Flash photolytic generation and study of 5-methoxy-o-quinone α-phenylethide† in aqueous solution: comparison of cis and trans isomer reactivity‡
Yvonne
Chiang
,
A. Jerry
Kresge
* and
Yu
Zhu
Department of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada. E-mail: akresge@chem.utoronto.ca; Fax: 1 (416) 978–7259
First published on 2nd January 2002
Abstract
Cis and trans isomeric 5-methoxy-o-quinone α-phenylethides were generated by flash photolysis of o-hydroxybenzyl alcohol and o-hydroxystyrene precursors and their rates of decay were measured in aqueous solutions of perchloric acid, sodium hydroxide, and biphosphate (dihydrogen phosphate) ion and carbonate ion buffers. The data so obtained gave two parallel rate profiles displaced from one another by about an order of magnitude, the faster of which could be assigned to the cis isomer and the slower to the trans isomer. Both rate profiles showed acid catalyzed, uncatalyzed, and hydroxide ion catalyzed portions, with acid catalysis becoming saturated at the high acidity end of the range investigated. This saturation implies a pre-equilibrium mechanism involving quinone ethide protonation on carbonyl oxygen for the acid catalyzed process, and analysis of the data shows the trans isomer to be a slightly stronger base than the cis isomer. The fact that two separate rate profiles are observed implies that the cationic intermediates formed by substrate protonation in these acid catalyzed processes do not interconvert on the time scale of the quinone ethide decay reactions, and that in turn indicates that the former ethide bonds of the cationic intermediates have substantial double-bond character; a lower limit of 10 kcal mol−1 may be estimated for the barrier to rotation about this bond.
Introduction
Quinone methides are synthetically useful reactive intermediates that also show pronounced biological activity; they have, for example, been implicated as the ultimate cytotoxins responsible for the effects of agents such as antitumor drugs, antibiotics and DNA alkylators.1 In biological systems where water is the ubiquitous medium this biological activity must operate against a background of wasteful quinone methide hydration reactions. In order to provide information about these hydrations, we have undertaken a detailed investigation of their kinetics and reaction mechanisms.We have found that hydration of the parent o-quinone methide, 1, occurs by an acid-catalyzed process in which rapid, pre-equilibrium protonation of the quinone methide on its carbonyl oxygen atom is followed by rate-determining nucleophilic capture of the ensuing o-hydroxybenzyl cation, 2, by a water molecule to give the o-hydroxybenzyl alcohol product, 3.2 We have also found that this acid catalysis can be saturated in the hydrolysis of more basic substrates such as o-quinone α-phenylmethide, 4, and o-quinone α-anisylmethide, 5, where increasing the hydronium ion concentration shifts the position of the pre-equilibrium protonation from quinone methide to benzyl cation.3

| (1) |

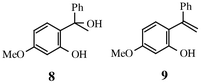
In this paper we report the results of our investigation of 5-methoxy-o-quinone α-phenylethide. This quinone ethide can exist in cis, 6, and trans, 7, isomeric forms; we have generated and studied both and in so doing have provided a comparison of cis and trans isomer reactivity. We produced these quinone ethides for the most part by photoelimination of water from the corresponding o-hydroxybenzyl alcohol, 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol, 8; this is a process well known to be a good source of quinone methides4 that we and others have employed before. We also generated the present quinone ethides in a few instances through photo-induced intramolecular proton transfer of 2-hydroxy-4-methoxy-α-phenylstyrene, 9, a process for which there is also precedent in other systems.5
It was once believed that quinone methide formation by photoelimination of o-hydroxybenzyl alcohols was facilitated by the markedly enhanced excited state acidity of phenols, and that the photoelimination occurred by excited state ionization of the phenolic group followed by expulsion of hydroxide ion from the benzyl alcohol moiety.4 The evidence upon which that view was based, however, has recently been discredited,3b and the mechanism of this reaction is consequently now not clear.
The reactions of the presently studied quinone ethides were fast, and we therefore employed flash photolytic techniques to follow their course.
Experimental
Materials
1-(2-Hydroxy-4-methoxyphenyl)-1-phenylethanol (8) was prepared by treating 2-hydroxy-4-methoxybenzophenone (Aldrich) with methylmagnesium chloride in anhydrous THF solution. The product, a colorless solid, mp 89–91 °C, was purified by recrystallization from pentane. The yield of pure material was 82%; it was characterized by its NMR and mass spectra: 1H NMR (200 MHz, CDCl3) δ(ppm) = 8.59 (s, 1H), 7.32–7.24 (m, 5H), 6.88–6.83 (m, 1H), 6.36–6.33 (m, 2H), 3.68 (s, 3H), 3.37 (s, 1H), 1.86 (s, 3H); 13C NMR (75 MHz, CDCl3): δ(ppm) = 160.64, 156.97, 146.30, 128.50, 128.00, 127.72, 125.71, 123.32, 105.73, 102.96, 79.12, 55.56, 31.44; HRMS: m/z = 244.1099 (calc), 244.1100 (found).2-Hydroxy-4-methoxy-α-phenylstyrene (9) was prepared by treating 2-hydroxy-4-methoxybenzophenone with the Wittig reagent made by dehydrohalogenating methyltriphenylphosphonium bromide with phenyllithium in THF solution. The product was purified by chromatography on silica gel using hexane as the eluent. This gave a 66% yield of purified material as a colorless, viscous liquid: 1H NMR (200 MHz, CDCl3): δ(ppm) = 7.33–7.30 (m, 5H), 7.03–6.99 (m, 1H), 6.50–6.46 (m, 2H), 5.76 (s, 1H), 5.36–5.34 (m, 2H), 3.76 (s, 3H); 13C NMR (75 MHz, CDCl3): δ(ppm) = 160.89, 154.44, 145.34, 140.08, 131.32, 128.84, 128.75, 127.42, 120.32, 116.25, 106.98, 101.38, 55.66; HRMS: m/z (M − 1) = 225.0916 (calc), 225.0916 (found).
Kinetics
Rate measurements were made using microsecond6 and nanosecond7 (λexc = 248 nm) flash photolysis systems that have already been described.6,7 Photolysis substrates were supplied at concentrations of the order of 10−4 M, and the temperature of all reacting solutions was controlled at 25.0 ± 0.05 °C. Reactions were monitored by following the decay of quinone methide absorbance at λ = 400 nm. The decay traces were biphasic, and the data were therefore analyzed by least squares fitting of double exponential expressions.Product analysis
Product analysis was conducted by HPLC using a Varian Vista 5500 instrument with a NovoPak C18 reverse-phase column and methanol–water (70 ∶ 30 = v/v) as the eluent. Reaction solutions, containing substrate at the concentration used for rate measurements, were subjected to a single flash from our microsecond flash system, and the compositions of these solutions were then determined by comparing retention times and UV spectra with those of authentic samples. The temperature of the reacting solutions was controlled at 25.0 ± 0.05 °C.Results
Photoelimination
Flash photolysis of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol (8) produced transient species with strong absorbance at λ = 400 nm, whose decay was biphasic and, as Fig. 1 illustrates, conformed to a double exponential rate law with good precision. Rates of this decay were measured in aqueous perchloric acid and sodium hydroxide solutions and in biphosphate ion and bicarbonate ion buffers. The data so obtained are summarized in Tables S1–S3.‡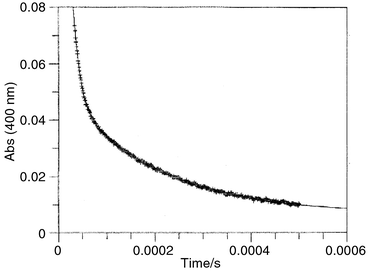 | ||
| Fig. 1 Decay of the transient absorbance formed upon flash photolysis of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol in 0.25 M aqueous perchloric acid at 25 °C. Least squares analysis using a double exponential rate law produced the rate constants k1 = (7.12 ± 0.22) × 104 s−1 and k2 = (5.14 ± 0.05) × 103 s−1. | ||
The measurements in buffers were made using series of solutions of constant buffer ratio and constant ionic strength (0.10 M), and therefore constant hydronium ion concentration, but varying total buffer concentration. The data were analyzed by least squares fitting of the buffer dilution expression shown in eqn. (2).
| kobs = kint + kcat[buffer] | (2) |
 | ||
| Fig. 2 Rate profiles for the decay of cis-5-methoxy-o-quinone α-phenylethide, ○, and trans-5-methoxy-o-quinone α-phenylethide, Δ, in aqueous solution at 25 °C. | ||
Photo-induced proton transfer
Flash photolysis of 2-hydroxy-4-methoxy-α-phenylstyrene (9) also gave transient species absorbing at λ = 400 nm whose decay was biphasic and conformed to a double exponential rate law well, just like the transients formed by flash photolysis of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol (8). Rates of decay of these transients obtained from the styrene were measured in perchloric acid solutions; the data so obtained are summarized in Table S4 and are displayed in Fig. 3. | ||
Fig. 3 Comparison of the rates of decay of the two transients produced by flash photolytic intramolecular photoprotonation of 2-hydroxy-4-methoxy-α-phenylstyrene (○ and Δ) with the rates of decay of the two transients produced by flash photolytic photoelimination of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol, — and ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) . These lines represent the relevant portions of the rate profiles shown in Fig. 2. . These lines represent the relevant portions of the rate profiles shown in Fig. 2. | ||
Discussion
Irradiation of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol (8) can be expected to lead to photoelimination of water giving two isomeric o-quinone ethides (6 and 7), which differ only in the disposition of methyl and phenyl groups about the ethide double bond, eqn. (3), and should consequently show similar but not exactly the same reactivity. This expectation is borne out by the present flash photolysis experiments, which produced transient species that decayed according to a double exponential rate law and gave the two closely similar rate profiles shown in Fig. 2.
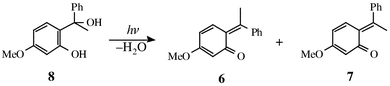
| (3) |
These two rate profiles, moreover, are characteristic of quinone methide decay. Just like the rate profiles for other quinone methides that we have studied,2,3 they have acid-catalyzed portions, attributable to reaction by the pre-equilibrium protonation mechanism of eqn. (1), and uncatalyzed and hydroxide-ion catalyzed portions, attributable to reaction of the quinone methide with water and hydroxide ion.
Fig. 2 shows that acid catalysis of the present reactions becomes saturated at the high acidity end of the range investigated. This, too, is characteristic of the reactions of the more basic quinone methides,3 such as the present substrates, and is caused by a shift of the position of the pre-equilibrium from unprotonated to protonated substrate.3Fig. 2 shows further that saturation of acid catalysis is followed by mild inhibition at the very highest acidities examined. This again has been observed in the reaction of other quinone methides3 and is caused by ion-pair formation between the carbocation reaction intermediates and perchlorate ion, supplied by the fully ionized perchloric acid catalyst, which shifts some of the carbocation into a less reactive ion pair form (R+ClO4−). Such ion-pair formation has been well documented in other systems.9
The reaction scheme that incorporates all of these features is shown in eqn. (4), and the rate law that applies relating observed rates of quinone methide decay, kobs, to the microscopic constants of eqn. (4) is given in eqn. (5). Least squares fitting of this expression gave the results listed in Table 1. These results were used to draw the rate profile lines shown in Fig. 2; it may be seen that the experimental data conform to the reaction model of eqns. (4) and (5) with good precision.

| (4) |

| (5) |
| cis | trans | cis/trans | |
|---|---|---|---|
| a The constants are defined by the reaction scheme of eqn. (4). | |||
| k w/105 s−1 | 2.71 ± 0.54 | 0.118 ± 0.012 | 23.0 |
| k uc/s−1 | 13.1 ± 0.9 | 0.459 ± 0.28 | 28.6 |
| k HO/102 M−1 s−1 | 3.26 ± 0.21 | 0.320 ± 0.014 | 10.2 |
| K SH/10−1 M | 5.51 ± 1.21 | 2.30 ± 0.27 | 2.40 |
| pKSH | 0.26 ± 0.10 | 0.64 ± 0.05 | |
| K IP/M−1 | 2.90 ± 0.89 | 2.21 ± 0.41 | 1.31 |
We found in our study of the parent unsubstituted o-quinone methide, 1, that substrate protonation was followed by nucleophilic capture of the ensuing carbocation by water to give a benzyl alcohol product, eqn. (1). In the present case, however, the carbocation formed, 10, has an α-methyl substituent, and that makes available another reaction channel involving proton elimination to give a styrene product, 9, eqn. (6). Examination of spent reaction mixtures by HPLC analysis shows that this does in fact take place. The corresponding benzyl alcohol, 8, however, is formed as well, in a roughly comparable amount. The rate constant, kw, for reaction of the carbocation with water is therefore the sum of individual rate constants for the two parallel elimination and nucleophilic capture processes.
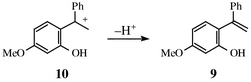
| (6) |
Further support for the interpretation of our experimental data according to the reaction scheme of eqn. (4) comes from flash photolysis experiments done using 2-hydroxy-4-methoxy-α-phenylstyrene, 9, as the substrate. Irradiation of an o-hydroxystyrene such as this is known to increase the basicity of the β-carbon atom of its ethylenic group markedly,10 to the point where proton transfer from the hydroxy group to this carbon atom readily takes place, producing an o-quinone ethide.5 As eqn. (7) demonstrates, this reaction with the present styrene would be expected to generate the same isomeric cis and trans quinone ethides as photoelimination of water from 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol, eqn. (3). Flash photolysis of this styrene does in fact produce two transient species, which, as Fig. 3 demonstrates, decay at the same rates as do the transients generated by flash photolysis of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol (8). This close correspondence of reaction rates shows that the transients produced by photo-induced proton transfer of 2-hydroxy-4-methoxy-α-phenylstyrene are the same as those generated by photoelimination of 1-(2-hydroxy-4-methoxyphenyl)-1-phenylethanol, and that reinforces identification of these transients as the cis and trans 5-methoxy-o-quinone α-phenylethides, 6 and 7.

| (7) |
Protonation of these quinone ethides on oxygen in the first step of their acid-catalyzed decay reactions produces cations in which the former ethide bonds have lost some of their double-bond character, and rotation about these bonds might therefore make these cations interconvertible. Because the protonation reactions are rapidly reversible, such cation interconversion would provide a mechanism for interconversion of the quinone ethides themselves, and that, by the Curtin–Hammett Winstein–Holness principle,11 would produce a single rather than a double rate profile. The fact that a double rate profile is observed, therefore, is evidence that such interconversion does not take place. The rate constant for bond rotation in the less stable of the two cations must therefore be smaller than the rate constant for capture of this cation by water, krot < kw (= 2.7 × 105 s−1). This corresponds to a cation rotational barrier greater than 10 kcal mol−1. This is consistent with the expectation that the positive charge of this cation will be stabilized by delocalization into the hydroxy and methoxy groups of the former quinoid ring, and that, as the resonance scheme of eqn. (8) demonstrates, confers double bond character on the former ethide bond.

| (8) |
This positive charge will, of course, be stabilized by delocalization into the unsubstituted phenyl group of these cations, as well. Because of steric crowding, such stabilization will be less effective in the cis cation, 6, than in the trans cation, 7, and that will make the cis isomer more reactive than the trans. The two values of kw determined from the profiles may thus be assigned to the two isomers on this basis: kw = 2.71 × 105 s−1 for product formation from the cis cation and kw = 1.18 × 104 s−1 for product formation from the trans cation.
This assignment of rate constants also determines distribution of the remaining profile parameters, inasmuch as all of the parameters determined from a given profile must belong to the same isomer. That means that all of the rate and equilibrium constants derived from the upper profile of Fig. 2 with the larger value of kw refer to the cis quinone ethide and all of the constants derived from the lower profile refer to the trans quinone ethide. These assignments are detailed in Table 1.
These results show the cis cation to be a stronger oxygen-acid than the trans cation; i.e., (KSH)cis > (KSH)trans. This is consistent with the assignment of a lower stability to the cis cation made above: this cation will be less easily formed and therefore more easily ionized. The ratio of equilibrium constants, however, (KSH)cis/(KSH)trans = 2.4, is considerably less than the ratio of product formation rate constants (kw)cis/(kw)trans = 23, which means that there must be an offsetting stability difference in the quinone ethide products of the cation ionization reactions governed by KSH, i.e., that the cis quinone ethide must be less stable than the trans quinone ethide. This is consistent with the fact that steric crowding between the unsubstituted phenyl group and the carbonyl oxygen atom in the cis quinone ethide can be expected to be more severe, and therefore more destabilizing, than the steric crowding between the methyl group and the carbonyl oxygen atom in the trans quinone ethide.
This lesser stability of the cis quinone methide makes it the more reactive isomer in the non-acid-catalyzed reactions of these quinone ethides with water and hydroxide ion, i.e., (kuc)cis/(kuc)trans = 28.6 and (kHO)cis/(kHO)trans = 10.2. The greater ratio for the uncatalyzed reaction than for the hydroxide-ion-catalyzed process is also consistent with the fact that the uncatalyzed reaction is the slower process with a later, more product-like transition state in which more of the initial state steric crowding of the cis cation is relieved.
The ion pair formation constants for the two cations, on the other hand, are much the same: (KIP)cis/(KIP)trans = 1.31. These constants are very probably determined only by the shapes and overall charges of the species involved, and these are not very different for the cis and trans cations.
Acknowledgement
We are grateful to the Natural Sciences and Engineering Research Council of Canada for financial support of this work.References
- (a) M. G. Peter, Chemical modification of biopolymers by quinones and quinone methides, Angew. Chem., Int. Ed. Engl., 1989, 28, 555–570 CrossRef; (b) J. L. Bolton, E. Pisha, F. Zhang and S. Qiu, Role of quinoids in estrogen carcinogenesis, Chem. Res. Toxicol., 1998, 10, 1113–1127 CrossRef CAS; (c) P. Pande, J. Shearer, J. Yang, W. A. Greenberg and S. E. Rokita, Alkylation of nucleic acids by a model quinone methide, J. Am. Chem. Soc., 1999, 121, 6773–6779 CrossRef CAS.
- Y. Chiang, A. J. Kresge and Y. Zhu, Flash photolytic generation of ortho-quinone methide in aqueous solution and study of its chemistry in that medium, J. Am. Chem. Soc., 2001, 123, 8089–8094 CrossRef CAS.
- (a) Y. Chiang, A. J. Kresge and Y. Zhu, Reactive intermediates. Some chemistry of quinone methides, Pure Appl. Chem., 2000, 72, 2299–2308 Search PubMed; (b) Y. Chiang, A. J. Kresge and Y. Zhu, Flash photolytic generation of o-quinone α-phenylmethide and o-quinone α-p-anisylmethide in aqueous solution and investigation of their reactions in that medium. Saturation of acid-catalyzed hydration, J. Am. Chem. Soc Search PubMed , in the press.
- (a) L. Diao, C. Yang and P. Wan, Quinone methide intermediates from the photolysis of hydroxybenzyl alcohols in aqueous solution, J. Am. Chem. Soc., 1995, 117, 5369–5370 CrossRef CAS; (b) P. Wan, B. Barker, L. Diao, M. Fischer, Y. Shi and C. Yang, Quinone methides: relevant intermediates in organic chemistry, Can. J. Chem., 1996, 74, 465–475 CAS.
- P. Kalanderopoulos and K. Yates, Intramolecular proton transfer in photohydration reactions, J. Am. Chem. Soc., 1986, 108, 6290–6295 CrossRef.
- Y. Chiang, M. Hojatti, J. R. Keeffe, A. J. Kresge, N. P. Schepp and J. Wirz, Vinyl alcohol: generation and decay kinetics in aqueous solution and determination of the tautomerization equilibrium constant and acid dissociation constants of the aldehyde and enol forms, J. Am. Chem. Soc., 1987, 109, 4000–4009 CrossRef CAS.
- J. Andraos, Y. Chiang, C.-G. Huang, A. J. Kresge and J. C. Scaiano, Flash photolytic generation and study of ketene and carboxylic acid enol intermediates formed by the photolysis of diazonaphthoquinones in aqueous solution, J. Am. Chem. Soc., 1993, 115, 10605–10610 CrossRef CAS.
- R. G. Bates, Determination of pH Theory and Practice, Wiley, New York, 1973, p. 49 Search PubMed.
- See J. Crugeiras and H. Maskill, The 4,4′-dimethoxytrityl carbenium ion by ionization of 4,4′-dimethoxytrityl alcohol in acetonitrile-aqueous perchloric and nitric acids containing electrolytes: kinetics, equilibria, and ion-pair formation, Can. J. Chem., 1999, 77, 530–536 Search PubMed; J. Crugeiras and H. Maskill, Kinetics and thermodynamics of ionization of 4,4′-dimethoxytrityl alcohol in acetonitrile–aqueous acids: determination of enthalpies and entropies of activation and of reaction, J. Chem. Soc., Perkin Trans. 2, 2000, 441–445 CrossRef CAS and references cited therein.
- P. Wan, S. Culshaw and K. Yates, Photohydration of aromatic alkenes and alkynes, J. Am. Chem. Soc., 1982, 104, 2509–2515 CrossRef CAS.
- J. I. Seeman, Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett/Winstein-Holness kinetics, Chem. Rev., 1983, 83, 83–134 CrossRef CAS.
Footnotes |
| † The IUPAC name for a quinone alkide is alkylidenecyclohexadienone. |
| ‡ Electronic supplementary information (ESI) available: Tables S1–S4. See http://www.rsc.org/suppdata/pp/b1/b107860g/ |
| This journal is © The Royal Society of Chemistry and Owner Societies 2002 |