids.11–16 Most photosensitizers for PDT are efficient producers of singlet oxygen in simple chemical systems, and it is assumed, although impossible to measure directly, that Type II photochemistry is the dominant mechanism for PDT in most circumstances in cells and tissues.10,11The first PDT photosensitizer to win approval by regulatory agencies in several countries was Photofrin, a complex mixture of the more active porphyrin oligomers that comprised hematoporphyrin derivative (HpD).17 Photofrin is now approved for treatment of lung and esophageal cancer in the US and for several other cancers worldwide.10,16,18 The limitations of Photofrin, including its complexity, its poor absorption of tissue-penetrating red light, and its tendency to be retained in skin and thus to produce cutaneous photosensitivity,16 have encouraged the development of many second-generation photosensitizers.19,20 Although most of the newer photosensitizers are porphyrin-like macrocycles, such as benzoporphyrins, purpurins, texaphyrins, phthalocyanines, and naphthalocyanines, or endogenously generated photosensitive metabolites, such as protoporphyrin IX (PPIX), a few have other types of structures, e.g., hypericin, rhodamine, methylene blue, and derivatives of these molecules (Fig. 1). In the case of PPIX, synthesis of this photosensitizing precursor of heme is greatly enhanced by supplying an earlier metabolic precursor, 5-aminolevulinic acid (ALA). In addition to Photofrin, regulatory approval has also been obtained for photodynamic treatment of age-related macular degeneration with Visudyne (formulated Verteporfin) and for treatment of actinic keratoses with Levulan (formulated ALA). In this review, we will attempt to identify the photosensitizer with which a particular observation was made, but it is not yet possible in many cases to assign a unique cellular response to a particular photosensitizer. Because investigators tend to study effects of one or at best a few photosensitizers, the conclusions that are drawn can sound as though the effect can be elicited only by that photosensitizer, when in fact, many of the responses that will be discussed here are a result of photodynamic damage to cells which can be produced by many different photosensitizers, so long as their amount and location and the fluence and wavelengths of light are appropriately adjusted. Emphasis will be on those factors, related both to the photosensitizer and to the cell properties, that can influence cellular responses to PDT.
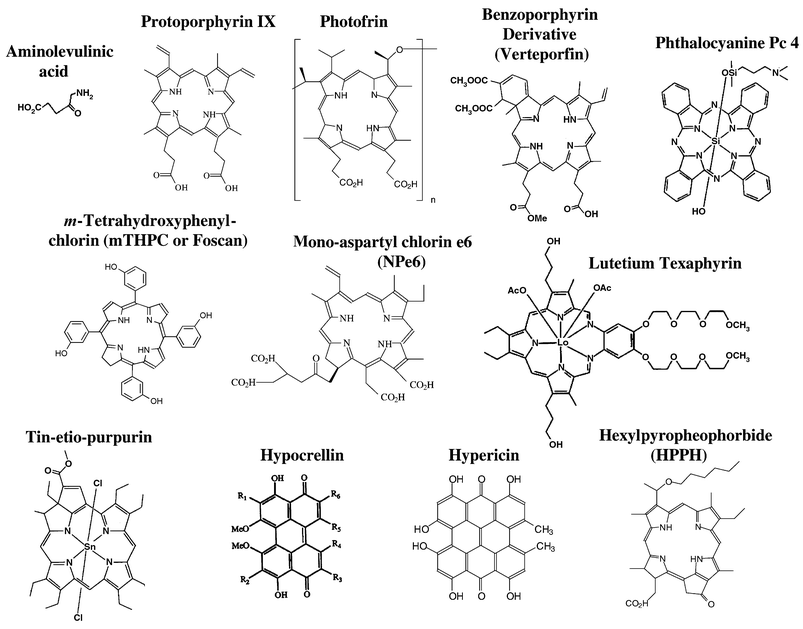 | ||
| Fig. 1 Names and structures of selected photosensitizers in clinical or pre-clinical studies. Three of these have obtained regulatory approval for clinical PDT: Photofrin; benzoporphyrin derivative (Verteporfin); and aminolevulinic acid (Levulan), which is not a photosensitizer but a metabolic precursor of protoporphyrin IX. | ||
In the vast majority of applications, the primary role of PDT is to kill unwanted cells. Since the term “apoptosis” was introduced in 1972,21 cell killing mechanisms are generally classified as occurring through apoptosis or necrosis. Apoptosis is a normal physiological process essential for the control of tissue development and involution and for tissue homeostasis.22 Apoptosis is a tightly regulated process of cell suicide, controlled by both intracellular and extracellular signals, terminating in a characteristic sequence of morphological and biochemical changes for the systematic dismantling of the cell and preparation of the residual cell components, known as apoptotic bodies, for engulfment by tissue macrophages or other neighboring cells. The process limits leakage of intracellular material to the immediate environment, and thereby prevents tissue inflammation. The loss of ability to regulate apoptosis can lead to disease.23–25 For example, many cancer cells have lost the ability to undergo apoptosis in response to normal signals or after damage from therapeutic interventions,26,27 whereas unrestrained apoptosis of neurons may contribute to Alzheimer's and other neurodegenerative diseases.28,29 In contrast, necrosis results from high levels of cell damage, in which plasma membrane integrity is lost, cells lyse, and tissue inflammation is triggered.30 Although PDT can produce apoptosis or necrosis, or a combination of the two mechanisms, in many cases PDT is highly efficient in inducing apoptosis.31 This property is of more than theoretical interest, because it implies that lower doses than those needed to produce necrosis are very effective in producing the desired cell killing result.
The past few years have produced a copious outpouring of research into the mechanisms of apoptosis, and many excellent reviews have appeared describing the steps involved in response to a wide variety of stimuli. An excellent series of five reviews appeared together in Science in 1998,22,23,32–34 and several others have appeared since then.24,25,30,35–37 Mechanisms of PDT have been reviewed as well, and the most recent contain brief capsules of research on PDT-induced apoptosis.10,20,38–40 Two recent reviews have focused on signaling mechanisms of PDT and their role in PDT-induced apoptosis41,42 and another two have reviewed PDT-induced apoptosis from the perspective of research on Verteporfin.43,44 This report aims to provide a critical evaluation of the state of the science on PDT-induced apoptosis, with the primary emphasis on elucidation of mechanisms. Accordingly, recent papers that have reported observations of apoptosis but without new mechanistic insights have been ignored.
2 Mechanisms of apoptosis
Apoptotic cells have been recognized since the 19th century by their distinctive morphological appearance in tissues as shrunken cells with condensed nuclear chromatin, retracted from surrounding cells. Initiation of apoptosis in developing tissues is asynchronous, and the biochemical pathways triggering the process depend on the inducing agent. During physiological cell turnover, apoptosis is initiated by depletion of a growth factor or by the interaction of cytokines or other ligands with cell surface receptors.22 However, cell damage by chemicals, radiation, heat, or other stresses can also trigger apoptosis.45–48 After initiation, signaling pathways from the cell surface or from the site of cell damage converge onto a small number of central pathways that lead to the final “execution” phase of apoptosis, during which hydrolytic enzymes, including proteases and nucleases, are activated, and the characteristic morphological changes become evident.25,34 Before discussing mechanisms of PDT-induced apoptosis, four general components of apoptosis mechanisms will be considered: (a) the role of caspases, (b) mechanisms initiated at the cell surface, (c) the role of mitochondria, and (d) the role of Bcl-2 family members.2.1 Caspases
The central hydrolytic reactions of apoptosis are catalyzed by a family of proteases, now termed “caspases” for “cysteine proteases acting on aspartic acid”.25,34,49,50 At least 12 of these enzymes are known. They have in common an active site cysteine and specificity for a short sequence of amino acids in their target site that terminates in aspartic acid. Caspases are synthesized as pro-enzymes that are activated by cleavage either in an autolytic fashion or by other caspases. For example, when multiple procaspase-9 molecules assemble on the apoptosomes, they can cleave one another to remove a leader sequence and generate a short and long peptide. Active caspases are composed of two short (∼p10) and two long (∼p20) peptides, i.e., a tetramer. Caspases are often classified as “initiator” or “effector” enzymes, depending on their placement in the pathway, either proximal to the initiating signal or at a late stage of cell disassembly, respectively. Effector caspases cleave and inactivate proteins that protect living cells from apoptosis, such as the DNA repair protein, poly(ADP-ribose) polymerase (PARP), ICAD/DFF45 (inhibitor of caspase-activated DNase, the nuclease responsible for DNA fragmentation), or the anti-apoptotic Bcl-2 proteins. Other actions of caspases in apoptosis include cleavage of cytoskeletal proteins, including the lamins, proteins forming the nuclear lamina; cytoplasmic intermediate filaments (vimentin, cytokeratins), and several proteins involved in cytoskeleton regulation (gelsolin, focal adhesion kinase and p21-activated kinase 2). This results in disassembly of cell structures that depend on the cytoskeleton.2.2 Cell surface “death receptors”
Apoptosis is rapidly induced by activation of “death-inducing signaling complexes” (DISCs) at the plasma membrane22,25 (Fig. 2). DISCs are formed when extracellular ligands interact with cell surface receptors belonging to the cysteine-rich tumor necrosis factor (TNF) receptor gene superfamily. The death receptors Fas (also called CD95 or Apo1) and TNFR1 (also called p55 or CD120a) are two of the best characterized. Fas and its ligand (FasL) are important for the elimination of activated mature T cells at the end of an immune response and of virus-infected cells, and for the attack of cancer cells by cytotoxic T cells and natural killer cells. Ligation of FasL to Fas causes the intracellular death domains of the receptor to cluster in groups of three. FADD (Fas-associated death domain) acts as the adaptor, binding the clustered death domains through its own death domain. Then, procaspase-8 is recruited to the adaptor, resulting in enzyme oligomerization and activation by self-cleavage. Caspase-8 activates downstream effector caspases, such as caspase-3, committing the cell to apoptosis. Alternatively, caspase-8 can cleave Bid, a Bcl-2 homolog, which then promotes apoptosis at the mitochondria (Fig. 2). Unlike Fas, the TNF pathway appears to require that protein synthesis be blocked for apoptosis to be induced. TNFα is produced by activated macrophages and T cells as a response to infection. Ligand binding causes trimerization of TNFR1 and a consequent association of the receptors' death domains. TRADD (TNFR-associated death domain) serves as the adaptor and binds through its own death domain to the clustered receptor death domains. TRADD is a platform adaptor, capable of recruiting several signaling molecules to activate divergent pathways that signal apoptosis or survival.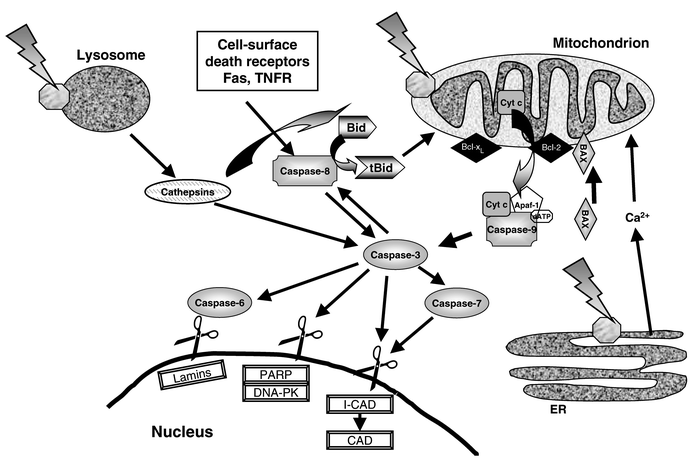 | ||
| Fig. 2 Some PDT-associated apoptosis pathways involving plasma membrane death receptors, mitochondria, lysosomes and ER, caspases, and Bcl-2 family proteins. Most photosensitizers for PDT bind to mitochondria, lysosomes, and/or other intracellular membranes, including the ER. Photoactivation of a mitochondrion-localized photosensitizer causes release of cytochrome c, which may or may not be accompanied by loss of the mitochondrial membrane potential and opening of the PTPC. The released cytochrome c becomes part of the “apoptosome” complex to generate active caspase-9, which then cleaves and activates caspase-3. Caspase-3 is the major effector caspase and is responsible for cleavage and activation of other caspases, especially caspases-6, -7, and -8. The effector caspases cleave numerous proteins, including nuclear lamins, leading to nuclear breakdown; PARP and DNA-PK, resulting in inhibition of DNA repair; ICAD, releasing active CAD to degrade DNA; and other proteins that affect cell structure and adhesion. In cases where the cell surface death receptors Fas and TNFR participate, binding to their respective ligands leads to activation of caspase-8, which can result in the activation of caspase-3 independent of mitochondrial involvement. Caspase-8 cleaves the Bcl-2 homolog Bid to produce the pro-apoptotic fragment tBid, which can act on mitochondria to cause cytochrome c release. Photoactivation of lysosome-bound photosensitizers can cause the release of cathepsins, which can cleave Bid to promote apoptosis and caspase-3 to inhibit apoptosis. Damage to the ER by PDT causes release of Ca2+, which can promote apoptosis. Apoptosis is controlled by members of the Bcl-2 family that either promote or inhibit the process. In addition to Bid, shown here are the anti-apoptotic proteins Bcl-2 and Bcl-xL, that block PDT-induced apoptosis by inhibiting caspase activation, and the pro-apoptotic Bax, that has been proposed to promote mitochondrial reactions, including cytochrome c release. | ||
2.3 Mitochondria
Mitochondria have emerged as the central processing organelles in the majority of apoptotic pathways. Signals from cell surface death receptors or from damaged sites converge on mitochondria, leading to permeabilization of both mitochondrial membranes, dissipation of the inner membrane transmembrane potential (ΔΨm), and release of apoptosis-related proteins, such as cytochrome c, apoptosis-inducing factor (AIF), SMAC (second mitochondria-derived activator of caspases), and certain procaspases from the intermembrane space. Although these changes in mitochondria are implicated in most apoptotic pathways, their ordering in the pathway remains in dispute.24,25,30,35–37,51One current model suggests that the release of apoptosis-related mitochondrial factors and the collapse of the mitochondrial transmembrane potential results from the opening of a large conductance channel known as the permeability transition pore complex (PTPC), which forms at contact sites of the outer and inner mitochondrial membranes. Models of the PTPC have been proposed in recent reviews.35,36,52–54 The PTPC is thought to be comprised of at least three transmembrane proteins: (a) the 30 kDa inner membrane adenine nucleotide translocator (ANT); (b) the 32 kDa outer membrane voltage-dependent anion channel (VDAC) or mitochondrial porin; and (c) the 18 kDa outer membrane peripheral benzodiazepine receptor (PBR). There is also evidence for the association of other proteins with the PTPC: members of the Bax/Bcl-2 family, cyclophilin D, and enzymes of energy metabolism, such as hexokinase and mitochondrial creatine kinase.
The potential across the inner mitochondrial membrane is a consequence of the pumping of protons to the cytoplasmic side of the membrane using the energy of electron transport. Under normal conditions, the passage of molecules and ions across the inner mitochondrial membrane is tightly regulated. In response to changes in redox state, pH, and local concentrations of ATP/ADP and ions, such as Ca2+ and Mg2+, the PTPC is thought to regulate membrane permeabilization. Opening of a non-selective channel in the inner membrane would allow ion equilibration between the matrix and intermembrane space, thus dissipating the H+ gradient across the inner membrane and uncoupling the respiratory chain. Another possible consequence of PTPC opening is mitochondrial volume dysregulation, as normally excluded ions and water expand the matrix space. Since the folded cristae of the inner mitochondrial membrane have a larger surface area than the outer membrane, matrix space expansion then causes outer membrane rupture and consequent release of caspase-activating proteins located within the intermembrane space into the cytosol.35,53
Therefore, cytochrome c may be released from mitochondria through opening of the PTPC and bursting of the outer membrane or through the creation of more selective channels in the outer membrane or by another mechanism. Once in the cytosol, cytochrome c binds to cytoplasmic apoptosis-activating factor-1 (APAF-1) to form the apoptosome complex and initiate activation of the cascade of caspases that carry out the final stages of apoptosis.37
2.4 Bcl-2 family proteins
Bcl-2 family members regulate apoptosis induced by many apoptotic stimuli, primarily at the level of the mitochondria.55 Anti-apoptotic members of this large protein family include Bcl-2, Bcl-xL, Bcl-w, Mcl-1, Al and Boo; whereas pro-apoptotic members include Bax, Bad, Bok, Bcl-xS, Bak, Bid, Bik, Bim, Krk, and Mtd.56 Apoptotic regulation by Bcl-2 family proteins has been extensively reviewed.32,33,52,55–57 In the absence of a death signal, pro-apoptotic Bcl-2 family members are often sequestered by cytoskeletal elements or cytoplasmic proteins (e.g., sequestration of phosphorylated Bad by 14–3–3 proteins) or are only loosely associated with membranes. In contrast, anti-apoptotic Bcl-2 family members are often integral membrane proteins found in the mitochondrial membrane, the nuclear envelope, and the endoplasmic reticulum.52The activity of Bcl-2-related proteins is regulated through several mechanisms, including their levels of expression, sequestration, and post-translational modifications, such as phosphorylation, cleavage, and translocation.52 Cleavage of Bcl-2 not only inactivates the protein but also produces a fragment that promotes apoptosis.58,59 Cytosolic Bid (p22) is cleaved by caspase-8 to generate truncated Bid (tBid), which then translocates to the mitochondria to integrate into the membrane and assist in the release of cytochrome c.52 A recent report demonstrated that proteases released from lysosomes, cathepsins, can also cleave Bid to generate a mitochondrion-targeted pro-apoptotic protein.60 In response to apoptotic stimuli, several pro-apoptotic proteins are translocated to the mitochondria, where they can interact with membrane-bound anti-apoptotic proteins, thereby inhibiting the survival functions of the latter.52 Bcl-2, Bcl-xL, and Bax can form ion channels in artificial membranes, suggesting regulation of apoptosis via the formation of pores.56,61 Other hypotheses for the inhibition of apoptosis by Bcl-2 include participation in an anti-oxidant pathway62 and blockage of the release of cytochrome c.52,55,63,64 Bcl-2 homologs often form homo- and heterodimers with each other and with other non-homologous proteins that are presumed to control the availability of their active forms.52,57 The ratio of pro- to anti-apoptotic members has been suggested to regulate cell life or death.32 Some examples of the interaction of these proteins at the mitochondrion to control apoptosis are shown in Fig. 2.
3 Measuring apoptosis
Quantification of the precise extent of apoptosis vs. necrosis and total cell death in cultured cells is difficult and several issues need to be considered. Apoptosis is a biochemical process that passes through several stages each with its own hallmarks and timing. The kinetics of the process depend upon a variety of factors, including the dose of the inducing agent and the ability of different cell types to carry out steps in the process. Thus, the percentage of cells in apoptosis will vary with time after initiation. Cells treated with doses higher than those needed to cause apoptosis may die without exhibiting the usual characteristics of apoptosis, and are generally regarded as undergoing necrosis. As noted above, during the later stages of apoptosis, cells disintegrate into membrane-enclosed apoptotic bodies that in vivo are phagocytosed by macrophages or adjacent tumor cells and further catabolized. Since phagocytic cells are not found in tissue cultures, apoptotic bodies may complete the breakdown of their cellular constituents or may simply become detached from the culture substrate. In addition, late apoptotic cells or bodies can eventually lose the ability to maintain their membrane pumps and as a result may be unable to exclude vital dyes, such as trypan blue. Such apoptotic cells or bodies would then appear as necrotic cells. Therefore, one cannot conclude that a particular cell type or treatment does not produce an apoptotic response without studying a range of doses of the initiating agent and assaying for apoptotic markers over a wide range of times.There are numerous assays for apoptosis. The two most common endpoints of apoptosis are morphological changes (cell shrinkage, condensation of nuclear chromatin, formation of apoptotic bodies) and DNA fragmentation [to large 300 kbp and 50 kbp fragments and then to oligonucleosome-sized fragments (multiples of ∼200 bp that appear as a “ladder” of DNA bands upon agarose gel electrophoresis)]. Although observation of these endpoints is an indicator of apoptosis, quantification of the percentage of cells in a population that are in apoptosis is difficult or impossible without more sophisticated methods. For this purpose, many investigators use the TUNEL assay (during which fluorescently tagged or biotinylated nucleotides are added to the ends of DNA fragments within fixed cells). The cells are then stained in their DNA with propidium iodide and can be viewed on a microscope stage, or they can be quantified by flow cytometry to indicate the presence of viable, apoptotic, necrotic, and late apoptotic cells. Apoptotic cells can also be detected with the protein Annexin V, which binds in a highly selective manner to phosphatidyl serine; this phospholipid flips from the inner to the outer leaflet of the plasma membrane during apoptosis. This assay is also adaptable to quantification by flow cytometry or to imaging.
Since many of the enzymatic steps in apoptosis are known, it is often desirable to follow the process by monitoring those steps. Antibodies are available commercially for all of the common caspases, so that procaspase levels and their proteolytic processing can be monitored by western blot analysis. In addition, model peptide substrates can also be purchased for many of the caspases in fluorogenic or chromogenic forms, permitting assay of cell extracts for the presence and level of individual active caspases. Inhibitors based on active site peptide motifs can be used to test the participation of suspected caspases. However, because caspases are not completely specific for the amino acid motifs, the model substrates and inhibitors cannot provide absolute identification of the involved caspase. Caspase action can also be monitored by assessing the cleavage of intracellular protein substrates; e.g., cleavage of PARP or lamin B is a common indicator of the action of caspase-3 or -7 in apoptosis. A listing of commercially available kits for these and other assays of apoptosis, including measurement of caspases, substrates, and inhibitors, can be found at the following web site: www.the-scientist.org.
With any of these or other assays, it is advisable to study sufficient doses and post-treatment times so that the progress of cell death may be observed. In addition, it is necessary to use more than one assay to confirm results. Finally, if some or all of the cells do not undergo apoptosis, one cannot conclude that the cells remain alive, without conducting an assay for total cell death. The best assay for this purpose is a clonogenic assay, which measures the ability of each cell in a population to grow, replicate, and form a colony. Unlike assays for apoptosis or for viability, such as dye exclusion or tetrazolium reduction, clonogenic assays register all cell death events integrated over time.65
4 Cell death in the response to PDT: apoptosis vs. necrosis
Since the first report of apoptosis in PDT-treated cells,66 apoptosis has been found to be a prominent form of cell death in response to PDT for many cells in culture, as judged by assays measuring either the fragmentation of DNA or the condensation of chromatin. There are by now numerous examples in which the ability of PDT-exposed cells to initiate the apoptotic process differs depending on the cell line, the photosensitizer and its subcellular location, the overall dose, and other conditions. In an early study, He et al.67 exposed three carcinoma cell lines to equitoxic doses of PDT with Photofrin and found apoptosis in two of the cell lines but not in the third. Noodt et al.68 compared responses of V79 Chinese hamster fibroblasts and WiDr human colon adenocarcinoma cells to equitoxic doses of PDT with ALA/PPIX, i.e., PPIX synthesized in the cells from exogenously supplied ALA. Using the TUNEL assay and flow cytometry to quantify the fraction of cells with fragmented DNA, they showed evidence for a dose- and time-dependent increase in the apoptotic fraction in PDT-treated V79 cells, with a maximum of 75–85% of the cells in apoptosis by 3–4 hours after a dose that resulted in 85% overall cell death. In contrast, under comparable conditions resulting in similar levels of overall cell death as judged by clonogenic assay, no evidence for apoptosis was found in WiDr cells. This study underscores the importance of verifying assays for apoptosis using an assay for total cell death.In a study of the ability of PDT sensitized by Pc 4 to purge viruses from red blood cell concentrates, Ben-Hur et al.69 found that T lymphocyte cell lines latently infected with HIV were more easily killed and more readily induced to undergo apoptosis than was a cell line with actively replicating virus. The authors attributed the enhanced elimination of HIV in latently infected cells to apoptosis. Lymphoid cell lines generally enter apoptosis more rapidly than those of non-lymphoid origin (e.g.,66,70,71). Furthermore, the rapid induction of apoptosis by PDT in murine lymphoma L5178Y-R cells appears to occur in all stages of the cell cycle, as revealed by flow cytometric analysis of TUNEL positivity vs. cell cycle stage,72,73 and murine leukemia L1210 cells arrested in the cell cycle by cryptophycin 52 were still able to undergo apoptosis within 60 min of exposure to CPO-sensitized PDT.74 Murine T cells, activated by a 48-hour treatment with anti-CD3 monoclonal antibody, were slightly more sensitive to the induction of apoptotic DNA fragmentation by Verteporfin-PDT than were resting T lymphocytes, which could have resulted from increased uptake of the photosensitizer, a higher level of Bcl-2, or other noted changes, including increased IL-2 and transferrin receptors.75 Moreover, murine thymocytes were modestly more sensitive to PDT than mature splenic T cells.76
The subcellular location of a photosensitizer has a strong influence on whether and to what extent cells undergo apoptosis in response to photoactivation (Fig. 2). Dellinger77 found apoptosis in CV-1 cells if they were photoirradiated 24 hours after introduction of Photofrin, when the photosensitizer was internalized, whereas after only 1 hour, when Photofrin was primarily in the plasma membrane, necrosis was the predominant form of cell death. The photosensitizer methylene blue derivative (MBD) localizes in mitochondria of V79 cells, as revealed by fluorescence microscopy and by the photodynamic loss of cytochrome c oxidase activity when enzymes of other organelles remained intact.78 When MBD was present at concentrations above 0.05 μg ml−1, photoactivation of the cells caused early (within 3 hours) apoptosis as the dominant mechanism of cell death. At 0.05 μg ml−1, the process was much slower, and apoptotic cells appeared only after 1 day. When PDT with the low concentration of MBD was combined with the inhibitor of glycolysis, 2-deoxyglucose, apoptotic cells appeared within 6 hours; in contrast, combination with the uncoupler of oxidative phosphorylation, CCCP, acted like PDT alone. The authors interpreted these data as indicating mitochondrial damage from MBD-PDT; at the higher concentrations, they suggested that damage to an apoptosis inhibitor, such as Bcl-2, might lead to release of apoptogenic factors, whereas at the low concentration of MBD, the induction of apoptosis may be indirect. Molecular evidence for the photochemical damage to Bcl-2 and for indirect pathways after low PDT doses has now been obtained (see Section 5.5).
With Rose Bengal, a photosensitizer that is distributed in cellular membranes, Kochevar et al.79 observed extensive apoptosis in HL-60 cells, if the dye was photoactivated by visible light, producing predominately singlet oxygen; in contrast, if the dye was activated by UVA radiation, producing Rose Bengal-derived radicals in addition to singlet oxygen, there was no further increase in the yield of apoptosis, in spite of a much greater yield of lipid peroxidation products. These results demonstrated that different reactive species produced at the same sites of photosensitizer localization can have markedly different cellular effects. Lin et al.80 studied the effects of confining Rose Bengal-PDT to the plasma membrane by irradiating bovine aortic endothelial cells attached to a glass substrate with evanescent wave visible radiation. In contrast to ordinary trans-illumination of the cells, evanescent waves penetrate only about 100 nm into the cell, limiting PDT to the plasma membrane and a very thin layer of cytoplasm. Under these conditions, Annexin V staining and nuclear morphology indicated that apoptosis was efficiently induced, demonstrating that singlet oxygen produced at the plasma membrane can induce apoptosis.
Kessel and Luo81 studied a series of photosensitizers in L1210 leukemia and other cells and demonstrated that photosensitizers that bind to mitochondria induce apoptosis upon photoirradiation, whereas those that bind to the plasma membrane or lysosomes, but not to mitochondria, kill cells less efficiently and by a non-apoptotic mechanism.81–83 The photosensitizer LuTex has been shown to bind to lysosomes of EMT6 cells in vitro and to produce apoptosis in EMT6 tumors in vivo, indicating that lysosomally bound photosensitizers can induce apoptosis upon photoactivation.84 To further evaluate the role of the subcellular localization of photosensitizers in the killing of V79 cells by PDT, Noodt et al.85 compared two lipophilic porphyrins (3THPP and Photofrin) that localize to intracellular membranes, including mitochondria, and two relatively hydrophilic sulfonated porphines (TPPS2a and TPPS4), that are taken up into lysosomes by endocytosis. PDT with either of the membrane-localizing photosensitizers resulted in increasing numbers of cells becoming apoptotic (TUNEL positive) during the first 12 hours, but apoptotic bodies were not observed. In contrast, after photoactivation of the lysosome-localized photosensitizers, apoptotic cells were not detected until after 12 hours but extensive fragmentation of the cells into apoptotic bodies was found. These data provide evidence for at least two distinct pathways by which PDT can induce apoptosis.
In some cases, the initial sites of photosensitizer binding may not be the most sensitive targets. Kessel and Poretz86 have reported that chlorin e6 triacetoxymethyl ester (CAME) accumulated within 10 minutes in acidic endosomes, but mitochondria were the preferential sites of photodamage at the early time. After hydrolysis and redistribution of CAME throughout the cells, which occurred by 18 hours, photodamage was found in mitochondria and lysosomes. The two conditions produced similar levels of apoptosis and loss of viability, although a higher concentration of CAME led to less apoptosis but more cell death, presumably by necrosis. This is in agreement with earlier evidence for a transition from apoptosis to necrosis with higher doses of PDT sensitized by any one of several photosensitizers, e.g., hypericin, dimethyl tetrahydroxyhelianthrone (DTHe), or phthalocyanines.87–90 Lavie et al.90 also found that the human leukemia cell line Hut-78 was resistant to DTHe-sensitized PDT in terms of morphological apoptosis, a phenomenon that may have been due to crosslinking of nuclear lamin.
Although PDT can produce apoptosis or necrosis, or evoke a combination of the two outcomes, in many cases PDT is highly efficient in inducing apoptosis.39 Thus, lower doses than those needed to produce necrosis may be very effective in eliciting cell killing. Furthermore, the efficient induction of apoptosis by PDT implies that PDT may be able to bypass mechanisms that make cells resistant to apoptosis in response to chemotherapeutic drugs and ionizing radiation.
A longstanding principle in studies of cell killing by many different types of toxic agents is that cell death occurs in those cells that are directly damaged by the treatment, whereas a neighboring cell that is not hit will remain alive. However, recent evidence has challenged this concept. A series of papers by Dahle et al.91–94 demonstrate that MDCK II and other cells treated with a photosensitizer and light die in clusters rather than as random individuals, producing an overabundance of colonies where either all or none of the cells are dead. This effect is known as the “bystander effect”. The experimental distribution of dead cells was best described by a mathematical model in which inactivated cells inactivated neighboring cells which could then inactivate their neighboring cells, i.e., a “propagated inactivation” model. When Photofrin served as photosensitizer in these studies, very few of the cells died through apoptosis. In contrast, for PDT with photosensitizers other than Photofrin, apoptosis was the main mechanism of cell death. However, the bystander effect was greater when the cells were dying by necrosis, possibly because necrotic cells had a better chance of leaking toxic substances through their damaged membranes. When MDCK II cells were treated with TPPS4 and light to various levels of overall cell killing, the fraction of cells dying by apoptosis was greater when the overall level of cell killing was low, as found for PDT with other photosensitizers (see above), and confluent cultures, with greater possibility for cell-to-cell interactions, were more sensitive than sub-confluent ones. In a comparison of PDT-induced killing of MDCK II cells vs. WiDr human colon adenocarcinoma cells, with the lipophilic photosensitizer 3THPP, the degree of the bystander effect was greater when the normal cells died by necrosis than by apoptosis and greater for normal cells than for the cancer cells. This potentially important component of PDT response remains unexplained at the molecular level. One possible mechanism for the bystander effect with PDT is the interaction of Fas ligand produced on one cell with Fas on the plasma membrane of an adjacent cell; such a mechanism is suggested by experiments described below (see Sections 6.3 and 7).
5 The role of mitochondria vs. other cellular targets
5.1 Mitochondrial binding sites for photosensitizers
Numerous reports have implicated mitochondria as important targets of PDT. Photosensitizers that localize to mitochondria are reported to be more efficient in killing cells than those that localize at other cellular sites81,82 (Fig. 2). Localization of drugs in mitochondria can be driven by the electrochemical potential gradient across the inner mitochondrial membrane. Examples of photosensitizers in this class are various cationic lipophilic compounds, such as the krytocyanine dye EDKC and rhodamine-12395 as well as MBD.78,96 Photosensitizers that are negatively charged, such as Photofrin, or neutral, such as Pc 4, have also been found to accumulate in mitochondria,97,98 and are thought to bind to various mitochondrial constituents. For instance, the inner membrane of mitochondria is rich in cardiolipins. Wilson et al.99 demonstrated the ability of a cardiolipin probe, 10N-nonyl acridine orange, to competitively inhibit the uptake of Photofrin into mitochondria, indicating that some photosensitizers might bind to cardiolipins of the inner mitochondrial membrane.Several investigations have examined components of the permeability transition pore complex (PTPC) as potential targets for PDT. PK11195, a selective high-affinity isoquinoline carboxamide ligand for the PBR, has been used to test the role of the PBR in PDT-induced apoptosis. An extensive series of studies by Verma and Snyder100–102 showed that porphyrins are endogenous ligands for the PBR; most prominent among these are dicarboxylic porphyrins, such as PPIX. Further, a porphyrin's binding affinity to the PBR correlated well with that porphyrin's ability to photoinactivate V79 cells, suggesting the PBR as an important molecular target for porphyrin-based photocytotoxicity. Morgan et al.103 found that a pyropheophorbide, also localized to mitochondria, can compete with PK11195 for binding to the PBR. In contrast, Kessel et al.104 demonstrated that PPIII and PPXIII, positional isomers of PPIX, had similar uptake and localization properties as PPIX and the same ability to photosensitize L1210 leukemia cells, as measured by a clonogenic assay, but significantly lower binding affinity to the PBR. Thus, a relationship between PBR affinity and photosensitizing efficacy may not extend beyond porphyrins with structures similar to PPIX. Ratcliffe et al.105 demonstrated that PK11195 inhibited PDT-induced apoptosis in rat pancreatoma cells treated with ALA to produce PPIX. For the phthalocyanine Pc 4, which localizes preferentially to mitochondria,97,98 competition with PK11195 for binding to the PBR demonstrated both mid- and low-affinity binding sites to the PBR. However, the observed inhibition of Pc 4-PDT-induced apoptosis of CHO cells by PK11195, which occurred as well in CHO cells overexpressing human Bcl-2, was likely due to inhibition of a downstream step in apoptosis, rather than to blockade of Pc 4 binding to the PBR.106
Other components of the PTPC have also been considered as targets of photosensitization. Salet et al.107 observed strong non-competitive inhibition of the ADP/ATP exchange carrier (ANT) and inhibition of PTPC opening in isolated rat liver mitochondria when challenged with hematoporphyrin-sensitized photodynamic action. Recently, Belzacq et al.108 loaded liposomes with semi-purified PTPC or with the purified ANT and showed that Verteporfin-PDT caused permeablization of the proteo-liposomes, but not of protein-free liposomes. The ANT ligands, ADP and ATP, inhibited the response. PDT using hypericin was reported to cause the release of mitochondrion-bound hexokinase into the cytosol and to inhibit hexokinase activity.109
Inhibition of respiration and oxidative phosphorylation is also common with PDT induced by mitochondrion-localized photosensitizers.78,110 Photoactivated Photofrin inhibited electron transport components, including succinate dehydrogenase and cytochome c oxidase, and also disrupted the mitochondrial electrochemical gradient.111–114 Morgan et al.115 compared PDT responses in a human ovarian carcinoma cell line 2008 and its mitochondrial DNA-deficient derivative ET3, which lacks several respiratory protein subunits. When photosensitizers that did not primarily localize in mitochondria (Nile Blue A or Photofrin after a short incubation) were studied, both cell lines responded similarly. For Photofrin after a long incubation, when it is found in various intracellular membranes including mitochondria, again the responses of the two cell lines were similar. However, with Victoria Blue BO (mitochondrial localization), ET3 cells were less sensitive than 2008 cells. The authors suggest that the study of cells deficient in mitochondrial DNA shows that inhibition of respiratory enzymes is a target of PDT with VBBO but much less so with the other photosensitizers. In response to PDT with various mitochondrion-bound photosensitizers, cytochrome c is released from the intermembrane space into the cytosol where it very likely forms a complex with cytoplasmic apoptosis-activating factor-1 (APAF-1) to initiate the activation of the cascade of caspases that carry out the final stages of apoptosis.110,116–118 In a study of the inhibition of respiration in L5178Y-R murine lymphoma cells that were treated with Pc 4-PDT then permeabilized, Varnes et al.110 were able to restore much of the loss in respiration with exogenous cytochrome c, suggesting that the inhibition of respiration by Pc 4-PDT primarily results from the release of cytochrome c, rather than from damage to the major enzyme complexes of the electron transport chain.
The importance of mitochondria as targets for the initiation of apoptosis by PDT has been convincingly demonstrated by Kessel and Luo.81,82,118 These investigators tested a series of photosensitizers for their preferential intracellular binding sites as well as their ability to photosensitize cells to apoptosis and overall cell death. They found that those photosensitizers that bound to mitochondria induced apoptosis upon photoirradiation, whereas those that bound to the plasma membrane or to lysosomes, but not to mitochondria, killed cells less efficiently and by a non-apoptotic mechanism. In a subsequent study, Kessel and Luo119 showed that lysosomally bound photosensitizers cause the release of cathepsins from the damaged organelles; once in the cytosol, these proteases cleaved procaspase-3, thus blunting the apoptosis pathway. Since lysosomal proteases can also cleave Bid to generate a mitochondrion-targeted pro-apoptotic protein,60 PDT damage to lysosomes can induce apoptosis as well as inhibit it.
Kessel et al.120 have recently reported the ability of the bile acid ursodeoxycholic acid (UDCA) to greatly potentiate the apoptotic response to PDT with CPO and SnET2, two photosensitizers that bind to lysosomes as well as other cytoplasmic membranes. UDCA binds to mitochondrial membranes and can protect cells from the apoptotic effects of a variety of cytotoxic agents. With PDT, UDCA potentiated the loss of mitochondrial membrane potential, release of cytochrome c into the cytosol, activation of caspase-3, and apoptotic cell death in murine leukemia L1210 or hepatoma 1c1c7 cells. Since UDCA did not enhance the intracellular accumulation of the photosensitizers, Kessel et al.120 proposed that UDCA sensitized mitochondrial membranes to photodamage. There are important clinical implications of this discovery, because UDCA is currently used to treat biliary cirrhosis and other conditions in gastroenterology. If the interaction observed in cell culture occurs in PDT-treated tissue in vivo, UDCA may offer a means to promote the efficacy of PDT with minimal adverse effects.
A novel approach to enhancing the endogenous production of PPIX in mitochondria has been devised by Gagnebin et al.121 With the ultimate goal of improving PDT targeting to tumor cells through gene therapy, these investigators generated an adenovirus vector that carries a mutated gene for ALA synthase, the rate-limiting enzyme in heme biosynthesis. ALA synthase is normally subject to feedback inhibition by heme through heme regulatory motifs (HRMs) that bind heme and prevent import of the proenzyme into mitochondria. The HRMs were mutated, permitting the virally expressed proenzyme to enter mitochondria and be processed to fully active enzyme independent of the heme concentration. Following infection of H1299 lung carcinoma or Saos-2 osteocarcinoma cells with the viral construct, PPIX production was increased and the cells became photosensitive with respect to the induction of apoptosis, as revealed by flow cytometric analysis of particles with less than the G1 content of DNA. As with all gene therapy approaches, the utility of this type of vector will depend upon the ability to target it selectively to tumors in patients.
5.2 Mitochondrial DNA
Mitochondria contain some 1000 proteins that are encoded in the nuclear DNA and are imported after synthesis on cytoplasmic ribosomes; only 13 proteins (and multiple RNAs) are encoded by mitochondrial DNA.122,123 The proteins that are specified by mitochondrial DNA are specific peptide components of respiratory complexes I, III, IV, and the ATP synthase. In the absence of those peptides, respiration is slowed. However, mitochondrial DNA does not appear to be necessary for efficient induction of apoptosis by PDT with a mitochondrion-localizing porphycene (CPO), as L1210 murine leukemia cells depleted of their mitochondrial DNA by prolonged exposure to ethidium bromide were more sensitive to apoptosis induction and loss of clonogenicity with CPO and light.124 With a lysosome-targeted photosensitizer, a chlorin e6 analog (LCP), L1210 cells with or without mitochondrial DNA showed equal sensitivity to PDT. The data suggest a protective role for mitochondrial DNA or its gene product(s) against the lethal effects of PDT, but only with photodamage directed to mitochondria. A different conclusion was drawn by Singh et al.123 based on their studies of HeLa cells (Rho+) and a mitochondrial DNA-deficient (Rhoo) derivative line. Using mesoporphyrin IX as photosensitizer, they reported a >2-log loss of clonogenicity for the PDT-treated Rhoo cells, as compared to no killing of the Rho+ cells, but only in a single condition in the presence of 80 μg ml−1 suramin, but not with less suramin. The Rhoo cells were also more sensitive to adriamycin than were the Rho+ cells. Although the report of Singh et al.123 may be valid for adriamycin, which was the main subject of the study and which was active in the absence of suramin, the conclusion regarding PDT is untenable, because mesoporphyrin IX uptake into the two cell lines was not quantified, the subcellular localization(s) of the photosensitizer was not identified, and appropriate conditions for killing of both lines by PDT, without the aid of suramin, were not established. Finally, the extent of apoptosis was low at 24 and 48 hours after introduction of adriamycin, but no data on apoptosis were reported for PDT. In another study, human B lymphoma cells (Namalwa) that were depleted of mitochondrial DNA were resistant to PDT with a boronated porphyrin.125 Similarly, human ovarian carcinoma cells that were depleted of their mitochondrial DNA were less susceptible to overall cell killing by PDT with the mitochondrion-localizing photosensitizer Victoria Blue BO.115 Thus, there is still insufficient information to predict the effect of mitochondrial DNA depletion on PDT response, which may vary with cell type and photosensitizer, and especially with the subcellular localization of the photosensitizer.5.3 Chronology of the mitochondrial events in response to PDT
Often the first event observed in PDT-induced apoptosis is a dissipation of the mitochondrial transmembrane potential (ΔΨm). Kessel et al.118 reported immediate loss of ΔΨm after PDT with porphycenes in leukemia P388 cells. Similarly, PDT with hypericin or hypocrellin, also mitochondrion-localized photosensitizers, induced rapid collapse of ΔΨm.126 The mechanisms responsible for the loss of ΔΨm remain unclear, as inhibitors of PTPC opening have provided contradictory results. The decrease in membrane potential after hypericin-PDT was not prevented by specific PTPC inhibitors, such as cyclosporin A and bongkrekic acid.127 In contrast, Lam et al.98 showed that cyclosporin A produced a partial inhibition of PTPC opening in human epidermoid carcinoma A431 cells exposed to Pc 4-PDT, suggestive of a transient inhibitory effect. With a combination of cyclosporin A and trifluoperazine, PTPC opening and apoptosis were strongly inhibited.98 In another study, dissipation of the membrane potential in isolated mitochondria exposed to PDT with Victoria Blue dyes was cyclosporin A-insensitive and was proposed to result from direct effects of the dye on inner membrane permeability to protons rather than on the mitochondrial permeability transition.128Contradictory results have also been obtained regarding the timing of the release of cytochrome c from mitochondria into the cytosol relative to the dissipation of the transmembrane potential. Kessel et al.118 demonstrated that cytochrome c was released simultaneously with the loss of ΔΨm, both events occurring immediately after PDT with porphycenes in leukemia P388 cells. In contrast, photodynamic treatment of HeLa cells with Verteporfin resulted in cytochrome c translocation from mitochondria into the cytosol and accessibility of the mitochondrial 7A6 epitope to the APO2.7 monoclonal antibody without any changes in transmembrane potential.129 Release of cytochrome c was shown to occur immediately upon Verteporfin-PDT.116 Interestingly, Chiu and Oleinick130 found the relationship between these critical events for Pc 4-PDT to be dose-dependent in mouse lymphoma L5178Y-R cells. Although high irradiation fluences resulted in a rapid dissipation of membrane potential concurrent with cytochrome c release, lower fluences produced cytochrome c release without loss of transmembrane potential. Furthermore, using an immunohistochemical method which does not require cell fractionation to monitor cytochrome c release, Chiu et al.131 demonstrated a dose-dependent and nearly complete release of cytochrome c about 15 minutes post Pc 4-PDT. In contrast, in A431 cells, Pc 4-PDT-induced efflux of cytochrome c from mitochondria followed the loss of ΔΨm.98
In the absence of mitochondrial depolarization, cytochrome c release appears to be independent of PTPC opening. However, transient opening of the PTPC as a result of mild (non-PDT) oxidative stress has been observed.54 A transient opening of the PTPC may cause limited swelling of the mitochondria, sufficient to disrupt the outer membrane and release cytochrome c while maintaining the inner mitochondrial membrane potential.54 Cytochrome c is only one of many proteins of the mitochondrial intermembrane space that are released into the cytosol during apoptosis.36,37,42 A recent study132 demonstrated the release of apoptosis-inducing factor (AIF) along with cytochrome c in smooth muscle cells exposed to Verteporfin-PDT.
5.4 Caspase activation in response to PDT
As noted above, photosensitizers that localize to the mitochondria are most efficient for inducing apoptosis following PDT. Cytochrome c released from the mitochondrial intermembrane space probably binds with cytoplasmic Apaf-1 and procaspase-9, leading to auto-cleavage and self-activation of this caspase. Caspase-9, in turn, activates caspase-3, which initiates the activation of the cascade of caspases. Activation of caspase-9 was reported following PDT with Verteporfin or Pc 4.133,134 Caspase-3, considered the major executioner caspase, is activated in most cases of PDT-induced apoptosis with a number of different photosensitizers.117,135–140 Strikingly, MCF-7 human breast cancer cells that lack endogenous procaspase-3 showed no significant difference in the degree of cell death induced by Pc 4-PDT, as measured by clonogenic assay, in comparison to MCF-7 cells stably transfected with procaspase-3.134 Cytochrome c release was similar in the two cell lines. However, in the absence of caspase-3, MCF-7 cells were not able to carry out the early apoptotic program involving caspases-9 and -3; but, they underwent delayed apoptosis, which suggests alternate mechanisms following release of cytochrome c or other apoptogenic mitochondrial factors. Activation of other caspases, such as 2, 6, 7, and 8, was also shown to occur downstream of cytochrome c release in response to Verteporfin photosensitization of HeLa cells or human endothelial cells.116,141 Caspase-8 was activated in response to Rose Bengal-sensitized PDT as well.142 However, HeLa cells overexpressing the serpin-like protease inhibitor CrmA, a selective inhibitor of caspases-1 and -8, still underwent significant apoptosis following PDT with hypericin, but not in response to TNFα.88,137 Ahmad et al.143 recently reported evidence for the increased expression and secretion of Fas protein into the medium, engagement of Fas Ligand and FADD, and cleavage of procaspase-8, providing evidence that this pathway may be linked to apoptosis in A431 human epidermoid carcinoma cells exposed to Pc 4-PDT. Inhibition of the pathway by recombinant human Fas:Fc fusion protein, which blocks Fas-dependent steps, or by z-VAD-fmk, a general caspase inhibitor, partially prevented the loss of cell viability, as monitored by the MTT assay. Thus, current evidence suggests that the most common pathway for apoptosis in PDT-treated cells is that involving cytochrome c, caspase-9, and caspase-3; however, other pathways, especially that through caspase-8, may also contribute, particularly in situations where the dominant pathway is suppressed.5.5 Role of Bcl-2 family proteins in PDT response
The Bcl-2 family of proteins exercises the most proximal and important controls over apoptosis. Some, like Bcl-2 and Bcl-xL, are anti-apoptotic, while others, like Bax and Bad, promote apoptosis. The focus of action of Bcl-2 family proteins is the mitochondrion, and depending upon the initiating agent and the cell type, different members are involved in the regulatory process. A role for Bcl-2 in suppressing PDT-induced apoptosis was first shown in 1996 by He et al.144 In that study, CHO cells overexpressing human Bcl-2 were found to be more resistant to apoptosis and to loss of clonogenicity caused by Pc 4-sensitized PDT than were the companion cells expressing only the endogenous hamster Bcl-2. Granville et al.145 subsequently confirmed the ability of overexpressed Bcl-2 to suppress PDT-induced apoptosis in HL60 cells treated with BPD-MA (Verteporfin) and light. This group also found that overexpressed Bcl-2 or Bcl-xL did not prevent the release of cytochrome c from mitochondria but instead blocked the activation of several caspases.129,146 In contrast to results from studies in various cell-free systems,53 cytochrome c release in response to PDT, at least with photosensitizers that preferentially target mitochondria, appears to be unaffected by the presence of Bcl-2 or Bcl-xL.The importance of Bcl-2 in regulating PDT-induced apoptosis has been tested by transfecting an antisense Bcl-2 sequence in a retrovirus vector into human gastric adenocarcinoma MGC803 cells followed by treatment with the photosensitizer 2-BA-2-DMHA and light.147 The antisense-treated cells retained about 20% of the level of Bcl-2 protein found in the control cell lines and were more sensitive to PDT, as determined by MTT assay and by the generation of oligonucleosomal DNA fragments, than were untransfected cells or those transfected with the neo gene. Antisense Bcl-2 also sensitized A431 cells to Pc 4-PDT.148
It has been proposed that a major influence over a cell's propensity to undergo apoptosis in response to a toxic stimulus is the ratio of Bax to Bcl-2;32 thus, elevated Bax promotes apoptosis even in the presence of Bcl-2. Kim et al.89 overexpressed Bcl-2 in human breast epithelial MCF-10A cells and found that overexpression was accompanied by the upregulation of Bax. A similar observation was made by Srivastava et al.148 in A431 cells exposed to Pc 4-PDT. When MCF-10A or A431 cells were exposed to AlPc- or Pc 4-sensitized PDT, respectively, in contrast to expectations, the cells overexpressing Bcl-2 were more sensitive to the induction of apoptosis than were the parental cells.89,148 However, the CHO cells overexpressing human Bcl-2 that were used in the study by He et al.144 were found to contain the same level of Bax as the cells from which they were derived,149 and there are other instances when Bax upregulation did not follow upon Bcl-2 overexpression.150 Thus, the conditions that result in the apparent control of Bax levels by Bcl-2 remain to be defined. Interestingly, in the study by Kim et al.,89 the level of Bcl-2 detected on western blots of the PDT-treated cells was much reduced in comparison to the untreated cells; this effect was found for both cell lines. The authors interpreted their results in terms of possible photodamage to Bcl-2, thereby increasing the ratio of the pro-apoptotic Bax to anti-apoptotic Bcl-2. In a recent study of Lewis lung carcinoma (LLC) cells, Usuda et al.151 found that stable overexpression of IL-6 conferred increased sensitivity to NPe6-mediated PDT, as measured by the MTT assay. Furthermore, apoptosis was observed only in those cells expressing IL-6, but not in various control cell lines. Interestingly, the IL-6-expressing cells contained elevated amounts of both Bax and cytochrome c and similar amounts of Bcl-2, as compared to untransfected LLC cells; at 6 and 12 hours post-PDT, Bcl-2 was lost and the level of cytochrome c was markedly reduced in both cell lines, whereas Bax expression was further elevated only in IL-6-overexpressing cells, implicating Bax in the increased sensitivity to PDT. Further study is needed to determine if the ability of Bcl-2 overexpression to produce PDT resistance vs. sensitization may be a function of the degree of Bax upregulation and if photodamaged Bcl-2 may promote apoptosis by more efficiently recruiting Bax to the mitochondria.
Xue et al.149 have studied the destruction of Bcl-2 by PDT in greater detail. They found that photodamage to Bcl-2, as observed on western blots, could be induced by Pc 4-PDT in several different cell lines, including human tumor lines. The effect was observed in cell samples recovered immediately after PDT and from cells photoirradiated in the cold. Study of an epitope-tagged Bcl-2 chimeric protein revealed that the entire protein is lost, rather than the photodamage being limited to the epitope for the anti-Bcl-2 antibody. In addition to a loss of the great majority of Bcl-2, a small amount of a 23 kDa cleavage product could be detected, as well as the apparent generation of high-molecular weight products that may be the result of the photochemical crosslinking of Bcl-2 to itself or to other proteins. The 23 kDa Bcl-2 fragment has been observed in various cells at late times (hours to days) after exposure to a variety of (non-PDT) apoptosis inducers;59,152–156 in some cases, it was shown that generation of the fragment was dependent on caspase-3. However, in the case of Pc 4-PDT, the fragment was found in MCF-7 cells that do not express this caspase.149 Additional evidence against the participation of a protease in the loss of Bcl-2 was obtained with a series of protease inhibitors, none of which blocked the response. Thus, it appears that the Bcl-2 protein is a target of PDT, at least with certain photosensitizers that localize in mitochondria,89,149 and probably with photosensitizers that bind to multiple cytoplasmic membranes.157 The damage appears to be photochemical, perhaps through the oxidation of key amino acid residues by photochemically generated singlet oxygen. Thus far, it appears that only Bcl-2, and not Bcl-xL, Bad, or Bax, is destroyed by PDT. Several other mitochondrial proteins remained intact after Pc 4-PDT: cytochrome c, a subunit of cytochrome oxidase, the voltage-dependent anion channel (VDAC1), and the adenine nucleotide translocator (ANT).149 However, as mentioned above, inhibition of ANT function as a pore-forming protein has been found in ANT-containing proteo-liposomal preparations treated with Verteporfin and light.108
Pro-apoptotic members of the Bcl-2 family may contribute to promotion of apoptosis in PDT-treated cells. During the first few hours after exposure of human umbilical vein endothelial cells (HUVEC) to Verteporfin-PDT, Bax was found to disappear from the cytosol, but not from whole cell lysates, suggesting it had redistributed to mitochondria.141 This response occurred simultaneously with the redistribution of cytochrome c from mitochondria to the cytosol. At the same time, Bid cleavage was observed, such that at times later than 2 hours post-PDT, Bid could not be detected in the cells. Bid cleavage was also reported for HL60 cells treated with Rose Bengal and light.158 This latter photosensitizer is thought to bind preferentially to the plasma membrane, so it would appear that apoptosis initiated at that site can also proceed through modification of Bcl-2 family members. Bid is usually cleaved by caspase-8, which was activated by Rose Bengal-sensitized PDT; the product of this cleavage, truncated Bid (tBid), is known to promote the release of cytochrome c from mitochondria. However, Bid can also be cleaved by lysosomal proteases,60 providing a potential mechanism for cases in which apoptosis occurs after PDT with lysosomally bound photosensitizers.
Ultimately, it will be important to determine the importance of the level of expression of Bcl-2 family members in the response of tumors to PDT. Kawaguchi et al.159 examined a series of squamous cell carcinomas of the bronchus that were treated with PDT (with Photofrin I or II) for their level of Bcl-2 and p53 expression, as determined immunohistochemically. Their analysis showed no correlation of expression of either protein with PDT response or cure. In the case of p53, the results of Gomer and co-workers (see below) would predict the observed result, i.e., no correlation. In the case of Bcl-2, all analyses were carried out on pre-treatment biopsies; thus, it is not known if the treatment eliminated some or all of the Bcl-2. If PDT destroys Bcl-2, then the pre-treatment level of this apoptosis suppressor may not be relevant to the ability of the tumor cells to undergo apoptosis in response to the treatment. The level of Bcl-2 has also been measured in biopsies of esophageal tumors treated with Photofrin-sensitized PDT.160 Biopsies were taken before delivering the photosensitizer, two days later before exposure to laser light, and then three days after that. Although the study was not designed to reveal early effects of PDT on Bcl-2, there was no apparent relationship between tumor response and level of Bcl-2. A recently reported study of tissue from esophageal cancer patients treated with HpD-sensitized PDT161 provides interesting data suggesting that tumors expressing higher levels of Bcl-2 were actually more sensitive to PDT. Although the study was small, Bcl-2 expression levels, but not p53 levels, were positively correlated with better relapse-free and overall survival of the patients. The critical question of the relevance of the Bcl-2 proteins to human tumor response to PDT needs considerable additional data from clinical studies.
6 Signaling pathways that can regulate PDT-induced apoptosis
PDT has been found to upregulate numerous signaling pathways (reviewed in ref. 39 and 42). The induction of apoptosis by most physiological stimuli or toxic agents proceeds through series of signaling pathways. Thus, DNA-damaging agents, such as ionizing radiation, require the signaling of damage by p53-dependent steps that block cell cycle progression and upregulate transcription of multiple genes eventually leading to apoptosis after one or more cell divisions.162,163 In contrast, Fas ligand acts by binding to its receptor and recruiting components of death-inducing complexes that directly activate caspase-8 (see above). PDT activates many signaling reactions that have been characterized in response to other oxidative stresses or physiological stimuli. It is therefore appropriate to ask which of the many reactions are mediators or promoters of apoptosis in PDT-treated cells, which are stress responses whose function is to promote repair or tolerance of damage, and which have no obvious relationship either to the cell death process or to its prevention. In order for a reaction to be a mediator of apoptosis, certain criteria must be met. First, the circumstances under which both apoptosis and the reaction are induced must be defined and agree with respect to dose and timing. Second, a simple demonstration of a correlation is insufficient to prove that the reaction is a cause or an effect of apoptosis. To demonstrate that an enzymatic reaction is necessary for PDT-induced apoptosis, the ability of PDT to induce apoptosis must be studied after inhibition of the enzyme either chemically or using dominant-negative, knock-out, or anti-sense technology. The goal in this review is to focus on those pathways that have been demonstrated to affect PDT-induced apoptosis. Therefore, reactions that are enhanced or inhibited by PDT but where no data are available to suggest an interplay of the pathway with apoptosis in PDT-treated cells will not be considered here.6.1 Ca2+
PDT causes both transient and more sustained increases in intracellular free calcium concentration [Ca2+]i, which can come from release of intracellular stores of Ca2+, mostly in the endoplasmic reticulum, and from an influx of extracellular Ca2+ (reviewed in ref. 164). The first demonstration of an association of a rapid, transient increase in [Ca2+]i with apoptosis was made by Agarwal et al.,165 studying PDT with the photosensitizer AlPc in mouse lymphoma L5178Y-R cells. The pulse of Ca2+, as observed with the Ca2+-sensitive fluorescent probe, Indo-1, reached a maximum within 1 min after PDT, followed an earlier increase in phospholipase C (PLC) activity, and was prevented by a PLC inhibitor but not by chelation of extracellular Ca2+ with EGTA. Inhibition of PLC also prevented the induction of apoptosis in these cells. The data showed that Ca2+ release from the endoplasmic reticulum was a result of PDT-induced stimulation of PLC activity and was necessary for the resultant apoptosis. In contrast, the addition of the chelator of intracellular free Ca2+, BAPTA-AM, did not alter the appearance of apoptotic DNA fragmentation in Pc 4-PDT-treated HIV-infected cells.69Several recent papers have further explored the relationship between the rise in [Ca2+]i and the induction of apoptosis by PDT.117,166,167 In human squamous cell carcinoma HSC-2 cells, PDT with Photofrin produced a small increase in [Ca2+]i by 30 min and a strong increase by 1–2 h post-PDT, as observed by confocal microscopy.166 The latter changes coincided with a dramatic increase in apoptotic cells, quantified by flow cytometry, which reached a maximum at 2 h. Inanami et al.117 observed a similar time-course for apoptosis in Chinese hamster V79 cells exposed to PDT with the photosensitizer pheophorbide a (PPa). In addition to detection of DNA fragmentation by flow cytometry and by agarose electrophoresis, these investigators documented the release of cytochrome c from mitochondria and the activation of caspase-3. Apoptosis was inhibited by azide, a scavenger of 1O2, by the [Ca2+]i chelator, BAPTA, by Ac-DEVD-CHO, an inhibitor of caspase-3-like activity, and by forskolin, which can increase the cellular level of cyclic AMP. Because increases in [Ca2+]i by a calcium ionophore can activate the apoptosis pathway through release of cytochrome c, these investigators concluded that [Ca2+]i was responsible for that mitochondrial step. However, because forskolin did not inhibit cytochrome c release, it was felt that cyclic AMP and Ca2+ were acting through different pathways to regulate apoptosis.
In a recent study by Rück et al.,167 apoptosis was induced in rat bladder epithelial cells by PDT with AlPcS4, and [Ca2+]i was followed by confocal microscopy using the calcium-sensitive dye Fluo-3. Under conditions in which the photosensitizer was located mainly in the endoplasmic reticulum (24 h incubation), low light fluences promoted cell growth, while higher fluences caused apoptosis, as indicated by Annexin V labeling of the plasma membrane. A transient elevation of [Ca2+]i, especially in the nucleus, occurred during the irradiation period, followed by a sustained increase. The antioxidant PDTC blocked the changes in [Ca2+]i and the appearance of apoptosis, indicating reactive oxygen species are involved in both endpoints. Thus, the available information implicates [Ca2+]i as one signal contributing to the induction of apoptosis by PDT with several different types of photosensitizer.
6.2 Lipid-derived second messengers: ceramide
Stress-induced activation of sphingomyelinase leads to generation of ceramide, an important lipid mediator of apoptosis.168 Recent evidence suggests that ceramide also plays a role in PDT-induced apoptosis. A series of studies by Separovic et al.72,169,170 showed that Pc 4-PDT resulted in elevated levels of ceramide in L5178Y-R cells that were rapid and correlated well with the fraction of cells undergoing apoptosis. Increased levels of ceramide following Pc 4-PDT were also shown for CHO and U937 cells, and the addition of N-acetylsphingosine, a cell-permeable C2-ceramide, mimicked the apoptotic response of PDT in both cell lines. Niemann-Pick disease lymphoblasts, which are deficient in acid sphingomyelinase, the enzyme that catalyzes the initial step in the sphingomyelin pathway, failed to respond to Pc 4-PDT with ceramide accumulation and apoptosis. Combined treatment of the cells with bacterial sphingomyelinase and Pc 4-PDT restored ceramide production and apoptotic response. Nevertheless, bacterial sphingomyelinase is not cell permeable and thus acts at the cell membrane. As Pc 4 does not localize to the plasma membrane,97,98 the mechanism by which the combined treatment restored apoptosis remains unclear.Matroule et al.171 reported acid sphingomyelinase activation, ceramide accumulation, and NF-κB activation after photosensitization of human colon carcinoma HCT-116 cells with pyropheophorbide-a methyl ester (PPME), again suggesting that acid sphingomyelinase may be another target for the cellular response to PDT. Using chloroquine, an acid sphingomyelinase inhibitor, these authors showed significant inhibition of PPME-PDT-induced activation of the NF-κB transcription factor, suggesting that sphingomyelinase activity and generation of ceramide are responsible for NF-κB activation. Nevertheless, these authors did not report chloroquine effects on the level of ceramide. Of note, chloroquine is a cell-permeable base that prevents acidosis which often precedes apoptosis in response to many stimuli (IgM anti-Fas antibody, cycloheximide, or ultraviolet irradiation)172 and also inhibits phospholipase A2 activity;173 hence non-specific effects cannot be excluded.
Nagy et al.174 demonstrated a rapid increase in ceramide levels in A431 cells exposed to Pc 4-PDT that correlated well with induction of apoptosis. Interestingly, a simultaneous inhibition of acid sphingomyelinase activity was also reported. With the aid of fumonisin B1, an inhibitor of ceramide synthase, the enzyme that catalyzes de novo synthesis of ceramide, the authors showed that despite complete inhibition of ceramide production, apoptosis following PDT was not blocked. Nevertheless, fumonisin B1 is also non-specific and can alone induce apoptosis.175 Embryonic fibroblasts from FADD-knockout mice were as efficient as those from mice with wild-type FADD in generating ceramide in response to Pc 4-PDT, although the FADD−/− cells were deficient in ceramide production in response to TNFα.176 Further, ceramide production was found to accompany but to be non-essential for apoptosis after Pc 4-PDT or TNFα.176 Although evidence to date is suggestive, the role of ceramide in PDT-induced apoptosis remains to be elucidated.
6.3 Cell surface death receptors
It is well established that PDT induces expression and secretion of TNFα.39,42 Recently, there has been some research into the possible involvement of the Fas or TNFR-mediated pathways in the induction of apoptosis in response to PDT. Jiang et al.76 found that either Verteporfin-PDT or anti-Fas antibody selectively depleted CD4+CD8+ cells from a pool of mature thymocytes, and the combined treatments augmented apoptosis in the pooled population, as assessed by caspase-3 activity, PARP cleavage, and DNA fragmentation. Ahmad et al.143 found a marked increase in expression of Fas in A431 cells during the first hour after Pc 4-PDT; this was followed by a decrease that corresponded with an increase of Fas in the culture medium. Evidence was also provided for the multimerization of Fas within the cells, for an increase in FasL and FADD expression, for the interaction of FasL with FADD, and for the activation of caspase-8. Exposure of the cells to either a blocking antibody or the general caspase inhibitor zVAD-fmk reduced the level of cell killing detected by the MTT assay within 6 or 12 hours post-PDT. It should be noted that caspase-8 is also activated by PDT downstream of cytochrome c release, presumably by caspase-3-dependent cleavage (see section 5.4). In a study of Hep-2 carcinoma cells, Yslas et al.177 demonstrated that PDT sensitized by 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin led to apoptosis, under which conditions Fas antigen was also expressed in the cells. These data argue for participation of Fas-mediated apoptosis in the response to PDT.In A431 cells, Azizuddin et al.178 found that Pc 4-PDT induced TNFα production but that recombinant human TNFα did not interact with PDT in the induction of apoptosis. A test of the importance of the death receptor pathways was conducted by Nagy et al.,176 using primary embryonic fibroblasts derived from mice expressing wild-type FADD and from FADD knock-out mice. They found that Pc 4-PDT induced similar levels of apoptosis (TUNEL-positive cells) and caspase-3 activity in both types of cells, although the FADD knock-out cells were resistant to the induction of apoptosis by TNFα, arguing that PDT can induce apoptosis in the absence of functional FADD. Thus, the role of pathways for apoptosis initiating with cell surface death responses in the response of cells to PDT is still controversial. It has been suggested that these pathways may be upregulated in response to cell damage and regulate stress responses (see Section 6.4).
6.4 Nuclear factor-kappa B (NF-κB)
NF-κB is a transcription factor responsible for promoting expression of genes for several proteins involved in immunoregulatory and proinflammatory processes.179,180 Evidence for regulation of apoptosis through transcription of anti-apoptotic genes has also been obtained. NF-κB normally exists as a dimer residing in the cytoplasm in association with an inhibitory subunit, one of several IκB factors. Stimulation by TNFα leads to phosphorylation of specific serine residues of IκB, ubiquitination of specific lysine residues, and targeting of the protein for degradation by the 26S proteasome. Upon release of the inhibitory subunit, the remaining dimer can enter the nucleus to promote transcription of genes containing NF-κB binding motifs. The most common NF-κB dimer is composed of a p50 and a p65 (RelA) species, of which there are several variants. One of the genes whose transcription is stimulated is that for IκB; resynthesis of the inhibitory subunit thus restores NF-κB to its baseline state.PDT produces an oxidative stress that can result in activation and translocation of NF-κB to the nucleus, as originally shown for treatment of L1210 murine leukemia cells with Photofrin and light.181 Although NF-κB activation was not observed in HeLa cells exposed to Photofrin-PDT,182 PDT with methylene blue was demonstrated to activate and translocate NF-κB to the nucleus.183 A similar NF-κB response was also found for treatment of HIV-infected lymphoid ACH-2 cells by photosensitization with the DNA-intercalating drug proflavine.184
Recently, new insights have been obtained into the importance of the NF-κB response in PDT-induced apoptosis. Using PPME as photosensitizer, Matroule et al.171 demonstrated that PDT recruits the IL-1 receptor-dependent signaling pathway to activate NF-κB in HCT-116 human colon cancer cells. This group then developed a new aminopyropheophorbide (APP) derived from PPME and showed that it bound to membranes but not to mitochondria of HCT-116 cells.185 Using an electrophoretic mobility shift assay with an oligonucleotide containing the NF-κB binding motif, they demonstrated that following PDT, nuclear extracts contained elevated levels of material that could retard migration of the oligonucleotide. The increase in nuclear NF-κB activity was biphasic, as demonstrated by an early (10–30 min) activation of NF-κB, which was followed by a decrease at 1 hour and a second wave of activity at 2–24 hours after PDT. Competition by various peptides confirmed that the activity was the p50/RelA heterodimer. To study the relationship of the NF-κB response to the induction of apoptosis, Matroule et al.185 constructed HCT-116 cell lines expressing mutated IκB; one such mutant (Y42F) expressed IκB with tyrosine 42 changed to phenylalanine, and the other (S32,36A) expressed IκB in which serines at positions 32 and 36, the serines known to be phosphorylated as a prerequisite for inactivation of the inhibitor, were converted to alanine. Activation of NF-κB by PDT was equally efficient in the Y42F-expressing line as in the parental cells, but there was no significant activation in the S32,36A-expressing line, demonstrating that phosphorylation of the two essential serines was necessary for release of the active NF-κB heterodimer from inhibition by IκB. HCT-116 S32,36A cells were much more sensitive to the induction of apoptosis by APP-PDT than were the parental cells, as demonstrated by caspase-3 activity, TUNEL assay, and chromatin condensation. Finally, activation of a κB-luciferase reporter construct provided further evidence for the PDT-induced activation of NF-κB. Thus, these studies confirmed that NF-κB provides an anti-apoptotic signal for cells exposed to PDT with a photosensitizer that does not localize in mitochondria.
In a follow-up study, Granville et al.133 extended the results to human promyelocytic leukemia HL-60 cells using the materials produced for the study of Matroule et al.185 and the photosensitizer Verteporfin. They also showed that NF-κB levels were increased in the nuclei of cells undergoing apoptosis, but that cellular IκB levels decreased; however, in spite of demonstrated activation of caspases-9 and -3 in the PDT-treated cells, it is unlikely that IκB was cleaved by caspase-3, since the loss of this inhibitory subunit was not prevented by the general caspase inhibitor zVAD-fmk. This may indicate that degradation of IκB occurred through the 26S proteasome.
A recent detailed study of the role of NF-κB activation in the response of HCT-116 cells to PPME-sensitized PDT has been reported.186 In this system, N-acetylcysteine and PDTC protected against apoptosis, but D2O (which enhances the lifetime of 1O2) increased necrosis but not apoptosis, implicating ROS other than 1O2 in the induced apoptosis. Evidence for the participation of mitochondria (activation of caspases-9 and -3, PARP cleavage, DNA fragmentation) was obtained; however, in spite of the localization of PPME in lysosomal and ER/Golgi membranes, no evidence was found for the participation of Ca2+, Bid, Bap31, or caspase-12. Likewise, IL-1 receptor signaling and ceramide appeared not to participate in PPME-PDT-induced apoptosis. Although Bcl-2 was converted by PDT to a more slowly migrating (phosphorylated?) form, transient transfection of the cells with either wild-type Bcl-2 or either of two dominant negative Bcl-2 mutants did not affect the appearance of apoptotic endpoints. This study confirms the earlier conclusion that NF-κB is activated by PPME-PDT but is distinct from the pathway leading to apoptosis.
6.5 Mitogen-activated protein kinases (MAPKs)
The MAPKs belong to a well-studied family of signaling pathways that function in all cells to convert various extracellular stimuli into intracellular responses, generally through regulation of genes controlling cell growth and responses to stress.187 There are at least three major pathways, each named for the terminal MAPK of a cascade of three kinases: ERK 1/2, SAPK/JNK, and p38/HOG. Each MAPK is phosphorylated on threonine and tyrosine by a dual-specificity MAP kinase kinase, which is activated on serine and threonine by a MAP kinase kinase kinase, itself activated through a variety of upstream signals. Extracellular signal-regulated kinase (ERK) is activated when certain growth factors engage their membrane-localized receptors; this pathway proceeds through sequential activation of Ras, Raf, and MEK, culminating in activation of ERK1 and ERK2. The other two major pathways are activated by a variety of cell stresses, including UV or ionizing radiation, osmotic stress, and withdrawal of certain growth factors. The stress-sensitive pathways are those culminating in stress-activated protein kinase (SAPK), also known as c-Jun N-terminal kinase (JNK), and p38/HOG (hyperosmotic glycerol) kinase. The upstream kinases in the SAPK/JNK pathway are MKK4/SEK1 and MKK7, whereas p38/HOG kinases are activated by MKK3 and 6.187 The identified substrates of each of the MAPKs are transcription factors: c-Jun, ATF2, and Elk1 in the case of SAPK; ATF2, Max, CREB, and Elk1 in the case of p38; and Elk1 and Stat3 in the case of ERKs.A link between the SAPK and p38 pathways and apoptosis has been suggested in several studies.188–190 For PDT, there is evidence linking the p38 pathway with apoptosis. The rapid activation of both the SAPK and p38 pathways, but not ERKs, has been observed in cells responding to PDT with BPD-MA,191 Pc 4,189 PPIX synthesized endogenously from ALA,192 and Rose Bengal.158 In three cases, the relationship of p38 activation to PDT-induced apoptosis was tested using an inhibitor of p38 phosphorylation. Inhibition of p38/HOG by SB202190 was found to partially inhibit apoptosis in L5178Y-R and CHO cells photosensitized with Pc 4.189 In contrast, Klotz found that inhibition by SB202190 promoted apoptosis in response to PPIX-PDT.192 With Rose Bengal as photosensitizer, PDT-induced activation of p38 led to activation of caspase-8 and cleavage of Bid, which suggests a mechanism for the intersection of this stress kinase pathway with the mitochondrial pathway for apoptosis.158 However, as with all chemical inhibitors, there is a question concerning their specificity. In the future, it will be necessary to address the role(s) of the stress kinases using genetic methods for eliminating selected activities in the stress kinase pathways.
Among the many known signal transduction pathways, phosphatidylinositol-3 kinase (PI3-kinase) has been shown to promote cell survival and resistance to apoptosis.193,194 The role of this pathway in apoptosis in response to Pc 4-PDT was investigated by modulating the activity of epithelial and endothelial derived tyrosine kinase (Etk), a newly discovered tyrosine kinase that is a substrate of PI3-kinase.195 Parental LNCaP human prostate cancer cells and two derivative cell lines, one overexpressing wild type Etk (Etkwt) and the other expressing a dominant-negative Etk (EtkDN), were used to study the function of Etk. As measured by DNA fragmentation and by cleavage of PARP, the extent of apoptosis was much reduced in Etkwt cells as compared to LNCaP or EtkDN cells. Assay of overall cell viability confirmed that Etkwt cells were considerably less sensitive to PDT than were the parental LNCaP or EtkDN cells. The results suggest that the PI3-kinase/Etk pathway is involved in the protection of prostate carcinoma cells from apoptosis in response to PDT. The mechanism for the protection is not yet known.
The phosphorylation of multiple cellular proteins has been observed on western blots using an anti-phosphotyrosine antibody. Two major phosphoprotein bands (p80 and p55) were observed in extracts of L5178Y-R cells immediately after Pc 4-PDT, one of which (p80) was identified as HS1, a substrate of nonreceptor-type src protein tyrosine kinases.73 Two other proteins (p>200 and p15) were found in murine P815 mastocytoma cells immediately following Verteporfin-PDT.196 In neither case could the protein tyrosine phosphorylations be linked directly with apoptosis, but evidence obtained with the protein tyrosine kinase inhibitor genistein suggested that the events in the L5178Y-R cells may be part of a stress response.73
6.6 p53
The response of many cells to DNA-damaging agents, such as ionizing radiation, is controlled by the product of the p53 tumor suppressor gene. Thus, in response to DNA damage, cells with wild-type p53 either become delayed in progression through the cell cycle to undergo repair of the damage or are directed into apoptosis. Cells with mutant or no p53 are deficient in these responses. With respect to PDT, it is known that cell lines with mutant p53 or lacking p53 can undergo apoptosis (e.g., L5178Y-R cells;66 V79 cells117). In order to determine whether the sensitivity of cells to the lethal effects of PDT or their ability to undergo apoptosis was affected by the expression of p53, Fisher et al.197–199 compared responses within several sets of closely related human tumor cells. The presence of functional p53 was demonstrated by the accumulation of p53 protein in PDT-treated cells, by the p53-mediated upregulation of p21, and by the dephosphorylation of the Rb protein. Human promyelocytic leukemia HL60 cells expressing wild-type p53 were more sensitive to cell killing by PDT, with either Photofrin or SnET2 as the photosensitizer, than were HL60 cells in which the p53 genes were deleted or inactive. All of these cell lines underwent rapid apoptosis in response to PDT.197 A human colon carcinoma cell line expressing wild-type p53 (LS513) was more sensitive to PDT than another colon carcinoma cell line with mutated p53.198 Although those studies provided evidence that p53 can sensitize cells to PDT-induced apoptosis, introduction of the viral oncoprotein E6 to abrogate wild-type p53 function of the LS513 cells or of MCF-7 cells did not alter their PDT sensitivity to loss of clonogenicity or induction of apoptosis, leading to the conclusion that PDT sensitivity is not p53-dependent.199The role of p53 in PDT response was further explored by Zhang et al.200 in the p53-null HT29 human colorectal carcinoma cell line using a hypocrellin A derivative as photosensitizer. When wild-type p53 was transfected into these cells, it caused growth inhibition but did not induce apoptosis; however, the transfected cells were more sensitive to the induction of apoptosis after PDT. As judged by an MTT assay, the p53-expressing cells were approximately two-fold more sensitive to PDT (i.e., 50% loss of viability required 9 J cm−2vs. 18 J cm−2 for the non-transfected cells or for cells transfected with the neo gene). However, in the absence of a clonogenic assay, it would appear that the apparent increase in sensitivity to killing of the p53-transfected cells may reflect a reduced rate of death that parallels their slower growth kinetics. Recently, Tong et al.201 reported a comparison of normal human fibroblasts that express wild-type p53, immortalized fibroblasts from a patient with Li-Fraumeni syndrome, in which the only p53 allele was mutated, and the Li-Fraumeni cells expressing wild-type p53 introduced by an adenovirus vector. The normal cells were significantly more sensitive than the Li-Fraumeni cells to PDT with Photofrin, as determined by clonogenic assay; transfection of p53 modestly enhanced the sensitivity of Li-Fraumeni cells. In this system, there was no apoptosis in the normal fibroblasts in response to PDT, and a low level of apoptosis in the Li-Fraumeni cells, which must have occurred through a p53-independent mechanism. The major conclusion from these data is that neither PDT-induced cell killing nor PDT-induced apoptosis has marked dependence on p53 function.
6.7 p21 and cell cycle controls
Mukhtar and co-workers have investigated a series of signaling reactions as potential explanations for the difference in response of two cell lines to Pc 4-PDT: murine RIF-1 fibrosarcoma cells, which often die without expressing apoptotic characteristics, and human A431 epidermoid carcinoma cells, which undergo apoptosis. The two cell lines were treated with the same external dose of Pc 4 and the same red light fluence. Gupta et al.202 found an early (maximum at 5 min) increase in nitric oxide formation that was greater in A431 cells than in RIF-1 cells, as well as a greater increase in the activity of nitric oxide synthase (NOS) in A431 cells after PDT. When they assayed for the amount of constitutive NOS on western blots, both cell lines revealed a strong upregulation of the protein in cells exposed to Pc 4 only, but little if any additional change upon photoirradiation. Further studies of A431 cells showed that the chosen PDT condition results in slow entry of the cells into apoptosis, such that the majority was apoptotic by 12 hours after treatment.203 During the progression into apoptosis, changes in the cell cycle distribution of the population were noted, i.e., an increase in the G0–G1 phase at the expense of cells in S phase by 12 hours. These changes were preceded by an increase in p21/WAF1/CIP1 and downregulation at both the protein and activity levels of cyclins D1 and E and cyclin-dependent kinases cdk2 and 6. Since these data suggested that Pc 4-PDT-treated A431 cells experienced a delay in passing the G1–S cell cycle checkpoint, Ahmad et al.204 explored subsequent steps in the pathway for regulating that checkpoint and found a decrease in the hyperphosphorylated form of pRb and downregulation of five members of the E2F family of transcription factors and their heterodimeric partners DP1 and DP2. Collectively, the data suggest that when A431 cells are exposed to Pc 4-PDT, the cells upregulate a pathway for delaying cell cycle progression prior to the G1–S border. The timing of the responses was consistent with the cells needing to delay at the G1 checkpoint prior to entering the final stages of apoptosis. It will be of interest in the future to study cells in which steps in the pathway are abrogated, e.g., by knockout of one or more of the proteins, to determine if the cells are prevented from undergoing apoptosis and further if they are rescued from overall cell death by disabling the pathway. Finally, Kalka et al.205 demonstrated that the glycoprotein clusterin was also upregulated in dying A431 cells but not in RIF-1 cells. Thus, this series of papers presents evidence for the activation of several pathways in response to Pc 4-PDT during the process of apoptosis. Which of these pathways are causative for the observed apoptosis is not clear. It is possible that all participate in sub-populations of the cells, or that they are stress responses turned on by lethally damaged cells in an attempt to recover from the damage.7 Apoptosis in PDT response in experimental tumors in vivo
Evidence for the occurrence of apoptosis in PDT-exposed tumors has been obtained for many different tumor types and photosensitizers, including human tumors grown as xenografts in athymic nude mice (Table 1). In addition to observation of apoptotic cells in stained tumor sections by light or electron microscopy, apoptosis has been verified by morphometric analysis,206 by TUNEL-positivity,174,207,208 by DNA fragmentation observed after agarose electrophoresis of extracted tumor DNA,84,209,210 and by western blot analysis of tumor proteins for PARP cleavage.208,209 Although the time course for the occurrence of apoptosis varies with the tumor, the photosensitizer, and the PDT dose, generally apoptosis has been found within the first several hours following PDT, and is considered an early event, preceding the appearance of extensive necrosis and tumor regression.| Tumor | Photosensitizer | Reference |
|---|---|---|
| RIF-1 fibrosarcoma | Pc 4 | 210 |
| HeLa cell xenografts | HpD | 206 |
| Chemically induced squamous papillomas | Pc 4 | 213 |
| Colon-26 | ALA-PPIX | 214 |
| MS-2 fibrosarcoma | Zn(II) Pc | 215 |
| EMT6 | LuTex | 84 |
| OVCAR-3 xenografts | Pc 4 | 209 |
| Human pancreatic carcinoma xenografts | PPa | 216 |
| A673 sarcoma xenografts | Photofrin | 211 |
| RIF-1 fibrosarcoma, murine skin papillomas | Pc 4 | 205 |
| VX2 | Photofrin, SnET2, ALA/PPIX, m-THPC | 212 |
| SW480 xenografts | Pc 4 | 208 |
| NR-S1 squamous carcinoma | Photofrin | 207 |
Recently, there have been several attempts to more carefully quantify and locate apoptotic cells in tumors following PDT. In human A673 sarcoma xenografts in athymic nude mice, Engbrecht et al.211 monitored the responses following Photofrin-sensitized PDT. They observed progressive blood vessel occlusion, as has been found for other tumors treated with Photofrin-PDT,10 and progressively increasing numbers of apoptotic (TUNEL-positive) cells from 4–36 hours after treatment. Patches of necrosis that were present before treatment did not increase during the same post-treatment period. The earliest evidence of apoptosis in this tumor model was associated with endothelial cells or in the vicinity of occluded vessels. Since Photofrin-PDT primarily targets the tumor vasculature, Engbrecht et al.211 suggested that tumor cell apoptosis was a secondary consequence of the hypoxia produced when blood flow was interrupted.
Yokota et al.207 exposed mouse NR-S1 squamous carcinomas implanted in C3H/HeNCrj mice to PDT sensitized by Photofrin. They excised, fixed, and sectioned the tumors at various times up to 48 hours after treatment, and identified apoptosis by the TUNEL assay and Fas and FasL by immunohistochemistry, in addition to locating areas of necrosis. Under the chosen conditions, PDT increased the area of necrosis, the number of TUNEL-positive tumor cells, and the number of tumor cells expressing Fas or FasL, reaching maxima at 24, 12, and 12 hours post-treatment, respectively. The time course was interpreted as suggesting that necrotic areas may derive from apoptotic ones. The Fas-expressing cells were found in the same non-necrotic tumor areas as the TUNEL-positive cells, and FasL-expressing cells were found in tumor cells surrounding the TUNEL-positive cells. A significant increase in FasL-positive lymphocytes was also observed at 12 hours, but these cells only rarely infiltrated into areas of TUNEL-positive tumor cells. Yokota et al.207 suggest that the Fas–FasL system may mediate the apoptotic response of tumors to PDT in an autocrine or paracrine fashion; however, in the tumor model, lymphocytes seemed to play only a minor role, if any.
The findings of Yokota et al.207 are important for relating in vivo observations to mechanisms of PDT response and deserve to be examined in other models and with other photosensitizers. Interestingly, the observation of a high incidence of apoptosis in areas of the tumor surrounding necrotic areas confirms similar findings by Lilge et al.,212 who quantified the incidence of apoptosis in normal brain and intracranial VX2 tumor 24 hours following PDT with 5 different photosensitizers (Photofrin, ALA/PpIX, AlPc, SnET2, and m-THPC). After staining apoptotic cells by the TUNEL assay, confocal microscopy was used to quantify the local incidence of apoptosis and its spatial distribution. The incidence of apoptosis was confirmed by histopathology. PDT with each photosensitizer except AlPc produced apoptotic cells near the region of coagulative necrosis by 24 hours. The apoptotic response did not appear to be related to photosensitizer dose. The results suggested that areas receiving high PDT doses became necrotic and surrounding tissue went through apoptosis.
Mechanisms for the induction of apoptosis by PDT in tumors in vivo may involve certain signal transduction pathways being identified in vitro. Studying human tumor xenografts in athymic nude mice, Colussi et al.209 found p21 upregulation in response to Pc 4-PDT in human ovarian cancer OVCAR-3 xenografts that express wild-type p53. However, in human colon cancer SW480 xenografts, that are p53 null, Whitacre et al.208 demonstrated rapid post-Pc 4-PDT phosphorylation (activation) of the p38 stress kinase, but no response of p21. Both tumors responded well to PDT. Thus, in agreement with data obtained on other tumor cells in vitro,197,199 p53-dependent responses may occur but appear to be unnecessary to elicit apoptosis in response to PDT in vivo.
8 Conclusions
1. It is unlikely that there is a single universal mechanism for the response of cells to PDT. The responses vary with the cell type and its genetic and metabolic potential, as well as the photosensitizer and, importantly, its sub-cellular localization. The responses also vary with the overall dose of PDT (e.g., sub-lethal vs. lethal vs. supra-lethal), since gene induction that requires many hours post-PDT is unlikely to occur or be relevant to cells that undergo apoptosis within a few hours.2. PDT stimulates multiple signal transduction reactions, some but not all of which have been discussed above. Current evidence implicates certain signals as promoters of cell death (e.g., Ca2+, p38), whereas others seem to provide a protective mechanism for cells (e.g., NF-κB). For still others, the roles appear to depend on the context. It is possible that multiple pathways can influence cell fate. Based on the available data, we have grouped the various responses and signals discussed above into 3 categories (Fig. 3): (a) those that are direct targets or modifiers of oxidative damage produced by PDT; (b) those that can potentially alter the ability of a PDT-damaged cell to live or die; and (c) those that determine whether a lethally damaged, “doomed” cell undergoes apoptosis or necrosis. Since the data needed to assign roles to these reactions are still limited, it is possible that certain reactions have more than one role; e.g., reactions in group b may also affect the choice of cell death pathway.
 | ||
| Fig. 3 A model of the controls over PDT-induced cell death. PDT produces initial photodamage to molecular targets, one of which can be Bcl-2. Scavengers of reactive oxygen species and other factors that can alter the amount or distribution of initial photoproducts determine the extent of the initial cellular damage. If damage is sufficiently high, the cell is doomed to die. However, various pathways (e.g., NFκB) can influence tolerance or repair of the damage that may allow some of the cells to survive the damage. Once a cell is doomed, it may die by apoptosis or necrosis, and the choice will depend upon the presence of the needed caspases, Bcl-2 family members, and other enzymes of apoptosis. Some pathways indicated as affecting the conversion of a damaged to a doomed cell may also affect the choice of pathway for eliminating the doomed cell. | ||
3. There are now numerous examples of cells that die in response to PDT without undergoing apoptosis. When very high PDT doses are involved, it is likely that excessive damage has occurred such that cells lyse or that components of the apoptotic pathway are damaged, preventing the process. In a limited number of cases, e.g., MCF-7 cells that lack the gene for procaspase-3, a component of the apoptosis pathway is known to be defective and accounts for the relative resistance to apoptosis. Even in this case, other caspases are present and may be responsible for the delayed appearance of apoptotic cells. Furthermore, it is well established that PDT can activate multiple caspases, including caspases-2, -6, -7, and -8, and these may be responsible for the apoptosis that can occur in the absence of caspase-3.
4. Photoactivation of a sensitizer in or influencing the mitochondria leads to rapid apoptosis, presumably as a result of the release of cytochrome c into the cytoplasm where it can interact with APAF-1 and activate the apoptotic caspases. In many cases, this appears to be the central pathway for induction of apoptosis by PDT. However, the observations of multiple pro- and anti-apoptotic pathways that are elicited by PDT suggest that the central pathway can be modulated strongly by such conditions as the relative levels of pro- and anti-apoptotic Bcl-2 homologs and by changes in components of mitochondrial membranes, such as the PTPC. For photosensitizers localized in lysosomes, cathepsins that are released upon photodamage can cleave Bid, which promotes apoptosis at the mitochondria, and/or cleave caspase-3, which inhibits apoptosis.
5. Resistance to apoptosis is not necessarily the same as resistance to death. Many workers in the field equate apoptosis with cell death. If that is true, than cells blocked in the ability to undergo apoptosis should remain alive, not just for a few days but for an unlimited time. In the few cases where this idea has been tested, the results suggest that apoptosis is not the process by which PDT-treated cells die, but rather the process by which lethally damaged cells are dismantled. Whether this conclusion is true for all photosensitizer and cell combinations is not clear at present. In order to evaluate the importance of any apoptosis-related reaction to overall cell death, knowing the total number of killed cells is important, and this requires using clonogenic assays for quantification of cell killing whenever possible, as viability assays are influenced by growth delays and cell loss post-treatment and are unreliable when the level of killing exceeds ∼90%.
6. PDT appears to be able to overcome or bypass many of the blocks to cell killing that limit the effectiveness of ionizing radiation and chemotherapy. One hypothesis to explain the apparent success of PDT in killing otherwise resistant cells is that PDT triggers apoptosis at a late stage in the process, often by directly damaging mitochondria. An important question for PDT is: what is the critical PDT-induced lesion(s) or step(s) which kill the cell and from which it cannot recover? One molecular target of PDT in vitro is the Bcl-2 protein. Photodamage to Bcl-2, which includes photocrosslinking as well as photo-induced cleavage, may limit the effectiveness of this important anti-apoptotic control. The direct targeting of Bcl-2 by PDT with certain photosensitizers can explain the ability of PDT to overcome the resistance to apoptosis afforded by overexpression of Bcl-2. For photosensitizers that do not cause photodamage to Bcl-2, there must be other (as yet unidentified) molecular targets. In some cases, control of PDT-induced apoptosis may depend on the ratio of Bax to Bcl-2 protein, although the universality of this notion is in question. Certainly, where cytochrome c release from mitochondria is an immediate response to PDT, a slower recruitment of Bax to the mitochondria cannot drive that response. However, when Bcl-2 is a phototarget, tumor cells with abnormally high levels of Bcl-2 may be sensitized to PDT even though they are resistant to most chemotherapeutic agents and radiation.
7. There are limited data from clinical studies to test the role of apoptosis and Bcl-2 or other molecular targets in PDT response. Where the dominant effect of PDT is vascular, the levels of molecular targets within the malignant cells may not be relevant; however, where some or all of the tumor response depends upon direct photodynamic action on the malignant cells, the possible sensitization of tumor cells to PDT by Bcl-2 may offer a distinct advantage. Because of the potential of PDT to overcome treatment resistance of tumors, further work on apoptosis and the identification of specific target molecules, such as Bcl-2, in PDT-treated human tumors may be exceptionally fruitful. It is suggested that pre-treatment clinical samples need to be compared to samples taken as soon after treatment as possible, so that the early responses can be recorded before major changes in the cellular content of the tumors blurs those responses.
9 Abbreviations
Abbreviations are given in Table 2.| AIF | Apoptosis inducing factor |
| ALA | 5-Aminolevulinic acid |
| ANT | Adenine nucleotide translocator (carrier) |
| APAF-1 | Apoptosis-activating factor-1 |
| APP | Aminopheophorbide |
| 2-BA-2-DMHA | 2-Butylamino-2-demethoxy-hypocrellin A |
| tBID | Truncated bid |
| BPD-MA | Benzoporphyrin derivative monoacid ring A (Verteporfin) |
| CAME | Chlorin e6 triacetoxymethyl ester |
| CCCP | Carbonylcyanide-m-chlorophenylhydrazone |
| CPO | 9-Capronyloxytetrakis(methoxyethyl)porphycene |
| DISC | Death-inducing signalling complex |
| 2-DOG | 2-Deoxyglucose |
| DTHe | Tetrahydroxyhelianthrone |
| EDKC | N,N′-Bis(2-ethyl-1,3-dioxolanyl)kryptocyanine |
| ER | Endoplasmic reticulum |
| ERK | Extracellular regulated kinase |
| FADD | Fas-associated death domain |
| FasL | Fas ligand |
| HIV | Human immunodeficiency virus |
| HOG | Hyperosmotic glycerol (kinase) |
| HRM | Heme regulatory motifs |
| ICAD/DFF45 | Inhibitor of caspase-activated dnase/dna fragmentation factor 45 |
| IL | Interleukin |
| IκB | Inhibitor of κB |
| JNK | C-jun N-terminal kinase |
| LuTex | Lutetium texaphyrin |
| MAPK | Mitogen-activated protein kinases |
| m-THPC | meta-Tetrakis(hydroxyphenyl)chlorin |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MBD | Methylene blue derivative |
| MDCK | Madison-Darby canine kidney cells |
| NF-κB | Nuclear factor-κB |
| PARP | Poly(ADP-ribose) polymerase |
| PBR | Peripheral (mitochondrial) benzodiazepine receptor |
| PDTC | Pyrrolidine dithiocarbamate |
| PPa | Pheophorbide a |
| Pc | Phthalocyanine |
| PDT | Photodynamic therapy |
| PLC | Phospholipase C |
| PPIX | Protoporphyrin IX |
| PPME | Pyropheophorbide-a methyl ester |
| PTPC | Permeability transition pore complex |
| ROS | Reactive oxygen species |
| SAPK | Stress-activated protein kinase |
| SMAC | Second mitochondrial activator of caspases |
| SnET2 | Tin ethyl etiopurpurin |
| 3THPP | Tetrakis(3-hydroxyphenyl)porphyrin |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| TPPS2a | Disulfonated meso-tetraphenylporphine |
| TPPS4 | Tetrasulfonated meso-tetraphenylporphine |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated d-UTP nick end-labeling |
| TRADD | TNF receptor-associated death domain |
| UDCA | Ursodeoxycholic acid |
| VDAC | Voltage-dependent anion channel |
10 Acknowledgements
Research in the authors' laboratory is supported by US Public Health Service Grants P01 CA48735, R01 CA83917, and P30 CA43703 from the National Cancer Institute, DHHS.11 References
- T. J. Dougherty, Photodynamic therapy, Photochem. Photobiol., 1993, 58, 895–900 Search PubMed.
- S. A. Khan, T. J. Dougherty and T. S. Mang, An evaluation of photodynamic therapy in the management of cutaneous metastases of breast cancer, Eur. J. Cancer, 1993, 29A, 1686–1690 CrossRef CAS.
- H. Lui and R. R. Anderson, Photodynamic therapy in dermatology: Recent developments, Dermatol. Clin., 1993, 11, 1–13 Search PubMed.
- H. I. Pass, Photodynamic therapy in oncology: Mechanisms and clinical use, J. Natl. Cancer Inst., 1993, 85, 443–456 CrossRef CAS.
- F. Sieber, Phototherapy, photochemotherapy, and bone marrow transplanation, J. Hematother., 1993, 2, 43–62 CAS.
- E. Ben-Hur, S. Rywkin, I. Rosenthal, N. E. Geacintov and B. Horowitz, Virus inactivation in red cell concentrates by photosensitization with phthalocyanines: Protection of red cells but not of vesicular stomatitis virus with a water-soluble analog of vitamin E, Transfusion, 1995, 35, 401–406 CrossRef CAS.
- P. Gonschlor, F. Gerbeuser, M. Fleuchaus, T. Y. Huehns, A. E. Goetz, U. Welsch, R. Sroka, M. Dellian, H. A. Lehr and B. Hofling, Local photodynamic therapy reduces tissue hyperplasia in an experimental restenosis model, Photochem. Photobiol., 1996, 64, 758–763 Search PubMed.
- J. G. Levy and M. Obochi, New applications in photodynamic therapy: Introduction, Photochem. Photobiol., 1996, 64, 737–739 Search PubMed.
- K. B. Trauner and T. Hasan, Photodynamic treatment of rheumatoid and inflammatory arthritis, Photochem. Photobiol., 1996, 64, 740–750 Search PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, Photodynamic Therapy: Review, J. Natl. Cancer Inst., 1998, 90, 889–902 CrossRef CAS.
- K. R. Weishaupt, C. J. Gomer and T. J. Dougherty, Identification of singlet oxygen as the cytotoxic agent in photoinactivation of a murine tumor, Cancer Res., 1976, 36, 2326–2329 Search PubMed.
- B. Halliwell and J. M. Gutteridge, Oxygen toxicity, oxygen radicals, transition metals and disease, Biochem. J., 1984, 219, 1–14 CAS.
- B. W. Henderson and T. J. Dougherty, How does photodynamic therapy work?, Photochem. Photobiol., 1992, 55, 145–157 Search PubMed.
- J. Moan and K. Berg, Photochemotherapy of cancer: experimental research, Photochem. Photobiol., 1992, 55, 931–948 Search PubMed.
- T. Hasan and J. A. Parrish, Photodynamic therapy of cancer, in Cancer Medicine, 4th edn., Williams & Wilkins, Baltimore, 1997, pp. 739–750 Search PubMed.
- M. Ochsner, Photodynamic therapy: the clinical perspective. Review on applications for control of diverse tumorous and non-tumorous diseases, Arzneim.-Forsch., 1997, 47, 1185–1194 Search PubMed.
- R. Lipson, E. Baldes and A. Olsen, The use of a derivative of hematoporphyrin in tumor detection, JNCI, 1961, 26, 1 Search PubMed.
- C. J. Lightdale, Role of photodynamic therapy in the management of advanced esophageal cancer, Gastrointest. Endosc. Clin. N. Am., 2000, 10, 397–408 Search PubMed.
- C. J. Gomer, Preclinical examination of first and second generation photosensitizers used in photodynamic therapy, Photochem. Photobiol., 1991, 54, 1093–1107 Search PubMed.
- C. M. Allen, W. M. Sharman and J. E. Van Lier, Current status of phthalocyanines in the photodynamic therapy of cancer, J. Porphyrins Phthalocyanines, 2001, 5, 161–169 CrossRef CAS.
- J. F. Kerr, A. H. Wyllie and A. R. Currie, Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics, Br. J. Cancer, 1972, 26, 239–257 CAS.
- A. Ashkenazi and V. M. Dixit, Death receptors: signaling and modulation, Science, 1998, 281, 1305–1308 CrossRef CAS.
- G. Evan and T. Littlewood, A matter of life and cell death, Science, 1998, 281, 1317–1322 CrossRef CAS.
- G. Kroemer and J. C. Reed, Mitochondrial control of cell death, Nat. Med., 2000, 6, 513–519 CrossRef CAS.
- J. C. Reed, Mechanisms of apoptosis, Am. J. Pathol., 2000, 157, 1415–1430 Search PubMed.
- R. J. Arceci, Tumor cell survival and resistance to therapy, Curr. Opin. Hematol., 1996, 3, 279–287 CrossRef CAS.
- H. J. Guchelaar, A. Vermes, I. Vermes and C. Haanen, Apoptosis: molecular mechanisms and implications for cancer chemotherapy, Pharm. World Sci., 1997, 19, 19–125 CrossRef.
- A. M. Gorman, A. McGowan, C. O'Neill and T. Cotter, Oxidative stress and apoptosis in neurodegeneration, J. Neurol. Sci., 1996, 139, Suppl 45–52 Search PubMed.
- R. Kinscherf, H. P. Deigner and U. Haberkorn, Apoptosis modulators in the therapy of neurodegenerative diseases, Expert Opin. Invest. Drugs, 2000, 9, 747–764 Search PubMed.
- G. Kroemer, B. Dallaporta and M. Resche-Rigon, The mitochondrial death/life regulator in apoptosis and necrosis, Annu. Rev. Physiol., 1998, 60, 619–642 CrossRef CAS.
- N. L. Oleinick, Apoptosis in response to photodynamic therapy, Photodynamics News, 1998, 6, 8–9 Search PubMed.
- J. M. Adams and S. Cory, The Bcl-2 protein family: arbiters of cell survival, Science, 1998, 281, 1322–1326 CrossRef CAS.
- D. R. Green and J. C. Reed, Mitochondria and apoptosis, Science, 1998, 281, 1309–1312 CrossRef CAS.
- N. A. Thornberry and Y. Lazebnik, Caspases: enemies within, Science, 1998, 281, 1312–1316 CrossRef CAS.
- P. Costantini, E. Jacotot, D. Decaudin and G. Kroemer, Mitochondrion as a novel target of anticancer chemotherapy, J. Natl. Cancer Inst., 2000, 92, 1042–1053 CrossRef CAS.
- R. A. Gottlieb, Mitochondria: execution central, FEBS Lett., 2000, 482, 6–12 CrossRef CAS.
- D. R. Green, Apoptotic pathways: paper wraps stone blunts scissors, Cell, 2000, 102, 1–4 CrossRef CAS.
- Q. Peng, J. Moan and J. M. Nesland, Correlation of subcellular and intratumoral photosensitizer localization with ultrastructural features after photodynamic therapy, Ultrastruct. Pathol., 1996, 20, 109–129 CrossRef CAS.
- N. L. Oleinick and H. H. Evans, The photobiology of photodynamic therapy: Cellular targets and mechanisms, Radiat. Res., 1998, 150, S146–S156 Search PubMed.
- I. J. Macdonald and T. J. Dougherty, Basic principles of photodynamic therapy, J Porphyrins Phthalocyanines, 2001, 105–129 CrossRef CAS.
- P. Agostinis, Z. Assefa, A. Vantieghem, J. R. Vandenheede, W. Merlevede and P. De Witte, Apoptotic and anti-apoptotic signaling pathways induced by photodynamic therapy with hypericin, Adv. Enzyme Regul., 2000, 40, 157–182 CrossRef CAS.
- A. C. Moor, Signaling pathways in cell death and survival after photodynamic therapy, J. Photochem. Photobiol., B, 2000, 57, 1–13 Search PubMed.
- D. Granville and D. Hunt, Porphyrin-mediated photosensitization-Taking the apoptosis fast lane, Curr. Opin. Drug Discovery Dev., 2000, 3, 232–243 Search PubMed.
- D. J. Granville, B. M. McManus and D. W. Hunt, Photodynamic therapy: shedding light on the biochemical pathways regulating porphyrin-mediated cell death, Histol. Histopathol., 2001, 16, 309–317 Search PubMed.
- J. D. Robertson and S. Orrenius, Molecular mechanisms of apoptosis induced by cytotoxic chemicals, Crit. Rev. Toxicol., 2000, 30, 609–627 Search PubMed.
- E. Solary, N. Droin, A. Bettaieb, L. Corcos, M. T. Dimanche-Boitrel and C. Garrido, Positive and negative regulation of apoptotic pathways by cytotoxic agents in hematological malignancies, Leukemia, 2000, 14, 1833–1849 CrossRef CAS.
- M. Verheij and H. Bartelink, Radiation-induced apoptosis, Cell Tissue Res., 2000, 301, 133–142 CrossRef CAS.
- L. Zhuang, B. Wang and D. N. Sauder, Molecular mechanism of ultraviolet-induced keratinocyte apoptosis, J. Interferon Cytokine Res., 2000, 20, 445–454 CrossRef CAS.
- V. J. Kidd, J. M. Lahti and T. Teitz, Proteolytic regulation of apoptosis, Semin. Cell Dev. Biol., 2000, 11, 191–201 CrossRef CAS.
- H. R. Stennicke and G. S. Salvesen, Caspases – controlling intracellular signals by protease zymogen activation, Biochim. Biophys. Acta, 2000, 1477, 299–306 CrossRef CAS.
- Z.-H. Qin, Y. Wang, K. K. Kikly, E. Sapp, K. B. Kegel, N. Aronin and M. DiFiglia, Pro-caspase-8 is Predominantly Localized in Mitochondria and Released into Cytoplasm upon Apoptotic Stimulation, J. Biol. Chem., 2001, 276, 8079–8086 CrossRef CAS.
- A. Gross, J. M. McDonnell and S. J. Korsmeyer, BCL-2 family members and the mitochondria in apoptosis, Genes Dev., 1999, 13, 1899–1911 CAS.
- M. Crompton, Mitochondrial intermembrane junctional complexes and their role in cell death, J. Physiol., 2000, 529, Pt 1, 11–21 Search PubMed.
- A. P. Halestrap, E. Doran, J. P. Gillespie and A. O'Toole, Mitochondria and cell death, Biochem. Soc. Trans., 2000, 28, 170–177 CAS.
- Y. Tsujimoto and S. Shimizu, Bcl-2 family: life-or-death switch, FEBS Lett., 2000, 466, 6–10 CrossRef CAS.
- B. Antonsson and J. C. Martinou, The Bcl-2 protein family, Exp. Cell Res., 2000, 256, 50–57 CrossRef CAS.
- J. C. Reed, Bcl-2 family proteins, Oncogene, 1998, 17, 3225–3236 CrossRef.
- B. W. Johnson and L. H. Boise, Bcl-2 and caspase inhibition cooperate to inhibit tumor necrosis factor-alpha-induced cell death in a Bcl-2 cleavage-independent fashion, J. Biol. Chem., 1999, 274, 18552–18558 CrossRef CAS.
- D. G. Kirsch, A. Doseff, B. N. Chau, D. S. Lim, N. C. de Souza-Pinto, R. Hansford, M. B. Kastan, Y. A. Lazebnik and J. M. Hardwick, Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c, J. Biol. Chem., 1999, 274, 21155–21161 CrossRef CAS.
- V. V. Stoka, B. Turk, S. L. Schendel, T. H. Kim, T. Cirman, S. J. Snipas, L. M. Ellerby, D. Bredesen, H. Freeze, M. Abrahamson, D. Bromme, S. Krajewski, J. C. Reed, X. M. Yin, V. V. Turk and G. S. Salvesen, Lysosomal protease pathways to apoptosis: cleavage of bid, not Pro-caspases, is the most likely route, J. Biol. Chem., 2001, 276, 3149–3157 CrossRef CAS.
- M. Lam, M. B. Bhat, G. Nunez, J. Ma and C. W. Distelhorst, Regulation of Bcl-xl channel activity by calcium, J. Biol. Chem., 1998, 273, 17307–17310 CrossRef CAS.
- D. M. Hockenbery, Z. N. Oltvai, X.-M. Yin, C. L. Milliman and S. J. Korsmeyer, Bcl-2 functions in an antioxidant pathway to prevent apoptosis, Cell, 1993, 75, 241–251 CrossRef CAS.
- J. C. Reed, Cytochrome c: Can't live with it – can't live without it, Cell, 1997, 91, 559–562 CrossRef CAS.
- J. Yang, K. Bhalla, C. N. Kim, A. M. Ibrado, T. I. Peng, D. P. Jones and X. Wang, Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked, Science, 1997, 275, 1129–1132 CrossRef CAS.
- J. M. Brown and B. G. Wouters, Apoptosis, p53, and tumor cell sensitivity to anticancer agents, Cancer Res., 1999, 59, 1391–1399 Search PubMed.
- M. L. Agarwal, M. E. Clay, E. J. Harvey, H. H. Evans, A. R. Antunez and N. L. Oleinick, Photodynamic therapy induces rapid cell death by apoptosis in L5178Y mouse lymphoma cells, Cancer Res., 1991, 51, 5993–5996 Search PubMed.
- X. Y. He, R. A. Sikes, S. Thomsen, L. W. K. Chung and S. L. Jacques, Photodynamic therapy with Photofrin II induces programmed cell death in carcinoma cell lines, Photochem. Photobiol., 1994, 59, 468–473 Search PubMed.
- B. B. Noodt, K. Berg, T. Stokke, Q. Peng and J. M. Nesland, Apoptosis and necrosis induced with light and 5-aminolaevulinic acid-derived protoporphyrin IX, Br. J. Cancer, 1996, 74, 22–29 CAS.
- E. Ben-Hur, J. Oetjen and B. Horowitz, Silicon phthalocyanine Pc 4 and red light causes apoptosis in HIV infected cells, Photochem. Photobiol., 1997, 65, 456–460 Search PubMed.
- Y. Luo, C. K. Chang and D. Kessel, Rapid initiation of apoptosis by photodynamic therapy, Photochem. Photobiol., 1996, 63, 528–534 Search PubMed.
- Y. Luo and D. Kessel, The phosphatase inhibitor calyculin antagonizes the rapid initiation of apoptosis by photodynamic therapy, Biochem. Biophys. Res. Commun., 1996, 221, 72–76 CrossRef CAS.
- D. Separovic, J. He and N. L. Oleinick, Ceramide generation in response to photodynamic treatment of L5178Y mouse lymphoma cells, Cancer Res., 1997, 57, 1717–1721 Search PubMed.
- L.-Y. Xue, J. He and N. L. Oleinick, Rapid tyrosine phosphorylation of HS1 in the response of mouse lymphoma L5178Y-R cells to photodynamic treatment sensitized by the phthalocyanine Pc 4, Photochem. Photobiol., 1997, 66, 105–113 Search PubMed.
- D. Kessel and Y. Luo, Cells in cryptophycin-induced cell-cycle arrest are susceptible to apoptosis, Cancer Lett., 2000, 151, 25–29 CrossRef CAS.
- D. W. Hunt, H. Jiang, D. J. Granville, A. H. Chan, S. Leong and J. G. Levy, Consequences of the photodynamic treatment of resting and activated peripheral T lymphocytes, Immunopharmacology, 1999, 41, 31–44 CrossRef CAS.
- H. Jiang, D. J. Granville, B. M. McManus, J. G. Levy and D. W. Hunt, Selective depletion of a thymocyte subset in vitro with an immunomodulatory photosensitizer, Clin. Immunol., 1999, 91, 178–187 CrossRef CAS.
- M. Dellinger, Apoptosis or necrosis following Photofrin photosensitization: influence of the incubation protocol, Photochem. Photobiol., 1996, 64, 182–187 Search PubMed.
- B. B. Noodt, G. H. Rodal, M. Wainwright, Q. Peng, R. Horobin, J. M. Nesland and K. Berg, Apoptosis induction by different pathways with methylene blue derivative and light from mitochondrial sites in V79 cells, Int. J. Cancer, 1998, 75, 941–948 CrossRef CAS.
- I. E. Kochevar, M. C. Lynch, S. Zhuang and C. R. Lambert, Singlet oxygen, but not oxidizing radicals, induces apoptosis in HL-60 cells, Photochem. Photobiol., 2000, 72, 548–553 Search PubMed.
- C. P. Lin, M. C. Lynch and I. E. Kochevar, Reactive oxidizing species produced near the plasma membrane induce apoptosis in bovine aorta endothelial cells, Exp. Cell Res., 2000, 259, 351–359 CrossRef CAS.
- D. Kessel and Y. Luo, Mitochondrial photodamage and PDT-induced apoptosis, J. Photochem. Photobiol., 1998, 42, 89–95 Search PubMed.
- D. Kessel, Y. Luo, Y. Deng and C. K. Chang, The role of subcellular localization in initiation of apoptosis by photodynamic therapy, Photochem. Photobiol., 1997, 65, 422–426 Search PubMed.
- Y. Luo and D. Kessel, Initiation of apoptosis versus necrosis by photodynamic therapy with chloroaluminum phthalocyanine, Photochem. Photobiol., 1997, 66, 479–483 Search PubMed.
- K. W. Woodburn, Q. Fan, D. R. Miles, D. Kessel, Y. Luo and S. W. Young, Localization and efficacy analysis of the phototherapeutic lutetium texaphyrin (PCI–0123) in the murine EMT6 sarcoma model, Photochem. Photobiol., 1997, 65, 410–415 Search PubMed.
- B. B. Noodt, K. Berg, T. Stokke, Q. Peng and J. M. Nesland, Different apoptotic pathways are induced from various intracellular sites by tetraphenylporphyrins and light, Br. J. Cancer, 1999, 79, 72–81 CrossRef CAS.
- D. Kessel and R. D. Poretz, Sites of photodamage induced by photodynamic therapy with a chlorin e6 triacetoxymethyl ester (CAME), Photochem. Photobiol., 2000, 71, 94–96 Search PubMed.
- J. He and N. L. Oleinick, in 5th International Photodynamic Association Meeting, 1995, Vol. 2371, pp. 92–96 SPIE, Amelia Island, Florida.
- A. Vantieghem, Z. Assefa, P. Vandenabeele, W. Declercq, S. Courtois, J. R. Vandenheede, W. Merlevede, P. de Witte and P. Agostinis, Hypericin-induced photosensitization of HeLa cells leads to apoptosis or necrosis. Involvement of cytochrome c and procaspase-3 activation in the mechanism of apoptosis, FEBS Lett., 1998, 440, 19–24 CrossRef CAS.
- H. R. Kim, Y. Luo, G. Li and D. Kessel, Enhanced apoptotic response to photodynamic therapy after bcl-2 transfection, Cancer Res., 1999, 59, 3429–3432 Search PubMed.
- G. Lavie, C. Kaplinsky, A. Toren, I. Aizman, D. Meruelo, Y. Mazur and M. Mandel, A photodynamic pathway to apoptosis and necrosis induced by dimethyl tetrahydroxyhelianthrone and hypericin in leukaemic cells: possible relevance to photodynamic therapy, Br. J. Cancer, 1999, 79, 423–432 CrossRef CAS.
- J. Dahle, O. Kaalhus, J. Moan and H. Steen, Cooperative effects of photodynamic treatment of cells in microcolonies, Proc. Natl. Acad. Sci. USA, 1997, 94, 1773–1778 CrossRef CAS.
- J. Dahle, H. B. Steen and J. Moan, The mode of cell death induced by photodynamic treatment depends on cell density, Photochem. Photobiol., 1999, 70, 363–367 Search PubMed.
- J. Dahle, S. Bagdonas, O. Kaalhus, G. Olsen, H. B. Steen and J. Moan, The bystander effect in photodynamic inactivation of cells, Biochim. Biophys. Acta, 2000, 1475, 273–280 CrossRef CAS.
- J. Dahle, S. O. Mikalsen, E. Rivedal and H. B. Steen, Gap junctional intercellular communication is not a major mediator in the bystander effect in photodynamic treatment of MDCK II cells, Radiat. Res., 2000, 154, 331–341 Search PubMed.
- A. R. Oseroff, D. Ohuoha, G. Ara, D. McAuliffe, J. Foley and L. Cincotta, Intramitochondrial dyes allow selective in vitro photolysis of carcinoma cells, Proc. Natl. Acad. Sci. USA, 1986, 83, 9729–9733 CAS.
- D. J. Ball, Y. Luo, D. Kessel, J. Griffiths, S. B. Brown and D. I. Vernon, The induction of apoptosis by a positively charged methylene blue derivative, J. Photochem. Photobiol., B, 1998, 42, 159–163 Search PubMed.
- N. S. Trivedi, H. W. Wang, A. L. Nieminen, N. L. Oleinick and J. A. Izatt, Quantitative analysis of Pc 4 localization in mouse lymphoma (LY-R) cells via double-label confocal fluorescence microscopy, Photochem. Photobiol., 2000, 71, 634–639 Search PubMed.
- M. Lam, N. L. Oleinick and A.-L. Nieminen, Photodynamic Therapy-Induced Apoptosis in Epidermoid Carcinoma Cells: Reactive Oxygen Species and Mitochondrial Inner Membrane Permeabilization, J Biol. Chem., 2001, in the press Search PubMed.
- B. C. Wilson, M. Olivo and G. Singh, Subcellular localization of Photofrin and aminolevulinic acid and photodynamic cross-resistance in vitro in radiation-induced fibrosarcoma cells sensitive or resistant to photofrin-mediated photodynamic therapy, Photochem. Photobiol., 1997, 65, 166–176 Search PubMed.
- A. Verma, J. Nye and S. H. Snyder, Porphyrins are endogenous ligands for the mitochondrial (periphera-type) benzodiazepine receptor, Mol. Pharmacol., 1987, 84, 2256–2260 Search PubMed.
- A. Verma and S. H. Snyder, Characterization of porphyrin interactions with peripheral type benzodiazepine receptors, Mol. Pharmacol., 1988, 34, 800–805 Search PubMed.
- A. Verma, S. L. Facchina, D. J. Hirsch, S.-Y. Song, L. F. Dillahey, J. R. Williams and S. H. Snyder, Photodynamic tumor therapy: Mitochondrial benzodiazepine receptors as a therapeutic target, Mol. Med., 1998, 4, 40–49 CAS.
- J. Morgan, I. J. MacDonald and A. R. Oseroff, The relationship of peripheral benzodiazepine receptor binding and mitochondrial localization to the phototoxicity of pyropheophorbide ethers, Photochem. Photobiol., 1998, 67, 26S Search PubMed.
- D. Kessel, M. Antolovich and K. M. Smith, The Role of the Peripheral Benzodiazepine Receptor in the Apoptotic Response to Photodynamic Therapy, Photochem. Photobiol., 2001, 74, 346–349 Search PubMed.
- S. L. Ratcliffe and E. K. Matthews, Modification of the photodynamic action of δ-aminolaevulinic acid (ALA) on rat pancreatoma cells by mitochondrial benzodiazepine receptor ligands, Br. J. Cancer, 1995, 71, 300–305 CAS.
- R. L. Morris, M. E. Varnes, M. E. Kenney, Y.-S. Li, M. W. McEnery and N. L. Oleinick, The Mitochondrial Benzodiazepine Receptor in Photodynamic Therapy: Atypical Binding of the Phthalocyanine Photosensitizer Pc 4 and Ligand Action in Apoptosis, Photochem. Photobiol., 2001, submitted Search PubMed.
- C. Salet, G. Moreno, F. Ricchelli and P. Bernardi, Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore, J. Biol. Chem., 1997, 272, 21938–21943 CrossRef CAS.
- A. S. Belzacq, E. Jacotot, H. L. Vieira, D. Mistro, D. J. Granville, Z. Xie, J. C. Reed, G. Kroemer and C. Brenner, Apoptosis induction by the photosensitizer verteporfin: identification of mitochondrial adenine nucleotide translocator as a critical target, Cancer Res., 2001, 61, 1260–1264 Search PubMed.
- L. Miccoli, A. Beurdeley-Thomas, G. De Pinieux, F. Sureau, S. Oudard, B. Dutrillaux and M. F. Poupon, Light-induced photoactivation of hypericin affects the energy metabolism of human glioma cells by inhibiting hexokinase bound to mitochondria, Cancer Res., 1998, 58, 5777–5786 Search PubMed.
- M. E. Varnes, S. M. Chiu, L. Y. Xue and N. L. Oleinick, Photodynamic therapy-induced apoptosis in lymphoma cells: Translocation of cytochrome c causes inhibition of respiration as well as caspase activation, Biochem. Biophys. Res. Commun., 1999, 255, 673–679 CrossRef CAS.
- S. L. Gibson and R. Hilf, Interdependence of fluence, drug dose and oxygen on hematoporphyrin derivative induced photosensitization of tumor mitochondria, Photochem. Photobiol., 1985, 42, 367–373 Search PubMed.
- D. S. Perlin, R. S. Murant, S. L. Gibson and R. Hilf, Effects of photosensitization by hematoporphyrin derivative on mitochondrial adenosine triphosphatase-mediated proton transport and membrane integrity of R3230AC mammary adenocarcinoma, Cancer Res., 1985, 45, 653–658 Search PubMed.
- A. Atlante, G. Moreno, S. Parsarella and C. Salet, Hematoporphyrin derivative (PF II) photosensitization of isolated mitochondria: Impairment of anion translocation, Biochem. Biophys. Res. Commun., 1986, 141, 584–590 CAS.
- G. Singh, W. P. Jeeves, B. C. Wilson and D. Jang, Mitochondrial photosensitization by Photofrin II, Photochem. Photobiol., 1987, 46, 645–649 Search PubMed.
- J. Morgan, W. R. Potter and A. R. Oseroff, Comparison of photodynamic targets in a carcinoma cell line and its mitochondrial DNA-deficient derivative, Photochem. Photobiol., 2000, 71, 747–757 Search PubMed.
- D. J. Granville, C. M. Carthy, H. Jiang, G. C. Shore, B. M. McManus and D. W. C. Hunt, Rapid cytochrome c release, activation of caspases 3, 6, 7 and 8 followed by Bap31 cleavage in HeLa cells treated with photodynamic therapy, FEBS Lett., 1998, 437, 5–10 CrossRef CAS.
- O. Inanami, A. Yoshito, K. Takahashi, W. Hiraoka and M. Kuwabara, Effects of BAPTA-AM and forskolin on apoptosis and cytochrome c release in photosensitized Chinese hamster V79 cells, Photochem. Photobiol., 1999, 70, 650–655 Search PubMed.
- D. Kessel and Y. Luo, Photodynamic therapy: A mitochondrial inducer of apoptosis, Cell Death Differ., 1999, 6, 28–35 CrossRef CAS.
- D. Kessel and Y. Luo, Intracellular Sites of Photodamage as a Factor in Apoptotic Cell Death, J. Porphyrin Phthalocyanines, 2001, 5, 181–184 Search PubMed.
- D. Kessel, J. A. Caruso and J. J. Reiners, Jr., Potentiation of photodynamic therapy by ursodeoxycholic acid, Cancer Res., 2000, 60, 6985–6988 Search PubMed.
- J. Gagnebin, M. Brunori, M. Otter, L. Juillerat-Jeanneret, P. Monnier and R. Iggo, A photosensitising adenovirus for photodynamic therapy, Gene Ther., 1999, 6, 1742–1750 CrossRef CAS.
- G. Schatz, Mitochondria: beyond oxidative phosphorylation, Biochim. Biophys. Acta, 1995, 1271, 123–126 CrossRef CAS.
- K. K. Singh, J. Russell, B. Sigala, Y. Zhang, J. Williams and K. F. Keshav, Mitochondrial DNA determines the cellular response to cancer therapeutic agents, Oncogene, 1999, 18, 6641–6646 CrossRef CAS.
- D. Kessel and H. H. Sun, Enhanced responsiveness to photodynamic therapy-induced apoptosis after mitochondrial DNA depletion, Photochem. Photobiol., 1999, 70, 937–940 Search PubMed.
- A. D. Munday, A. Sriratana, J. S. Hill, S. B. Kahl and P. Nagley, Mitochondria are the functional intracellular target for a photosensitizing boronated porphyrin, Biochim. Biophys. Acta, 1996, 1311, 1–4 CrossRef CAS.
- R. Chaloupka, T. Obsil, J. Plasek and F. Sureau, The effect of hypericin and hypocrellin-A on lipid membranes and membrane potential of 3T3 fibroblasts, Biochim. Biophys. Acta, 1999, 1418, 39–47 CrossRef CAS.
- R. Chaloupka, P. X. Petit, N. Israel and F. Sureau, Over-expression of Bcl-2 does not protect cells from hypericin photo-induced mitochondrial membrane depolarization, but delays subsequent events in the apoptotic pathway, FEBS Lett., 1999, 462, 295–301 CrossRef CAS.
- A. J. Kowaltowski, J. Turin, G. L. Indig and A. E. Vercesi, Mitochondrial effects of triarylmethane dyes, J. Bioenerg. Biomembr., 1999, 31, 581–590 CrossRef CAS.
- C. M. Carthy, D. J. Granville, H. Jiang, J. G. Levy, C. M. Rudin, C. B. Thompson, B. M. McManus and D. W. Hunt, Early release of mitochondrial cytochrome c and expression of mitochondrial epitope 7A6 with a porphyrin-derived photosensitizer: Bcl-2 and Bcl-xL overexpression do not prevent early mitochondrial events but still depress caspase activity, Lab. Invest., 1999, 79, 953–965 Search PubMed.
- S.-M. Chiu and N. L. Oleinick, Dissociation of mitochondrial depolarization from cytochrome c release during apoptosis induced by photodynamic therapy, Br. J. Cancer, 2001, 84, 1099–1106 CrossRef CAS.
- S. Chiu, H. H. Evans, M. Lam, A. Nieminen and N. L. Oleinick, Phthalocyanine 4 photodynamic therapy-induced apoptosis of mouse L5178Y-R cells results from a delayed but extensive release of cytochrome c from mitochondria, Cancer Lett., 2001, 165, 51–58 CrossRef CAS.
- D. J. Granville, B. A. Cassidy, D. O. Ruehlmann, J. C. Choy, C. Brenner, G. Kroemer, C. van Breemen, P. Margaron, D. W. Hunt and B. M. McManus, Mitochondrial release of apoptosis-inducing factor and cytochrome c during smooth muscle cell apoptosis, Am. J. Pathol., 2001, 159, 305–311 Search PubMed.
- D. J. Granville, C. M. Carthy, H. Jiang, J. G. Levy, B. M. McManus, J. Y. Matroule, J. Piette and D. W. Hunt, Nuclear factor-kappaB activation by the photochemotherapeutic agent verteporfin, Blood, 2000, 95, 256–262 CAS.
- L.-Y. Xue, S.-M. Chiu and N. L. Oleinick, Photodynamic Therapy-Induced Death of MCF-7 Human Breast Cancer Cells: A Role for Caspase-3 in the Late Steps of Apoptosis but Not for the Critical Lethal Event, Exp. Cell Res., 2001, 263, 145–155 CrossRef CAS.
- D. J. Granville, J. G. Levy and D. W. C. Hunt, Photodynamic therapy induces caspase-3 activation in HL-60 cells, Cell Death Differ., 1997, 4, 623–628 CrossRef CAS.
- J. He, C. M. Whitacre, L.-Y. Xue, N. A. Berger and N. L. Oleinick, Protease activation and cleavage of Poly(ADP-ribose) polymerase: An integral part of apoptosis in response to photodynamic treatment, Cancer Res., 1998, 58, 940–946 Search PubMed.
- Z. Assefa, A. Vantieghem, W. Declercq, P. Vandenabeele, J. R. Vandenheede, W. Merlevede, P. de Witte and P. Agostinis, The activation of the c-Jun N-terminal kinase and p38 mitogen-activated protein kinase signaling pathways protects HeLa cells from apoptosis following photodynamic therapy with hypericin, J. Biol. Chem., 1999, 274, 8788–8796 CrossRef CAS.
- W. H. Chan, J. S. Yu and S. D. Yang, Apoptotic signalling cascade in photosensitized human epidermal carcinoma A431 cells: involvement of singlet oxygen, c-Jun N-terminal kinase, caspase-3 and p21-activated kinase 2, Biochem. J., 2000, 351, 221–232 CrossRef CAS.
- R. Z. Renno, F. C. Delori, R. A. Holzer, E. S. Gragoudas and J. W. Miller, Photodynamic therapy using Lu-Tex induces apoptosis in vitro, and its effect is potentiated by angiostatin in retinal capillary endothelial cells, Invest. Ophthalmol. Vis. Sci., 2000, 41, 3963–3971 Search PubMed.
- F. Gad, G. Viau, M. Boushira, R. Bertrand and R. Bissonnette, Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant t cells, J. Cutan. Med. Surg., 2001, 5, 8–13 CAS.
- D. J. Granville, J. R. Shaw, S. Leong, C. M. Carthy, P. Margaron, D. W. Hunt and B. M. McManus, Release of cytochrome c, Bax migration, Bid cleavage, and activation of caspases 2, 3, 6, 7, 8, and 9 during endothelial cell apoptosis, Am. J. Pathol., 1999, 155, 1021–1025 Search PubMed.
- S. Zhuang, M. C. Lynch and I. E. Kochevar, Caspase-8 mediates caspase-3 activation and cytochrome c release during singlet oxygen-induced apoptosis of HL–60 cells, Exp. Cell Res., 1999, 250, 203–212 CrossRef CAS.
- N. Ahmad, S. Gupta, D. K. Feyes and H. Mukhtar, Involvement of Fas (APO–1/CD–95) during photodynamic-therapy-mediated apoptosis in human epidermoid carcinoma A431 cells, J. Invest. Dermatol., 2000, 115, 1041–1046 CrossRef CAS.
- J. He, M. L. Agarwal, H. E. Larkin, L. R. Friedman, L.-Y. Xue and N. L. Oleinick, The induction of partial resistance to photodynamic therapy by the protooncogene BCL-2, Photochem. Photobiol., 1996, 64, 845–852 Search PubMed.
- D. J. Granville, H. Jiang, M. T. An, J. G. Levy, B. M. McManus and D. W. Hunt, Bcl-2 overexpression blocks caspase activation and downstream apoptotic events instigated by photodynamic therapy, Br. J. Cancer, 1999, 79, 95–100 CrossRef CAS.
- D. J. Granville, H. Jiang, M. T. An, J. G. Levy, B. M. McManus and D. W. Hunt, Overexpression of Bcl-X(L) prevents caspase-3-mediated activation of DNA fragmentation factor (DFF) produced by treatment with the photochemotherapeutic agent BPD-MA, FEBS Lett., 1998, 422, 151–154 CrossRef CAS.
- W. G. Zhang, L. P. Ma, S. W. Wang, Z. Y. Zhang and G. D. Cao, Antisense bcl-2 retrovirus vector increases the sensitivity of a human gastric adenocarcinoma cell line to photodynamic therapy, Photochem. Photobiol., 1999, 69, 582–586 Search PubMed.
- M. Srivastava, H. Ahmad, S. Gupta and H. Mukhtar, Involvement of Bcl-2 and Bax in photodynamic therapy mediated apoptosis, J. Biol. Chem., 2001, 276, 15481–15488 CrossRef CAS.
- L.-Y. Xue, S.-M. Chiu and N. L. Oleinick, Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4, Oncogene, 2001, 20, 3420–3427 CrossRef CAS.
- A. M. Ibrado, Y. Huang, G. Fang, L. Liu and K. Bhalla, Overexpression of Bcl-2 or Bcl-xL inhibits Ara-C-induced CPP32/Yama protease activity and apoptosis of human acute myelogenous leukemia HL-60 cells, Cancer Res., 1996, 56, 4743–4748 Search PubMed.
- J. Usuda, T. Okunaka, K. Furukawa, T. Tsuchida, Y. Kuroiwa, Y. Ohe, N. Saijo, K. Nishio, C. Konaka and H. Kato, Increased Cytotoxic Effects of Photodynamic Therapy in IL-6 Gene Transfected Cells via Enhanced Apoptosis, Int. J. Cancer, 2001, 93, 475–480 CrossRef CAS.
- E. H. Cheng, D. G. Kirsch, R. J. Clem, R. Ravi, M. B. Kastan, A. Bedi, K. Ueno and J. M. Hardwick, Conversion of Bcl-2 to a Bax-like death effector by caspases, Science, 1997, 278, 1966–1968 CrossRef CAS.
- N. Fujita and T. Tsuruo, Involvement of Bcl-2 cleavage in the acceleration of VP–16-induced U937 cell apoptosis, Biochem. Biophys. Res. Commun., 1998, 246, 484–488 CrossRef CAS.
- D. Grandgirard, E. Studer, L. Monney, T. Belser, I. Fellay, C. Borner and M. R. Michel, Alphaviruses induce apoptosis in Bcl-2-overexpressing cells: evidence for a caspase-mediated, proteolytic inactivation of Bcl-2, EMBO J., 1998, 17, 1268–1278 CrossRef CAS.
- B. Fadeel, Z. Hassan, E. Hellstrom-Lindberg, J. I. Henter, S. Orrenius and B. Zhivotovsky, Cleavage of Bcl-2 is an early event in chemotherapy-induced apoptosis of human myeloid leukemia cells, Leukemia, 1999, 13, 719–728 CrossRef CAS.
- G. Tudor, A. Aguilera, D. O. Halverson, N. D. Laing and E. A. Sausville, Susceptibility to drug-induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels, Cell Death Differ., 2000, 7, 574–586 CrossRef CAS.
- D. Kessel and M. Castelli, Evidence that Bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response, Photochem. Photobiol., 2001, 74, 318–322 Search PubMed.
- S. Zhuang, J. T. Demirs and I. E. Kochevar, p38 mitogen-activated protein kinase mediates bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide, J. Biol. Chem., 2000, 275, 25939–25948 CrossRef CAS.
- T. Kawaguchi, S. Yamamoto, N. Naka, K. Okishio, S. Atagi, M. Ogawara, S. Hosoe, M. Kawahara and K. Furuse, Immunohistochemical analysis of Bcl-2 protein in early squamous cell carcinoma of the bronchus treated with photodynamic therapy, Br. J. Cancer, 2000, 82, 418–423 CrossRef CAS.
- T. J. McGarrity, L. P. Peiffer, D. J. Granville, C. M. Carthy, J. G. Levy, M. Khandelwal and D. W. Hunt, Apoptosis associated with esophageal adenocarcinoma: influence of photodynamic therapy, Cancer Lett., 2001, 163, 33–41 CrossRef CAS.
- M. I. Koukourakis, L. Corti, J. Skarlatos, A. Giatromanolaki, B. Krammer, S. Blandamura, M. Piazza, T. Verwanger, G. Schnitzhofer, J. Kostandelos and K. Beroukas, Clinical and experimental evidence of Bcl-2 involvement in the response to photodynamic therapy, Anticancer Res., 2001, 21, 663–668 Search PubMed.
- W. G. Kaelin, The p53 gene family, Oncogene, 1999, 18, 7701–7705 CrossRef CAS.
- M. S. Sheikh and A. J. Fornace, Role of p53 family members in apoptosis, J. Cell Physiol., 2000, 182, 171–181 CrossRef CAS.
- E. Ben-Hur and T. M. Dubbelman, Cytoplasmic free calcium changes as a trigger mechanism in the response of cells to photosensitization, Photochem. Photobiol., 1993, 58, 890–894 Search PubMed.
- M. L. Agarwal, H. E. Larkin, S. I. A. Zaidi, H. Mukhtar and N. L. Oleinick, Phospholipase activation triggers apoptosis in photosensitized mouse lymphoma cells, Cancer Res., 1993, 53, 5897–5902 Search PubMed.
- H. Tajiri, A. Hayakawa, Y. Matsumoto, I. Yokoyama and S. Yoshida, Changes in intracellular Ca2+ concentrations related to PDT-induced apoptosis in photosensitized human cancer cells, Cancer Lett., 1998, 128, 205–210 CrossRef CAS.
- A. Rück, K. Heckelsmiller, R. Kaufmann, N. Grossman, E. Haseroth and N. Akgun, Light-induced apoptosis involves a defined sequence of cytoplasmic and nuclear calcium release in AlPcS4-photosensitized rat bladder RR 1022 epithelial cells, Photochem. Photobiol., 2000, 72, 210–216 Search PubMed.
- D. K. Perry, Ceramide and apoptosis, Biochem. Soc. Trans., 1999, 27, 399–404 CAS.
- D. Separovic, K. J. Mann and N. L. Oleinick, Association of ceramide accumulation with photodynamic treatment-induced cell death, Photochem. Photobiol., 1998, 68, 101–109 Search PubMed.
- D. Separovic, J. J. Pink, N. L. Oleinick, M. Kester, D. A. Boothman, M. McLoughlin, L. A. Pena and A. Haimovitz-Friedman, Niemann-Pick human lymphoblasts are resistant to phthalocyanine 4-photodynamic therapy-induced apoptosis, Biochem. Biophys. Res. Commun., 1999, 258, 506–512 CrossRef CAS.
- J. Y. Matroule, G. Bonizzi, P. Morliere, N. Paillous, R. Santus, V. Bours and J. Piette, Pyropheophorbide-a methyl ester-mediated photosensitization activates transcription factor NF-kappaB through the interleukin-1 receptor-dependent signaling pathway, J. Biol. Chem., 1999, 274, 2988–3000 CrossRef CAS.
- R. A. Gottlieb, J. Nordberg, E. Skowronski and B. M. Babior, Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification, Proc. Natl. Acad. Sci. USA, 1996, 93, 654–658 CrossRef CAS.
- R. Zidovetzki, I. W. Sherman, M. Cardenas and D. B. Borchardt, Chloroquine stabilization of phospholipid membranes against diacylglycerol-induced perturbation, Biochem. Pharmacol., 1993, 45, 183–189 CrossRef CAS.
- B. Nagy, S. M. Chiu and D. Separovic, Fumonisin B1 does not prevent apoptosis in A431 human epidermoid carcinoma cells after photosensitization with a silicon phthalocyanine, J. Photochem. Photobiol. B, 2000, 57, 132–141 Search PubMed.
- W. H. Tolleson, L. H. Couch, W. B. Melchior, G. R. Jenkins, M. Muskhelishvili, L. Muskhelishvili, L. J. McGarrity, O. Domon, S. M. Morris and P. C. Howard, Fumonisin B1 induces apoptosis in cultured human keratinocytes through sphinganine accumulation and ceramide depletion, Int. J. Oncol., 1999, 14, 833–843 Search PubMed.
- B. Nagy, W.-C. Yeh, T. W. Mak, S.-M. Chiu and D. Separovic, FADD Null Mouse Embyonic Fibroblasts Undergo Apoptosis after Photosensitization with the Silicon Phthalocyanine Pc 4, Arch. Biochem. Biophys., 2001, 385, 194–202 CrossRef CAS.
- I. Yslas, M. G. Alvarez, C. Marty, G. Mori, E. N. Durantini and V. Rivarola, Expression of Fas antigen and apoptosis caused by 5,10,15, 20-tetra(4-methoxyphenyl)porphyrin (TMP) on carcinoma cells: implication for photodynamic therapy, Toxicology, 2000, 149, 69–74 CrossRef CAS.
- K. Azizuddin, K. Kalka, S. Chiu, N. Ahmad, H. Mukhtar and D. Separovic, Recombinant human tumor necrosis factor alpha does not potentiate cell killing after photodynamic therapy with a silicon phthalocyanine in A431 human epidermoid carcinoma cells, Int. J. Oncol., 2001, 18, 411–415 Search PubMed.
- B. B. Aggarwal, Apoptosis and nuclear factor-kappa B: a tale of association and dissociation, Biochem. Pharmacol., 2000, 60, 1033–1039 CrossRef CAS.
- V. Bours, M. Bentires-Alj, A. C. Hellin, P. Viatour, P. Robe, S. Delhalle, V. Benoit and M. P. Merville, Nuclear factor-kappa B, cancer, and apoptosis, Biochem. Pharmacol., 2000, 60, 1085–1089 CrossRef CAS.
- S. W. Ryter and C. J. Gomer, Nuclear factor kappa B binding activity in mouse L1210 cells following photofrin II-mediated photosensitization, Photochem. Photobiol., 1993, 58, 753–756 Search PubMed.
- G. Kick, G. Messer, A. Goetz, G. Plewig and P. Kind, Photodynamic therapy induces expression of interleukin 6 by activation of AP-1 but not NF-κB DNA binding, Cancer Res., 1995, 55, 2373–2379 Search PubMed.
- B. Piret, S. Legrand-Poels, C. Sappey and J. Piette, NF-kappa B transcription factor and human immunodeficiency virus type 1 (HIV–1) activation by methylene blue photosensitization, Eur. J. Biochem., 1995, 228, 447–455 CAS.
- S. Legrand-Poels, V. Bours, B. Piret, M. Pflaum, B. Epe, B. Rentier and J. Piette, Transcription factor NF-kappa B is activated by photosensitization generating oxidative DNA damages, J. Biol. Chem., 1995, 270, 6925–6934 CrossRef CAS.
- J. Y. Matroule, A. C. Hellin, P. Morliere, A. S. Fabiano, R. Santus, M. P. Merville and J. Piette, Role of nuclear factor-kappa B in colon cancer cell apoptosis mediated by aminopyropheophorbide photosensitization, Photochem. Photobiol., 1999, 70, 540–548 Search PubMed.
- J.-Y. Matroule, C. M. Carthy, D. J. Granville, O. Jolois, D. W. Hunt and J. Piette, Mechanism of colon cancer apoptosis mediated by pyropheophorbide-a methylester photosensitization, Oncogene, 2001, 20, 4070–4084 CrossRef CAS.
- T. S. Lewis, P. S. Shapiro and N. G. Ahn, Signal transduction through MAP kinase cascades, Adv. Cancer. Res., 1998, 74, 49–139 Search PubMed.
- X. Wang, J. L. Martindale, Y. Liu and N. J. Holbrook, The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival, Biochem. J., 1998, 333, 291–300 CAS.
- L.-y. Xue, J. He and N. L. Oleinick, Promotion of photodynamic therapy-induced apoptosis by stress kinases, Cell Death Differ., 1999, 6, 855–864 CrossRef CAS.
- Y. Zhang, Y. Huang, A. K. Rishi, M. S. Sheikh, B. Shroot, U. Reichert, M. Dawson, G. Poirer and J. A. Fontana, Activation of the p38 and JNK/SAPK mitogen-activated protein kinase pathways during apoptosis is mediated by a novel retinoid, Exp. Cell Res., 1999, 247, 233–240 CrossRef CAS.
- J.-S. Tao, J. S. Sanghera, S. L. Pelech, G. Wong and J. G. Levy, Stimulation of stress-activated protein kinase and p38 HOG1 kinase in murine keratinocytes following photodynamic therapy with benzoporphyrin derivative, J. Cell Biochem., 1996, 271, 27107–27115 CAS.
- L. O. Klotz, C. Fritsch, K. Briviba, N. Tsacmacidis, F. Schliess and H. Sies, Activation of JNK and p38 but not ERK MAP kinases in human skin cells by 5-aminolevulinate-photodynamic therapy, Cancer Res., 1998, 58, 4297–4300 Search PubMed.
- M. E. Burow, C. B. Weldon, L. I. Melnik, B. N. Duong, B. M. Collins-Burow, B. S. Beckman and J. A. McLachlan, PI3-K/AKT regulation of NF-kappaB signaling events in suppression of TNF-induced apoptosis, Biochem. Biophys. Res. Commun., 2000, 271, 342–345 CrossRef CAS.
- A. H. Kim, G. Khursigara, X. Sun, T. F. Franke and M. V. Chao, Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1, Mol. Cell. Biol., 2001, 21, 893–901 CrossRef CAS.
- L. Y. Xue, Y. Qiu, J. He, H. J. Kung and N. L. Oleinick, Etk/Bmx, a PH-domain-containing tyrosine kinase, protects prostate cancer cells from apoptosis induced by photodynamic therapy or thapsigargin, Oncogene, 1999, 18, 3391–3398 CrossRef CAS.
- D. J. Granville, J. G. Levy and D. W. Hunt, Photodynamic treatment with benzoporphyrin derivative monoacid ring A produces protein tyrosine phosphorylation events and DNA fragmentation in murine P815 cells, Photochem. Photobiol., 1998, 67, 358–362 Search PubMed.
- A. M. R. Fisher, K. Danenberg, D. Banerjee, J. R. Bertino, P. Danenberg and C. J. Gomer, Increased photosensitivity in HL60 cells expressing wild-type p53, Photochem. Photobiol., 1997, 66, 265–270 Search PubMed.
- A. M. Fisher, N. Rucker, S. Wong and C. J. Gomer, Differential photosensitivity in wild-type and mutant p53 human colon carcinoma cell lines, J. Photochem. Photobiol., 1998, 42, 104–107 Search PubMed.
- A. M. R. Fisher, A. Ferrario, N. Rucker, S. Zhang and C. J. Gomer, Photodynamic therapy sensitivity is not altered in human tumor cells after abrogation of p53 function, Cancer Res., 1999, 59, 331–335 Search PubMed.
- W. G. Zhang, X. W. Li, L. P. Ma, S. W. Wang, H. Y. Yang and Z. Y. Zhang, Wild-type p53 protein potentiates phototoxicity of 2-BA–2-DMHA in HT29 cells expressing endogenous mutant p53, Cancer Lett., 1999, 138, 189–195 CrossRef CAS.
- Z. Tong, G. Singh and A. J. Rainbow, The role of the p53 tumor suppressor in the response of human cells to photofrin-mediated photodynamic therapy, Photochem. Photobiol., 2000, 71, 201–210 Search PubMed.
- S. Gupta, N. Ahmad and H. Mukhtar, Involvement of nitric oxide during phthalocynine (Pc 4)-photodynamic therapy mediated apoptosis, Cancer Res., 1998, 58, 1785–1788 Search PubMed.
- N. Ahmad, D. K. Feyes, R. Agarwal and H. Mukhtar, Photodynamic therapy results in induction of WAF1/CIP1/p21, leading to cell cycle arrest and apoptosis, Proc. Natl. Acad. Sci. USA, 1998, 95, 6977–6982 CrossRef CAS.
- N. Ahmad, S. Gupta and H. Mukhtar, Involvement of retinoblastoma (Rb) and E2F transcription factors during photodynamic therapy of human epidermoid carcinoma cells A431, Oncogene, 1999, 18, 1891–1896 CrossRef CAS.
- K. Kalka, N. Ahmad, T. Criswell, D. Boothman and H. Mukhtar, Up-regulation of clusterin during phthalocyanine 4 photodynamic therapy-mediated apoptosis of tumor cells and ablation of mouse skin tumors, Cancer Res., 2000, 60, 5984–5987 Search PubMed.
- S. Hotta, H. Kashimura, S. Hirai, A. Nakahara, H. Fukutomi, T. Osuga and Y. Uchiyama, Immediate changes in subcellular structures of transplanted tumors following photodynamic and laser hyperthermic therapy, Lasers Surg. Med., 1995, 16, 262–271 CAS.
- T. Yokota, H. Ikeda, T. Inokuchi, K. Sano and T. Koji, Enhanced cell death in NR-S1 tumor by photodynamic therapy: possible involvement of Fas and Fas ligand system, Lasers Surg. Med., 2000, 26, 449–460 CrossRef CAS.
- C. M. Whitacre, D. K. Feyes, T. Satoh, J. Grossmann, J. W. Mulvihill, H. Mukhtar and N. L. Oleinick, Photodynamic therapy with the phthalocyanine photosensitizer Pc 4 of SW480 human colon cancer xenografts in athymic mice, Clin. Cancer Res., 2000, 6, 2021–2027 Search PubMed.
- V. C. Colussi, D. K. Feyes, J. W. Mulvihill, Y. S. Li, M. E. Kenney, C. A. Elmets, N. L. Oleinick and H. Mukhtar, Phthalocyanine 4 (Pc 4) photodynamic therapy of human OVCAR-3 tumor xenografts, Photochem. Photobiol., 1999, 69, 236–241 Search PubMed.
- S. I. A. Zaidi, N. L. Oleinick, M. T. Zaim and H. Mukhtar, Apoptosis during photodynamic therapy-induced ablation of RIF-1 tumors in C3H mice: Electron microscopic, histopathologic, and biochemical evidence, Photochem. Photobiol., 1993, 58, 771–776 Search PubMed.
- B. W. Engbrecht, C. Menon, A. V. Kachur, S. M. Hahn and D. L. Fraker, Photofrin-mediated photodynamic therapy induces vascular occlusion and apoptosis in a human sarcoma xenograft model, Cancer Res., 1999, 59, 4334–4342 Search PubMed.
- L. Lilge, M. Portnoy and B. C. Wilson, Apoptosis induced in vivo by photodynamic therapy in normal brain and intracranial tumour tissue, Br. J. Cancer, 2000, 83, 1110–1117 CrossRef CAS.
- R. Agarwal, N. J. Korman, R. R. Mohan, D. K. Feyes, S. Jawed, M. T. Zaim and H. Mukhtar, Apoptosis is an early event during phthalocyanine photodynamic therapy-induced ablation of chemically induced squamous papillomas in mouse skin, Photochem. Photobiol., 1996, 63, 547–552 Search PubMed.
- J. Webber, Y. Luo, R. Crilly, D. Fromm and D. Kessel, An apoptotic response to photodynamic therapy with endogenous protoporphyrin in vivo, J. Photochem. Photobiol., B. Biol., 1996, 35, 209–211 CrossRef CAS.
- C. Zhou, C. Shunji, D. Jinsheng, L. Junlin, G. Jori and C. Milanesi, Apoptosis of mouse MS-2 fibrosarcoma cells induced by photodynamic therapy with Zn(II)-phthalocyanine, J. Photochem. Photobiol., B. Biol., 1996, 33, 219–223 CrossRef CAS.
- A. Hajri, S. Coffy, F. Vallat, S. Evrard, J. Marescaux and M. Aprahamian, Human pancreatic carcinoma cells are sensitive to photodynamic therapy in vitro and in vivo, Br. J. Surg., 1999, 86, 899–906 Search PubMed.
| This journal is © The Royal Society of Chemistry and Owner Societies 2002 |