Photochemical and photophysical properties of poly(propylene amine) dendrimers with peripheral naphthalene and azobenzene groups†
Fritz
Vögtle
*a,
Marius
Gorka
a,
Richard
Hesse
a,
Paola
Ceroni
b,
Mauro
Maestri
b and
Vincenzo
Balzani
*b
aKekulé-Institut für Organische Chemie und Biochemie der Universität Bonn, Gerhard-Domagk Strasse 1, D-53121, Bonn, Germany. Fax: 49 228 735662; Tel: 49 228 735672E-mail: voegtle@uni-bonn.de
bDipartimento di Chimica “G. Ciamician”, Università di Bologna, via Selmi 2, I-40126, Bologna, Italy. Fax: 39 051 2099560; Tel: 39 051 2099456E-mail: balzani@ciam.unibo.it
First published on 2nd January 2002
Abstract
We report the preparation, the absorption spectra, and the photophysical and photochemical properties in dichloromethane solution of four dendrimers of the poly(propylene amine) family (indicated by POPAM or PPI) functionalised with naphthalene and trans-azobenzene units. Each dendrimer Gn, where n = 1 to 4 is the generation number, comprises 2n + 1 − 2 (i.e., 30 for G4) tertiary amine units in the interior and 2n + 1 (i.e., 32 for G4) naphthalene and trans-azobenzene units in the periphery. For comparison purposes, the photophysical and photochemical properties of model compounds of the peripheral units have also been investigated. We have found that the fluorescence of the naphthalene units is quenched by the tertiary amines (via electron transfer) as well as by the trans-azobenzene units (via energy transfer). The quantum yields of the trans→cis and cis→trans photoisomerisation of the azobenzene units have been measured at various excitation wavelengths. Quenching of the fluorescence of the excited naphthalene unit by the trans- and cis-azobenzene units is accompanied by the sensitisation of the cis→trans (but not of the trans→cis) isomerisation. The rate constant of the thermal cis→trans isomerisation of the azobenzene units has also been measured. Comparison of the results obtained for model compounds and for the G4 dendrimer shows that the dendritic structure favours the trans configuration of the azobenzene units (“dendritic effect”), presumably because it is less demanding in terms of space and causes less crowding on the dendrimer surface.
Introduction
Dendrimers1 are complex, but well defined chemical compounds, that can contain selected functions in predetermined sites of their tree-like multi-branched structure and host molecules or ions in their internal cavities. Because of such unique properties, dendrimers are currently attracting increasing attention for a wide range of potential applications in such different fields as medicine, biology, chemistry, physics, and engineering.1 Dendrimers containing photoactive components (for some recent papers, see refs. 2–10) are particularly interesting since (i) luminescence signals offer a handle to better understand the dendritic structures and superstructures, (ii) cooperation among the photoactive components can allow the dendrimer to perform useful functions such as light harvesting, (iii) changes in the photophysical properties can be exploited for sensing purposes with signal amplification, and (iv) photochemical reactions can change the structure and other properties of dendrimers.Continuing our studies in this field,4 we have prepared four dendrimers of the poly(propylene amine) family (usually indicated by POPAM or PPI) functionalised with both naphthalene and azobenzene units (Fig. 1). Each dendrimer Gn, where n = 1 to 4 is the generation number, contains 2n + 1 − 2 (i.e., 30 for G4) tertiary amine units in the branches and 2n + 1 (i.e., 32 for G4) naphthalene and azobenzene units in the periphery. We have investigated the absorption spectra and the photophysical and photochemical properties of these dendrimers in dichloromethane solution. For comparison purposes, model compounds of the luminescent and photoreactive units of the dendrimers have also been investigated. We have shown that several photochemical and photophysical processes, including electron transfer and energy transfer quenching, photoisomerisation reactions, and energy transfer sensitisation reactions, take place in these systems. Particularly interesting are the trans→cis photoisomerisation reaction of the azobenzene units and the back cis→trans isomerisation that can take place both thermally and photochemically (Fig. 2). Such reactions, which involve a large structural rearrangement and a big change in the dipole moment,11 have been thoroughly investigated and extensively used for a variety of purposes.11,12 Several papers on dendrimers containing azobenzene units have also appeared.13
 | ||
| Fig. 1 Structural formulas of the dendrimers. | ||
 | ||
| Fig. 2 Isomerisation reaction of azobenzene. | ||
Results and discusssion
Model compounds
In order to better understand the properties of the Gn dendrimers, we have investigated the TA-NA-tAZ triad (Fig. 3: TA, NA, and tAZ stand for tertiary amine, naphthalene and trans-azobenzene moieties, respectively), which is the model compound of the peripheral units of the dendrimers. We have also investigated the properties of the TA-NA dyad and of trans-4-methylazobenzene (tMeAZ), which are model compounds of the TA-NA and tAZ moieties of the TA-NA-tAZ triad.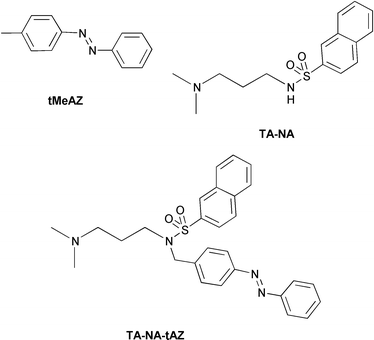 | ||
| Fig. 3 Structural formulas of model compounds of the peripheral units of the dendrimers. | ||
trans-4-Methylazobenzene (tMeAZ)
trans-4-Methylazobenzene exhibits a very intense π→π* absorption band in the UV region, a less intense n→π* absorption band in the visible, no emission, and trans→cis photoisomerisation. In dichloromethane solution, we have found the following data: λmax = 328 nm, ε = 23400 M−1 cm−1; λmax = 442 nm, ε = 700 M−1 cm−1; Φt→c 0.20 and 0.18 for 287 and 365 nm excitation, respectively (Table 1).| Photochemical isomerisation (λirr/nm)a | Thermal isomerisation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 287 | 313 | 365 | 436b | 323 K | |||||
| Φ t→c | % trans | Φ t→c | % trans | Φ t→c | % trans | Φ c→t | % trans | k c→t/10−5 s−1 | |
| a Quantum yield and molar fraction of the trans isomer at the photostationary state. b Photochemical cis→trans isomerisation performed on solutions that had previously been brought to the photostationary state by irradiation at 365 nm. | |||||||||
| MeAZ | 0.20 | 62 | — | — | 0.18 | 7 | 0.70 | 93 | 4.5 |
| TA-NA-AZ | 0.16 | 69 | 0.15 | 36 | 0.18 | 7 | 0.71 | 91 | 4.8 |
| G4 | 0.14 | 73 | 0.11 | 48 | 0.14 | 13 | 0.90 | 91 | 7.5 |
The cis-azobenzene species obtained by irradiation of the corresponding trans isomers are known to undergo a back cis→trans photoisomerisation reaction, which also occurs, slowly, in the dark.14 Therefore continuous irradiation of a trans (or cis) azobenzene species leads to a photostationary state in which the concentrations of the two isomers, t and c, depend on the photoisomerisation quantum yields and molar absorption coefficients of the two isomers at the excitation wavelength (eqn. (1)).
| [t]εtΦt→c = [c]εcΦc→t | (1) |
The concentration of the two isomers at the photostationary state (Table 1) can be measured with good accuracy, whereas the quantum yields of photoisomerisation are affected by larger uncertainties caused by the fact that both isomers absorb light and undergo photoisomerisation. The highest molar fraction (93%) for the cis isomer was obtained by 365 nm excitation. Starting from this solution, the quantum yield of the back cis→trans isomerisation was measured for irradiation at 436 nm (Table 1), where the absorbance of the cis isomer is higher than that of the trans isomer. We have also measured the rate constant of the thermal cis→trans isomerisation reaction, which in our solvent is 4.5 × 10−5 s−1 at 323 K.
TA-NA dyad
The absorption spectrum of TA-NA (Fig. 4a) shows the characteristic bands of the naphthalene chromophoric group, whereas the emission spectrum (Fig. 4b) consists of a relatively weak naphthalene-type fluorescence band (λmax = 345 nm, Φf = 0.016) and a much weaker, broad band with λmax = 530 nm (Fig. 4b, inset). The overall emission shows a two-exponential decay (lifetimes 0.3 and 7.2 ns), with the shorter lifetime corresponding to the band at longer wavelength. Upon addition of trifluoroacetic acid, significant spectral changes were observed, which mostly took place during the addition of the first equivalent of acid and reached plateaus before addition of two equivalents. In the absorption spectrum (Fig. 4a), a small decrease in absorbance was observed in the 250–280 nm spectral region; in the emission spectrum (Fig. 4b), the intensity of the emission band with λmax = 345 nm became much stronger (Φf = 0.13), the very weak band with λmax = 530 nm disappeared, and a monoexponential decay of the emission intensity was observed with τ = 7.3 ns. These results can straightforwardly be interpreted as follows. The protonated TAH+-NA species exhibits a simple photophysical behaviour because the naphthalene-based chromophoric unit is unperturbed. In the unprotonated TA-NA species, however, the potentially fluorescent excited state of the naphthalene-based moiety is quenched by the amine group by electron transfer and formation of exciplexes.15 The latter species are responsible for the broad and weak emission with λmax = 530 nm. | ||
| Fig. 4 Absorption (a) and emission (b) spectra of the TA-NA dyad in dichloromethane solution before (full line) and after (dashed line) addition of trifluoroacetic acid. | ||
TA-NA-tAZ triad
The absorption spectrum of the TA-NA-tAZ triad is dominated by the bands of the tAZ moiety. The emission spectrum shows a weak band with λmax = 370 nm with lifetime <1 ns. Such an emission is partly reabsorbed by the strong azobenzene absorption band with λmax = 324 nm. After correction for reabsorption, the emission maximum moves to 345 nm, showing that the observed band is the naphthalene-type fluorescence of the NA moiety previously observed for the TA-NA dyad (Fig. 4). Contrary to what happens for the fluorescence of the TA-NA dyad, however, the fluorescence quantum yield of the triad is hardly affected by addition of acid and the corrected quantum yield (Φf = 5 × 10−4) is much smaller than that exhibited by the unprotonated (0.016) and protonated (0.13) TA-NA dyad. This result can be accounted for by considering that the triad contains not only the TA, but also the tAZ moiety, and that the fluorescent excited state of the NA moiety is quenched by the tAZ unit much faster than by the TA one.Energy transfer quenching by the Förster-type mechanism16 is indeed expected to be very fast because of the strong overlap between the NA fluorescence and the singlet ππ* absorption of the tAZ unit. Judging from the energy of the fluorescent excited state of the NA moiety (3.8 eV), the literature oxidation potential of naphthalene (+1.54 V vs. SCE in acetonitrile)17 and the measured reduction potential of tAZ in the triad (−1.33 V vs. SCE in acetonitrile), electron transfer quenching is also allowed.
Upon light excitation of the TA-NA-tAZ triad, strong spectral changes are observed as expected for the trans→cis isomerisation of the tAZ moiety. On continued irradiation, a photostationary state is reached. The quantum yield values, the molar fractions of the trans isomer at the photostationary states, and the rate constant of the thermal cis→trans isomerisation reaction in the triad are displayed in Table 1. Clearly, the photoisomerisation behaviour of the AZ moiety of the TA-NA-AZ triad is practically the same as that of the MeAZ model compound. The lower quantum yield of trans→cis photoisomerisation of the TA-NA-tAZ triad upon 287 nm irradiation can be accounted for by the fact that at this wavelength the absorbed light is distributed between the tAZ (72%) and NA (28%) components. This result also shows that, although the fluorescence of the NA unit is quenched by the tAZ unit (vide supra), quenching does not cause sensitisation of the trans→cis isomerisation. By contrast, the higher molar fraction of trans isomer upon 287 nm excitation compared with MeAZ suggests that the cis→trans isomerisation in the triad is sensitised by the NA unit. Selected sensitisation of the cis→trans isomerisation has previously been found in a dansyl–azobenzene dyad.18 Finally, it can be noted that the rate of the thermal cis→trans isomerisation is practically the same for MeAZ and TA-NA-AZ.
Gn dendrimers
 | ||
| Fig. 5 Molar absorption coefficients of the dendrimers at 325 and 443 nm. | ||
 | ||
| Fig. 6 Spectral changes observed upon irradiation of dendrimer G4 (1.34 × 10−6 M, dichloromethane solution, 298 K) with 365 nm light. Curve a is the initial spectrum; curve b is the spectrum at the photostationary state. Inset shows the corresponding change in the molar fractions of the trans and cis isomers during irradiation. | ||
Comparison of the results obtained for MeAZ, TA-NA-AZ, and G4 (Table 1) shows some peculiar features: (i) at each irradiation wavelength, the quantum yield of the trans→cis photoisomerisation is smaller for G4 compared with MeAZ and TA-NA-AZ; (ii) conversely, the quantum yield of the cis→trans isomerisation is larger for the dendrimer than for the model compounds; (iii) the molar fraction of trans-azobenzene units at the photostationary state is larger for the dendrimer than for the model compounds (more specifically, the ratio of the trans molar fraction between G4 and TA-NA-AZ increases as the photostationary state contains a lower trans molar fraction); (iv) the rate of the thermal cis→trans isomerisation reaction is faster for the azobenzene units of the dendrimer than for those of the model compounds. All these results indicate that the dendritic structure stabilizes the trans isomer of the peripheral AZ units. This result is not surprising since the isomerisation reaction of the azobenzene unit (Fig. 2) is known to involve a large structural rearrangement and a big change in the dipole moment. The trans form is planar, with a distance of 9.0 Å between the para carbon atoms, and has no dipole moment, whereas the cis form is nonplanar, with the para carbon atoms at 5.5 Å, and exhibits a dipole moment of 3.0 D.11 Both steric crowding and dipole–dipole repulsion can therefore destabilize the formation of the cis isomer in the peripheral moieties of the dendrimer.
Conclusion
We have prepared poly(propylene amine) dendrimers (POPAM, PPI) bearing two chemical functions in each peripheral branch. One of the functions is the fluorescent naphthalene unit and the other one the photoactive azobenzene unit. The largest dendrimer, G4, contains 32 naphthalene and 32 azobenzene units, besides 30 tertiary amine functions (which are known to be relatively strong reductants) in its interior. Because of their proximity, the various functional groups of the dendrimer may interact, so that the properties of the dendrimers are different from those exhibited by the separated functional units. Both the naphthalene fluorescence and the azobenzene photoisomerisation can be observed in the dendrimer, but it has been shown that (i) the fluorescent excited state of the naphthalene units is substantially quenched by both the amines and the azobenzene units, and (ii) in the latter case the fluorescence quenching is accompanied by the photosensitised isomerisation of the cis→trans but not of the trans→cis reaction. Furthermore, we have found a clear, even if not very large, dendritic effect which disfavours the trans→cis photoisomerisation of the AZ units present in the dendrimer periphery. Such an effect is likely to be related to the larger space occupied by the cis isomeric units in the crowded periphery of the dendrimer and/or to electrostatic repulsions among the dipolar cis species. These results suggest that the trans and cis forms of G4 could exhibit different capacities to host other molecules, thereby functioning as photoswitchable dendritic boxes. This hypothesis is currently under investigation in our laboratories.Experimental
Synthesis and characterization of the Gn dendrimers (Fig. 1)
All synthetic experiments were routinely carried out under a dry nitrogen atmosphere. Starting materials (POPAM-dendrimers, generation 1–4) were purchased from Aldrich.4-Methylazobenzene (tMeAZ) was prepared according to published procedure.20
4-Bromomethylazobenzene
A solution containing 6.10 g (31.10 mmol) of 4-methylazobenzene (tMeAZ), 5.60 g (31.20 mmol) of NBS and a few crystals of AIBN in CCl4 was refluxed for 2 h and irradiated with 300 W light source. The precipitated succinimide was filtered off and the filtrate was evaporated under reduced pressure. Recrystallisation from toluene yielded 4.00 g (47%) of an orange solid. Mp 114 °C. FAB-MS: m/z = 274.2 (M+, 10%), 195 (M − Br+, 13%). C13BrH11N2 (274.01).General synthetic procedure for the naphthalenesulfonamide-dendrimers 1–4
1/n equivalents of the starting poly(propylene amine)-(POPAM)-dendrimer-(NH2)n and x equivalents of triethylamine (Scheme 1) were dissolved in 150 ml of dry dichloromethane (n is the number of primary amino groups of the dendrimer and x corresponds to the 1.5–3-fold excess of naphthalene-2-sulfonyl chloride). To a refluxing mixture a solution of x equivalents of naphthalene-2-sulfonyl chloride in 50 ml dichloromethane was added dropwise. After stirring for three days under reflux the mixture was stirred at 25 °C for two days. The solvent was removed in vacuo and the residue was collected in dichloromethane. After washing with water, aq. NaHCO3 and again with water the organic phase was dried with MgSO4. Further purification was achieved by column chromatography (SiO2, 40–63 μm, 1. CH2Cl2, 2. CH2Cl2–MeOH–Et3N: 100 ∶ 5–12 ∶ 1–2) yielding a bright yellow solid. | ||
| Scheme 1 | ||
R f: 0.28 (SiO2, 40–63 μm, 1. CH2Cl2, 2. CH2Cl2–MeOH–Et3N: 100 ∶ 5 ∶ 1). FAB-MS: m/z = 1049.3 (M + H+, 90%). C54H60N6O8S4 (1048.34).
General synthetic procedure for the azobenzene–naphthalenesulfonamide-dendrimers G1–G4
1/n equivalents of the monosubstituted POPAM-dendrimer (naphthalenesulfonamide-dendrimer 1–4) and 4–5-fold excess of potassium carbonate (Scheme 1) were dissolved in 150 ml of dry DMF. To this suspension a solution of x equivalents of 4-bromomethylazobenzene in 50 ml DMF was added dropwise (n is the number of secondary amino groups of the dendrimer and x is the 1.5–3-fold excess of 4-bromomethylazobenzene). The mixture was stirred at 25 °C for five days. After filtering the undissolved K2CO3 the solvent was removed in vacuo and the residue was collected in dichloromethane. After washing with water, aq. NaHCO3 and again with water the organic phase was dried with MgSO4. Further purification was achieved by column chromatography (SiO2, 40–63 μm, 1. CH2Cl2, 2. CH2Cl2–MeOH–Et3N: 100 ∶ 2–10 ∶ 1–2) yielding a bright yellow solid.Photochemical and photophysical experiments
Experiments were performed in air equilibrated dichloromethane solution. Spectroscopic equipment and techniques have been described elsewhere.4aContinuous irradiation experiments were performed in a 1 cm spectrophotometric cell by a medium pressure mercury lamp. Interference filters (Oriel) were used to select the irradiation wavelength. The intensity of the incident light (3.2 × 10−8 einstein min−1 at 287 nm, 1.4 × 10−7 einstein min−1 at 313 nm and 1.9 × 10−7 einstein min−1 at 365 nm; 2.5 × 10−7 einstein min−1 at 436 nm) was measured with the ferrioxalate actinometer.16 The photoisomerisation quantum yield (Φc→t or Φt→c) was calculated by extrapolation to zero time of the values obtained for short irradiation periods.
The estimated experimental errors are: ±2 nm on the band maximum, ±5% on the molar absorption coefficient, ±10% on the fluorescence and photoisomerisation quantum yields, ±5% on the fluorescence lifetime, and ±5% on the molar fractions of the two isomers at the photostationary state.
Electrochemical experiments
The electrochemical experiments were carried out in argon-purged 0.05 M tetraethylammonium hexafluorophosphate (TEAPF6) acetonitrile (Romil Hi-Dry™) solutions at 298 K. The equipment used has been previously described.4dAcknowledgements
This work has been supported by MURST (Artificial Photosynthesis), University of Bologna (Funds for Selected Topics), and CNR (Sensori Fluorescenti Supramolecolari).Fritz Vögtle and Marius Gorka would like to thank the Fonds der Chemischen Industrie (Frankfurt/Main) and DECHEMA e.V. (Frankfurt/Main) for support.
References
- (a) R. Newkome, C. Moorefield and F. VögtleDendritic Molecules: Concepts, Syntheses, Perspectives, VCH, Weinheim, 1996; 2nd edn., 2001 Search PubMed; (b) A. W. Bosman, H. M. Janssen and E. W. Meijer, About dendrimers: structure, physical properties, and applications, Chem. Rev., 1999, 99, 1665 CrossRef CAS; (c) G. R. Newkome, E. He and C. Moorefield, Suprasupermolecules with novel properties: metallodendrimers, Chem. Rev., 1999, 99, 1689 CrossRef CAS; (d) S. Hecht and J. M. J. Fréchet, Dendritic encapsulation of function: applying nature's site isolation principle from biomimetics to materials science, Angew. Chem., Int. Ed., 2001, 40, 75; (e) A. Juris, M. Venturi, P. Ceroni, V. Balzani, S. Campagna and S. Serroni, Dendrimers based on electroactive metal complexes. Recent advances, Collect. Czech. Chem. Commun., 2001, 66, 1 CrossRef CAS; (f) A. Archut, S. Gestermann, R. Hesse, C. Kauffmann and F. Vögtle, Selective Activation, Different Branching and Grafting of Poly(propyleneimine)-Dendrimers, Synlett, 1998, 546–548 CAS.
- (a) V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Designing dendrimers based on transition-metal complexes. Light-harvesting properties and predetermined redox patterns, Acc. Chem. Res., 1998, 31, 26 CrossRef CAS; (b) V. Balzani, P. Ceroni, A. Juris, M. Venturi, S. Campagna, F. Puntoriero and S. Serroni, Metal-containing photoactive dendrimers. Recent advances, Coord. Chem. Rev., 2001, 219–221, 545 CrossRef CAS.
- S. F. Swallen, Z. Zhengguo, J. S. Moore and R. Kopelman, Correlated excimer formation and molecular rotational dynamics in phenylacetylene dendrimers, J. Phys. Chem. B, 2000, 104, 3988 CrossRef CAS.
- (a) V. Balzani, P. Ceroni, S. Gestermann, C. Kauffmann, M. Gorka and F. Vögtle, Dendrimers as fluorescent sensors with signal amplification, Chem. Commun., 2000, 853 RSC; (b) F. Vögtle, S. Gestermann, C. Kauffmann, P. Ceroni, V. Vicinelli and V. Balzani, Coordination of Co2+ ions in the interior of poly(propylene amine) dendrimers containing fluorescent dansyl units in the periphery, J. Am. Chem. Soc., 2000, 122, 10398 CrossRef; (c) V. Balzani, P. Ceroni, C. Gestermann, M. Gorka, C. Kauffmann and F. Vögtle, Effect of protons and metal ions on the fluorescence properties of a polylysin dendrimer containing twenty four dansyl units, J. Chem. Soc., Dalton Trans., 2000, 3765 Search PubMed; (d) P. Ceroni, V. Vicinelli, M. Maestri, V. Balzani, W. M. Müller, U. Müller, U. Hahn, F. Osswald and F. Vögtle, Dendrimers with a 4,4′-bipyridinium core and electron-donor branches. Electrochemical and spectroscopic properties, New J. Chem., 2001, 25, 989 RSC.
- T. Nishioka, K. Tashiro, T. Aida, J.-Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, Molecular design of a novel dendrimer porphyrin for supramolecular fullerene/dendrimer hybridization, Macromolecules, 2000, 33, 9182 CrossRef CAS.
- (a) A. Adronov, S. L. Gilat, J. M. J. Fréchet, K. Ohta, F. V. R. Neuwahl and G. R. Fleming, Light harvesting and energy transfer in laser-dye-labeled poly(aryl ether) dendrimers, J. Am. Chem. Soc., 2000, 122, 1175 CrossRef CAS; (b) L. A. Chrisstoffels, A. Adronov and J. M. J. Fréchet, Surface-confined light harvesting, energy transfer, and amplification of fluorescence emission in chromophore-labeled self-assembled monolayers, Angew. Chem., Int. Ed., 2000, 39, 2163 CrossRef CAS; (c) A. Adronov and J. M. J. Fréchet, Light-harvesting dendrimers, Chem. Commun., 2000, 1701 RSC; (d) F. V. R. Neuwahl, R. Righini, A. Adronov, P. R. L. Malenfant and J. M. J. Fréchet, Femtosecond transient absorption studies of energy transfer within chromophore-labeled dendrimers, J. Phys. Chem. B, 2001, 105, 1307 CrossRef CAS.
- M. Cardona, J. Alvarez, A. E. Kaifer, T. D. McCarley, S. Pandey, G. A. Baker, N. J. Bonzagni and F. V. Bright, Dendrimers functionalized with a single fluorescent dansyl group attached “off center”: synthesis and photophysical studies, J. Am. Chem. Soc., 2000, 122, 6139 CrossRef CAS.
- R. K. Lammi, A. Ambrois, T. Balasubramanian, R. W. Wagner, D. F. Bocian, D. Holten and J. S. Lindsey, Structural control of photoinduced energy transfer between adjacent and distant sites in multiporphyrin arrays, J. Am. Chem. Soc., 2000, 122, 7579 CrossRef CAS.
- L.-Z. Gong, Q. Hu and L. Pu, Optically active dendrimers with a binaphthyl core and phenylene dendrons: light harvesting and enantioselective fluorescent sensing, J. Org. Chem., 2001, 66, 2358 CrossRef CAS.
- G. De Belder, G. Schweitzer, S. Jordens, M. Lor, S. Mitra, J. Hofkens, S. De Feyter, M. Van der Auweraer, A. Herrmann, T. Weil, K. Müllen and F. C. De Schryver, Singlet-singlet annihilation in multichromophoric peryleneimide dendrimers, determined by fluorescence upconversion, ChemPhysChem, 2001, 49 CrossRef CAS.
- V. Balzani and F. Scandola, Supramolecular Photochemistry, Horwood, Chichester, England, 1991, ch. 7 Search PubMed.
- (a) For some recent examples, see: D. G. Walter, D. J. Campbell and C. A. Mirkin, Photon-gated transfer in two component self-assembled monolayers, J. Phys. Chem. B, 1999, 103, 402 Search PubMed; (b) M. S. Vollmer, T. D. Clark, C. Steinem and M. R. Ghadiri , Photoswitchable hydrogen-bonding in self-organized cylindrical peptide systems, Angew. Chem., Int. Ed., 1999, 38, 1598 CrossRef CAS; (c) I. Willner, V. Pardo-Yissar, E. Katz and K. T. Ranjit, A photoactivated “molecular train” for optoelectronic applications: light stimulated translocation of a beta-cyclodextrin receptor within a stoppered azobenzene-alkyl chain supramolecular monolayer assembly on a Au-electrode, J. Electroanal. Chem., 2001, 497, 172 CrossRef CAS.
- (a) For some recent papers, see: A. Archut, G. C. Azzellini, V. Balzani and L. De Cola, Toward photoswitchable dendritic hosts. Interaction between azobenzene-functionalised dendrimers and eosin, J. Am. Chem. Soc., 1998, 120, 12187 Search PubMed; (b) D. M. Junge and D. V. McGrath, Photoresponsive azobenzene-containing dendrimers with multiple discrete states, J. Am. Chem. Soc., 1999, 121, 4912 CrossRef CAS; (c) S. Li and D. V. McGrath, Effect of macromolecular isomerism on the photomodulation of dendrimer properties, J. Am. Chem. Soc., 2000, 122, 6795 CrossRef CAS; (d) K. Tsuda, G. C. Dol, T. Gensch, J. Hofkens, L. Latterini, J. W. Weener, E. W. Meijer and F. C. De Schryver, Fluorescence from azobenzene functionalised poly(propylene imine) dendrimers in self-assembled supramolecular structures, J. Am. Chem. Soc., 2000, 122, 3445 CrossRef CAS; (e) J.-W. Weener and E. W. Meijer, Photoresponsive dendritic monolayers, Adv. Mater., 2000, 12, 741 CrossRef CAS; (f) G. C. Dol, K. Tsuda, J.-W. Weener, M. J. Bartels, T. Asavei, T. Gensh, J. Hofkens, L. Latterini, A. P. H. J. Schenning, B. W. Meijer and F. C. De Schryver, Merging of hard spheres by phototriggered micromanipulation, Angew. Chem., Int. Ed., 2001, 40, 1710 CrossRef CAS.
- H. Rau, in Photochromism, Molecules and Systems, eds. H. Dürr and H. Bouas-Laurent, Elsevier, Amsterdam, 1990, ch. 4 Search PubMed.
- For the quenching of the naphthalene fluorescence by amines, see: S. Quici, A. Manfredi, M. Maestri, I. Manet, P. Passaniti and V. Balzani, Synthesis and photophysical properties of polyazacrown ethers with appended naphthyl or anthracenyl units, Eur. J. Org. Chem., 2000, 2041 Search PubMed.
- A. Gilbert and J. Baggot, Essentials of Molecular Photochemistry, Blackwell Scientific Publications, Oxford, England, 1991 Search PubMed.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, 2nd edn., Dekker, New York, USA, 1993 Search PubMed.
- P. Ceroni, I. Laghi, M. Maestri, V. Balzani, S. Gestermann, M. Gorka and F. Vögtle, Photochemical, photophysical and electrochemical properties of six dansyl-based dyads, New J. Chem., in the press Search PubMed.
- M. W. P. L. Baars, R. Kleppinger, M. H. J. Koch, S. L. Yeu and E. W. Meijer, The Localization of Guests in Water-Soluble Oligoethyleneoxy-Modified Poly(propylene imine) Dendrimers, Angew. Chem., 2000, 112(7), 1341–1344 CrossRef.
- C. Mills, Some new Azo-compounds, J. Chem. Soc., 1895, 67, 925–933 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR data. See http://www.rsc.org/suppdata/pp/b1/b106813j/ |
| ‡ This naming bears an inconsistency in its combination of the IUPAC nomenclature with dendrimer nomenclature.1 Yet, it seems necessary to first use azabutylidene according to Newkome's nomenclature to define the type of branch. After having done this, it is difficult not to refer again to this nitrogen in the course of the sulfonyl group attachment in order to show where the sulfonyl group is located. It seems that this problem for the case of peripherally disubstituted POPAM has not been clearly solved hitherto, but avoided in the same way as here,1f but we have the impression that even if there remains an inconsistency, the name characterizes the structure well. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2002 |