Cyclic thiatetraynes: syntheses and structural properties
Bernhard J. Rausch, Daniel B. Werz, Stefan Rittinger, Rolf Gleiter*, Thomas Oeser and Frank Rominger
Organisch-Chemisches Institut der Universität Heidelberg, Im Neuenheimer Feld 270, D-69120 Heidelberg, Germany. E-mail: rolf.gleiter@urz.uni-heidelberg.de; Fax: +49-6221-544205; Tel: +49-6221-548400
First published on 27th November 2001
Abstract
The syntheses of three cyclic dithiatetraynes 4–6 with the sulfur atoms in propargylic positions are reported. Their structural features have been studied by means of X-ray analysis on single crystals. Additionally, six cyclic octathiatetraynes 15–20 with sulfur atoms in α,α′-positions to the triple bond have been prepared. The structures of 15 and 17 have been elucidated by means of X-ray analysis. These macrocycles exhibit an ellipsoidal geometry dominated by the stiff triple bond moieties. In the case of 4 a three-dimensional columnar structure is found, demonstrating the unique features of the potential cavities.
1 Introduction
Alkyne units, benzene rings or ethylene moieties impart rigidity to ring systems. This concept can be used to construct large, stiff ring systems such as pericyclynes,1 the higher annulenes,2 cyclic oligoalkynes3 and various cyclophanes4 to name but a few. There are two main strategies for including alkyne units into a ring system. Ours is to perform a ring closure starting with an acyclic precursor containing triple bonds. This approach is usually limited to larger rings as the four carbon atoms of a C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C unit prefer an almost linear arrangement. In constructing strained systems with alkyne units one has to rely on elimination procedures like that shown in Scheme 1 for the synthesis of cyclodeca-1,6-diyne.5
C–C unit prefer an almost linear arrangement. In constructing strained systems with alkyne units one has to rely on elimination procedures like that shown in Scheme 1 for the synthesis of cyclodeca-1,6-diyne.5 | ||
| Scheme 1 Synthesis of cycloocta-1,6-diyne. | ||
To obtain medium and larger rings one usually employs a cyclisation of two components as shown in Scheme 2 for the synthesis of cyclotetradeca-1,8-diyne.6
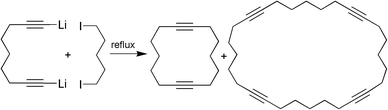 | ||
| Scheme 2 Two component cyclisation reaction. | ||
As a side reaction of this two component cyclisation hydrocarbons with four triple bonds emerge in moderate yields. Lately, tetraynes have received increasing attention, but reactions leading to heterosubstituted derivatives have been poorly investigated.1,7 In particular, the generation of thiatetraynes has only been reported for the case of pericyclynes as shown in Scheme 3 for dithia[4]pericyclyne.1
![Synthesis of dithia[4]pericyclyne.](/image/article/2002/P2/b108403h/b108403h-s3.gif) | ||
| Scheme 3 Synthesis of dithia[4]pericyclyne. | ||
To further utilize the unique geometry of these macrocycles that incorporate four stiff triple bond units we investigated the synthesis of several thiatetraynes. They could be of considerable interest for the formation of intramolecular complexes with ions and neutral molecules.7–11 Herein we present the syntheses and structural features of several dithia- and octathiatetraynes.
2 Results and discussion
To correlate the effect of sulfur introduction in the tetrayne moieties with its position in the macrocycle we investigated two groups of tetraynes: (a) dithiatetraynes with two sulfur atoms in propargylic† positions; (b) octathiatetraynes with eight sulfur atoms in α,α′-positions to the four triple bonds.The syntheses of the acyclic starting materials were carried out according to literature methods. The dithiatetraynes 4 and 6 were obtained by reacting the corresponding dibromides 1 and 312,13 with sodium sulfide under phase transfer conditions utilizing Bu4NBr in a methylene chloride–water mixture. Reacting the dibromide 214 with bis(tributyltin) sulfide in a THF–acetonitrile mixture in the presence of caesium fluoride–18-crown-6 produced the corresponding tetrayne 5. The yields varied between 8 and 24%, respectively (Scheme 4).
 | ||
| Scheme 4 Syntheses of dithiatetraynes 4, 5 and 6. | ||
To introduce eight sulfur atoms into the tetrayne moieties we reacted acyclic dithiadiynes 7, 8 and 915 with α,ω-dithiocyanatoalkanes 10–1416–20 to obtain the octathiacycles 15–20 in moderate yields (2–5%) (Scheme 5). A correlation between the ring size and the yields obtained was not found.
 | ||
| Scheme 5 Syntheses of octathiatetraynes 15–20. | ||
The structural properties of the eighteen- to thirty-membered tetraynes have been examined by means of X-ray investigations on single crystals. All of the structures show ellipsoidal shapes and, with the exception of 6, possess a molecular centre of inversion. Therefore they can be characterized by the distances a and b, defined as the transannular distance of the centres of the bridging units as shown in Fig. 1. The number of ring atoms and the most relevant distances a and b are given in Table 1.
 | ||
| Fig. 1 Definition of the transannular distances a and b in tetraynes. | ||
| Compound | Ring atoms | a/pm | b/pm |
|---|---|---|---|
| 4 | 18 | 889 | 789 |
| 5 | 18 | 854 | 661 |
| 6 | 22 | 678 | 1026 |
| 15 | 24 | 501 | 1072 |
| 17 | 30 | 720 | 1046 |
A comparison between the molecular structures in the solid state of the 18-membered tetraynes 4 and 5 is presented in Fig. 2. A common feature of the two structures with sulfur in propargylic positions is the ellipsoidal shape. This is determined not only by the bond angles of the heteroatoms but also by the four stiff C–CC–C units. Introduction of the two disila bridges in 4 increases the rigidity of the macrocycle.
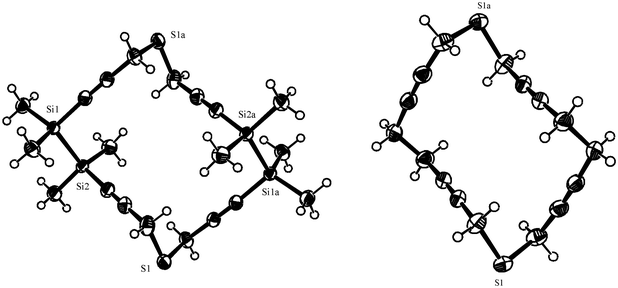 | ||
| Fig. 2 X-Ray structures of 4 (left) and 5 (right). The plots are shown at 50% probability of the thermal ellipsoids. | ||
The size of the potential cavity of dithiadisilatetrayne 4 is demonstrated in its three-dimensional columnar structure which is presented in Fig. 3. The intermolecular distance between the rings in the direction of the stack is 735 pm; the intermolecular separation of the triple bond moieties, marked as dashed lines, is about 547 pm. The formation of columnar structures has not been reported before for tetraynes.
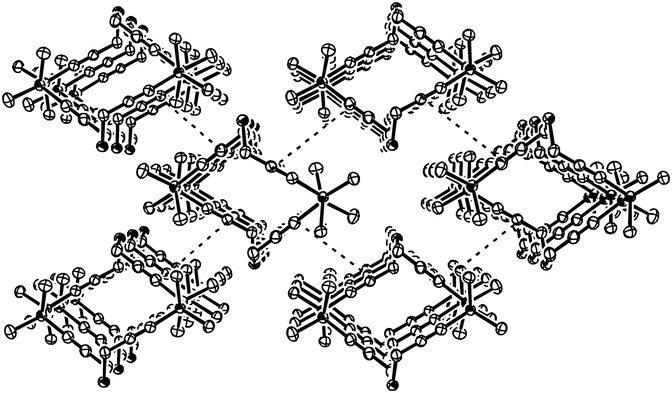 | ||
| Fig. 3 Three-dimensional columnar structure of 4 with shaded sulfur and silicon heteroatoms. The plot is shown at 50% probability of the thermal ellipsoids and hydrogens are omitted for clarity. The intermolecular distance between the triple bonds (dashed line) is about 547 pm. | ||
To investigate the properties of tetraynes with electron rich triple bonds we examined the solid state structures of the octathia derivatives 15 and 17 (Fig. 4). The most salient feature of these structures is the large torsion angle between the CH2–S⋯S–CH2 bonds tied to the triple bond which varies between 84.2 and 87.6°. These values are due to repulsion between the 3p lone pairs of the sulfurs and the π-MOs of the triple bond which finally leads to the observed values of nearly 90°.15,21
 | ||
| Fig. 4 X-Ray structure of octathiatetrayne 17. The plot is shown at 50% probability of the thermal ellipsoids. The intramolecular distances of the S1–CC–S4 units are marked as dashed lines. | ||
Furthermore, the X-ray analysis of 17 reveals an almost linear arrangement of the triple bonds with an intramolecular separation between the S1–CC–S4 units of 398 pm (dashed line). The size of the ellipsoid-shaped macrocycle can be characterized by its transannular distances a and b as defined in Fig. 1. They were found to be 720 and 1046 pm, respectively (Table 1).
3 Conclusion
A series of macrocyclic tetraynes have been synthesized with sulfur atoms in the propargylic positions or α,α′-positions to the triple bonds, respectively. To introduce the sulfur in the propargylic position we chose either a four component cyclization with an α,ω-bis(propargyl bromide) with sodium sulfide under phase transfer conditions (4, 6) or the reaction of the bromide with (Bu3Sn)2S (5). The octathiatetraynes 15–20 could be obtained by reaction of the lithium salts of the thiaacetylides 7–9 with the α,ω-dithiocyanatoalkanes 10–14. All three procedures yielded the desired macrocycles in moderate yields. X-Ray analysis shows that the rings form an ellipsoidal geometry dominated by the stiff triple bond moieties. The dithiadisilatetrayne 4 exhibits in the solid state a special columnar structure which is unique for these sulfur substituted macrocycles. This has not been reported before for tetraynes. Such compounds might find use in the formation of intramolecular complexes with ions and neutral molecules.7–115 Experimental
5.1 General methods
All melting points are uncorrected. Elemental analyses were carried out by Mikroanalytisches Laboratorium der Universität Heidelberg. IR spectra were recorded with a Bruker Vector 22. The NMR spectra were measured with a Bruker WH 300 (13C NMR at 75.47 MHz) and a Bruker Avance 500 (13C NMR at 125.77 MHz) using the solvent as internal standard (δ). The high-resolution mass spectra (HRMS) were recorded in the EI (70 eV) and positive-ion FAB mode in m-nitrobenzyl alcohol. All reactions were carried out in dried glassware under a nitrogen or argon atmosphere using dried and oxygen-free solvents. Materials used for column chromatography: silica gel (Macherey–Nagel), Celite (Fluka). Acyclic diynes12–15 and dithiocyanatoalkanes16–20 were prepared according to literature methods.5.2 X-Ray crystallography and structure solution‡
The crystallographic data were recorded with a Bruker Smart CCD diffractometer at 200 K except for compound 5 (Nonius-CAD-diffractometer at 295 K). Relevant crystal and data collection parameters are given in Table 2. The structures were solved by using direct methods, least-squares refinement, and Fourier techniques. Structure solution and refinement were performed with SHELXTL (5.10) software package.22Table 2 contains the crystallographic data and details of the data collection and the refinement procedure. ORTEP drawings were obtained using the ORTEP-3 for Windows program by Farrugia.23| Compound | 4 | 5 | 6 | 15 | 17 |
|---|---|---|---|---|---|
| Chemical formula | C20H32S2Si4 | C16H16S2 | C20H24S2 | C16H16S8 | C22H28S8 |
| Formula weight | 448.94 | 272.40 | 328.51 | 464.77 | 548.92 |
| Crystal colour | Colourless | Colourless | Colourless | Colourless | Colourless |
| Crystal shape | Polyhedron | Polyhedron | Polyhedron | Polyhedron | Polyhedron |
| Crystal size/mm3 | 0.21 × 0.20 × 0.18 | 0.50 × 0.50 × 0.25 | 0.56 × 0.20 × 0.11 | 0.17 × 0.14 × 0.06 | 0.36 × 0.32 × 0.28 |
| Temperature/K | 200 | 295 | 200 | 200 | 200 |
| Wavelength/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/n | Pbca | P21/n | P21/c | Pbca |
| Z | 2 | 4 | 4 | 2 | 4 |
| Unit cell dimensions | |||||
| a/Å | 7.3483(1) | 9.846(2) | 17.0312(1) | 11.6465(4) | 9.8778(2) |
| b/Å | 12.1868(2) | 9.030(2) | 5.0495(1) | 9.3011(3) | 9.5740(2) |
| c/Å | 15.0313(2) | 16.153(3) | 22.4681(1) | 10.0017(4) | 28.6050(2) |
| α/° | 90 | 90 | 90 | 90 | 90 |
| β/° | 98.228(1) | 90 | 106.752(1) | 101.196(1) | 90 |
| γ/° | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 1332.23(3) | 1436.2(5) | 1850.23(4) | 1062.82(7) | 2705.18(8) |
| Dcalc/g cm−3 | 1.119 | 1.260 | 1.179 | 1.452 | 1.348 |
| Absorpt. coeff. μ/mm−1 | 0.383 | 0.350 | 0.283 | 0.837 | 0.669 |
| θ range for data coll./° | 2.16–27.41 | 2.50–27.90 | 1.33–27.48 | 1.78–24.09 | 2.51–27.49 |
| Index ranges | −9 ≤ h ≤ 9 | 0 ≤ h ≤ 12 | −22 ≤ h ≤ 22 | −13 ≤ h ≤ 13 | −12 ≤ h ≤ 12 |
| −15 ≤ k ≤ 15 | 0 ≤ k ≤ 11 | −6 ≤ k ≤ 6 | −10 ≤ k ≤ 10 | −12 ≤ k ≤ 12 | |
| −19 ≤ l ≤ 19 | 0 ≤ l ≤ 20 | −29 ≤ l ≤ 28 | −11 ≤ l ≤ 11 | −37 ≤ l ≤ 37 | |
| Reflections collected | 13420 | 1992 | 18002 | 8114 | 25957 |
| Independent reflections | 3032 | 1712 | 4229 | 1686 | 3097 |
| Max. and min. transmission | 0.95 and 0.74 | 1.00 and 0.92 | 0.97 and 0.84 | 0.96 and 0.44 | 0.86 and 0.69 |
| Data/parameters | 3032/122 | 1229/114 | 4229/208 | 1686/109 | 3097/192 |
| Goodness-of-fit on F2 | 1.07 | 1.07 | 1.02 | 1.05 | 1.12 |
| R(F) | 0.036 | 0.041 | 0.036 | 0.040 | 0.029 |
| Rw(F2) | 0.091 | 0.103 | 0.088 | 0.099 | 0.069 |
| (Δρ)max, (Δρ)min/e Å−3 | 0.64, −0.45 | 0.25, −0.54 | 0.37, −0.18 | 0.43, −0.30 | 0.23, −0.27 |
5.3 General procedure for the preparation of dithiacycloalkatetraynes 4 and 6
To a solution of the dibromide and nBu4NBr in methylene chloride was added Na2S·9H2O in water. The mixture was vigorously stirred for 20 h. After separating the two phases the aqueous layer was extracted with methylene chloride (3 × 50 ml). The combined organic layers were washed with water (2 × 100 ml). After drying (MgSO4) the solvent was removed and the crude product was purified by column chromatography on silica with light petroleum–methylene chloride (4 ∶ 1) to yield the desired product.Recrystallisation from ether yielded 135 mg (24%) of 6 as colourless plates. Mp 124 °C. IR (KBr): 2942, 2861, 2223, 1630, 1457, 1431, 1407, 1369, 1330 cm−1. 1H NMR (CDCl3): 1.60–1.64 (m, 8H, CH2), 2.24 (t, 8H, CH2), 3.42 (t, 8H, CH2). 13C NMR (CDCl3): 18.9 (CH2), 19.1 (CH2), 27.8 (CH2), 75.1 (C), 83.2 (C). EI-HRMS: m/z 328.1325. C20H24S2 requires M, 328.1330.
5.4 Synthesis of 1,10-dithiacyclooctadeca-3,7,12,16-tetrayne (5)
Two solutions of 2.49 g of 1,8-dibromoocta-2,6-diyne 2 in 300 ml of acetonitrile and 5.77 g of bis(tributyltin) sulfide in 150 ml of THF and 150 ml of acetonitrile were added at 60 °C over a period of 30 h to a mixture of 5.88 g of caesium fluoride and 960 mg of 18-crown-6 in 300 ml of acetonitrile. After complete addition the reaction mixture was adsorbed on Celite and filtered through a pad of Celite eluting with chloroform. Column chromatography of the crude product with light petroleum–ether (10 ∶ 1) as eluent afforded 100 mg (8%) of 5 as colourless crystals. Mp 170 °C. Elem. anal.: found C, 70.40; H, 5.75; S, 23.25. C16H16S2 requires C, 70.54; H, 5.92; S, 23.54%. IR (KBr): 2920, 2829, 2279, 2225, 1635, 1419, 1319 cm−1. 1H NMR (CDCl3): 2.40 (s, 8H, CH2), 3.50 (s, 8H, CH2). 13C NMR (CDCl3): 19.7 (CH2), 19.9 (CH2), 77.3 (C), 82.8 (C). EI-HRMS: m/z 272.0652. C16H16S2 requires M, 272.0610.5.5 General procedure for the preparation of octathiacycloalkatetraynes 15–20
To a solution of the dithiaalkadiyne in anhydrous THF was added n-butyllithium at −25 °C over a period of 15 min. The solution was stirred for 1 h at −25 °C and then allowed to warm up to room temperature. To 800 ml of anhydrous THF were added solutions of the dilithiated dithiaalkadiyne in THF and the dithiocyanatoalkane in THF dropwise at −40 °C over a period of 5 h. After complete addition, the reaction mixture was allowed to warm up to room temperature overnight. After rotary evaporation of the solvent the crude product was absorbed on Celite. Polymers and salts were removed by flash filtration {SiO2 [3% NEt3 (v/v)], toluene}. The solvent was removed by rotary evaporation and the tetrayne was isolated by column chromatography {SiO2 [3% NEt3 (v/v)], mixtures of n-hexane and toluene}.Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, and BASF Aktiengesellschaft, Ludwigshafen, for financial support. D. B. Werz thanks the Studienstiftung des deutschen Volkes for a graduate fellowship.References
- L. T. Scott and M. J. Cooney, Modern Acetylene Chemistry, eds. P. J. Stang and F. Diederich, VCH, Weinheim, 1995, pp. 321–351 Search PubMed.
- W. B. Wan and M. M. Haley, J. Org. Chem., 2001, 66, 3893–3901 CrossRef CAS.
- R. Gleiter and R. Merger, Modern Acetylene Chemistry, eds. P. J. Stang and F. Diederich, VCH, Weinheim, 1995, pp. 285–319 Search PubMed.
- F. Diederich, Cyclophanes, The Royal Society of Chemistry, Cambridge, 1991 Search PubMed.
- R. Gleiter, M. Karcher, R. Jahn and H. Irngartinger, Chem. Ber., 1988, 121, 735–740 Search PubMed.
- R. Gleiter, R. Merger, B. Treptow, W. Wittwer and G. Pflaesterer, Synthesis, 1993, 6, 558–560 CrossRef.
- J. D. Ferrara, C. Tessier-Youngs and W. J. Youngs, Organometallics, 1987, 6, 676–678 CrossRef CAS.
- T. Nishinaga, T. Kawamura and K. Komatsu, Chem. Commun., 1998, 2263–2264 RSC.
- J. D. Ferrara, C. Tessier-Youngs and W. J. Youngs, Inorg. Chem., 1988, 27, 2201–2202 CrossRef CAS.
- R. Gleiter, K. Hoevermann, F. Rominger and T. Oeser, Eur. J. Org. Chem., 2001, 725–728 CrossRef CAS.
- A. Kunze, R. Gleiter and F. Rominger, Chem. Commun., 1999, 2, 171–172 RSC.
- G. Haberhauer, F. Rominger and R. Gleiter, J. Chem. Soc., Perkin Trans. 2, 1999, 5, 947–950 RSC.
- K. C. Nicolaou, G. Zuccarello, C. Riemer, V. A. Estevez and W. M. Dai, J. Am. Chem. Soc., 1992, 114, 7360–7371 CrossRef CAS.
- L. Trabert and H. Hopf, Liebigs Ann. Chem., 1980, 1786–1800 Search PubMed.
- C. Benisch, S. Bethke, R. Gleiter, T. Oeser, H. Pritzkow and F. Rominger, Eur. J. Org. Chem., 2000, 2479–2488 CrossRef CAS.
- H. L. Buff, Liebigs Ann. Chem., 1856, 100, 219–242 Search PubMed.
- L. Hagelberg, Chem. Ber., 1890, 23, 1083–1092 Search PubMed.
- J. v. Braun and G. Lemke, Chem. Ber., 1922, 55, 3536–3559 Search PubMed.
- J. v. Braun and A. Trümpler, Chem. Ber., 1910, 43, 545–551 Search PubMed.
- H. D. Vogelsang, T. Wagner-Jauregg and R. Rebling, Liebigs Ann. Chem., 1950, 569, 183–198 Search PubMed.
- D. B. Werz, F. Rominger, I. Hyla-Kryspin and R. Gleiter, J. Org. Chem., 2001, 66, 3416–3422 CrossRef CAS.
- G. M. Sheldrick, Bruker Analytical X-ray Division, Madison, WI, 1997.
- ORTEP-3 for Windows—A Version of ORTEP-III with a Graphical User Interface (GUI): L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 Search PubMed.
Footnotes |
| † The IUPAC name for propargyl is prop-2-ynyl. |
| ‡ Complete crystallographic data (excluding structure factors) for all structural analyses have been deposited with the Cambridge Crystallographic Data Centre. CCDC reference numbers 172996–173000. See http://www.rsc.org/suppdata/p2/b1/b108403h/ for crystallographic files in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2002 |