Vibrational spectroscopy of a tetraureidocalix[4]arene based molecular capsule†
Jörg Dormanna, Andreas Ruoffa, Jürgen Schatz*b, Myroslav O. Vysotskyc and Volker Böhmer*c
aSection of Vibrational Spectroscopy, Albert-Einstein-Allee 11, D-89081 Ulm, Germany
bDivision of Organic Chemistry I, Albert-Einstein-Allee 11, D-89081 Ulm, Germany. E-mail: juergen.schatz@chemie.uni-ulm.de; Fax: +49 731 50 22803
cFachbereich Chemie und Pharmazie, Abteilung Lehramt Chemie, Johannes-Gutenberg-Universität, Duesbergweg 10–14, D-55099 Mainz, Germany. E-mail: vboehmer@mail.uni-mainz.de; Fax: +49 6131 39 25419
First published on 11th December 2001
Abstract
Structural models for self-assembled dimers composed of two urea calix[4]arenes which entrap benzene or cyclohexane are developed using Fourier transform infrared (FTIR) spectroscopy. Based on the host–guest ratio determined by 1H NMR spectroscopy in solution, and confirmed for the solid state by a thermogravimetric analysis, it is possible to prove by a comparison of the FTIR data of host, guest, complex and model compounds, that the capsule is held together by a cyclic array of weak and strong hydrogen bonds between the urea units attached at the wide rim of the calixarenes. The dimerization of the two urea units leads to a loss of symmetry, and an averaged C4 symmetrical arrangement is probable. Guest molecules, such as benzene or cyclohexane, are enclosed inside the container rotating fast on the IR timescale around a longitudinal axis of the guest. From the observed splitting of absorption bands upon dimerization and inclusion it follows that either two crystallographically independent types of capsules exist in the crystal lattice or that the guests are occupying two major orientations in the capsule. As indicated by a higher complexation induced shift for cyclohexane, this guest exhibits a tighter interaction with the host molecules compared to benzene.
Introduction
Molecular capsules based on calixarenes1–3 or resorcinarenes can be formed by metal–ligand interactions4,5 or by hydrogen bonds. Calix[4]arenes substituted by four urea functions at the wide rim represent an especially interesting example.6–9 In apolar solvents they form dimeric capsules held together by a seam of hydrogen bonds between the interlocking urea moieties (Fig. 1). A guest molecule—often the solvent—is included in a cavity of about 200 Å3 and serves as a template for the capsule formation. Kinetically stable capsules in apolar solvents (e.g. benzene) were accessible using sterically demanding urea residues.8 The kinetic stability is in general drastically increased in cyclohexane10 in comparison to benzene, and in a special example it is observed also in DMSO.11 Such (weak) molecular assemblies can be studied by single crystal structure determination in the solid state,7,9 NMR spectroscopy in solution,6,8,12,13 or mass spectrometry in the gas phase.14We are interested in the development of alternative methods to determine structural models of calix[n]arene–guest complexes for those cases where a single crystal structure determination is not accessible.15–17 Capsules based on tetra-urea calixarenes are interesting candidates for the use of a thorough Fourier transform infrared (FTIR) spectroscopic analysis. To gain a deeper insight into the solid state structure we have chosen for a first study dimeric capsules formed from calix[4]arene 1 with enclosed benzene or cyclohexane, respectively. Results gained by a FTIR spectroscopic analysis have already proved to give structural information for calixarene complexes in solution,18–23 hydrogen bonded,24–27 or surface-bound systems,28,29 and structures in the solid state, which are comparable with results obtained by X-ray structure determination.16,30
Results and discussion
Materials
The two complexes 1·C6H6·1 and 1·C6H12·1 were prepared as colourless powders by dissolving 1 in benzene or cyclohexane, respectively, evaporating the clear solution and drying the residue at 100 °C at <1 Torr. Monomeric 1 was obtained by recrystallisation of 1 from chloroform–methanol. From 1H NMR spectra of 1·guest·1 measured in C6D12, where these capsules show a high kinetic stability,10 it could be concluded that the samples did not contain free guest (C6H6 or C6H12), and that one guest molecule (δ = 3.87 for benzene; δ = −1.44 for cyclohexane) is included per dimeric capsule.![Tetraureido calix[4]arene 1 (R1
= C5H11, R2
= Me), Top right: schematic representation of the hydrogen bonds keeping the capsule together. Bottom: calculated structure for the dimer of tetraureido calix[4]arene 1 with included benzene (R1 and R2 are omitted for clarity).](/image/article/2002/P2/b108055p/b108055p-f1.gif) | ||
| Fig. 1 Tetraureido calix[4]arene 1 (R1 = C5H11, R2 = Me), Top right: schematic representation of the hydrogen bonds keeping the capsule together. Bottom: calculated structure for the dimer of tetraureido calix[4]arene 1 with included benzene (R1 and R2 are omitted for clarity). | ||
This host–guest ratio was confirmed by thermogravimetric analysis.15 From the loss of weight during heating of the crystalline samples (Fig. 2) host–guest ratios of 2 ∶ 0.82 for cyclohexane and 2 ∶ 0.86 for benzene as guest molecule, respectively, can be deduced. This is in reasonable agreement with the data obtained by 1H NMR spectroscopy in solution. Most guest molecules leave the crystal lattice when the samples are heated ca. 110–120 K above the boiling point of the free solvent (ΔTbp). Similar ΔTbp values can be found for p-tert-butylcalix[4]arene clathrates with THF (121 K), chloroform (121 K), or benzene (110 K) as guest molecules.15
At ca. 200 °C the interdigitating hydrogen bonds start to break and the entrapped guest molecules leave the interior of the capsule irrespective of their nature. Cooling the sample back to room temperature and re-heating up to 400 °C did not show any loss of weight, indicating that all guest molecules have left the capsule during the first heating cycle. The temperature interval ΔT in which the solvent is completely lost is somewhat broader for 1·C6H6·1 (83 K) compared to 1·C6H12·1 (36 K) and p-tert-butylcalix[4]arene·benzene (32 K). As expected, the loss of the entrapped molecule is endothermic (ΔH = 3.2 J g−1 was determined by differential scanning calorimetry for 1·C6H12·1).
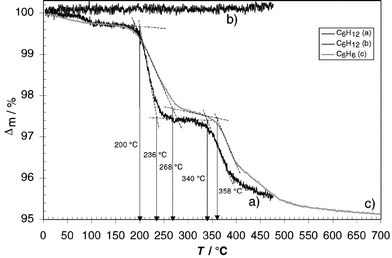 | ||
| Fig. 2 Thermogravimetric analysis of calixarene-based capsules 1·C6H12·1 (a and b) and 1·C6H6·1 (c) (heating rate 10 K min−1 under nitrogen, curve b) was obtained by heating 1·C6H12·1 up to 450 °C (a), cooling to room temperature and heating again to 470 °C. | ||
General procedure for the vibrational analysis of calixarene-based molecular capsules
To gain a deeper insight into the structural properties of the urea-based capsules, FTIR spectra of monomer 1, the capsules 1·guest·1, and free guest were recorded both at room and at low temperature (80 K). The measurements at low temperature show better resolution of individual absorption bands because the molecular motion is decreased significantly at 80 K. After full assignment of all absorptions observed, structural information can be deduced by comparing the obtained spectra. Information about symmetry and interaction with the surrounding capsule of the included guests are accessible when the data of the free guest and signals observable for the guest in the dimers (1·guest·1) are compared (Tables 1 and 2). From the line width of significant absorptions conclusions for the dynamic behaviour of the guests can be drawn. In case the capsule itself comes into focus, a comparison of the signals observed for the calixarene monomer 1 and dimer 1·guest·1 instructs about type, strength and symmetry of intermolecular forces involved in the formation process of the capsule. Observed band splittings, changes of intensities and shifts of absorption are important experimental data for this purpose (Table 3).| T = 293 K | T = 80 K | Int.a | C6H6 | Assignment33 |
|---|---|---|---|---|
| a Interpretation of band splitting.b Superposition of guest and host bands.c Different orientations of the guest molecule in the crystal lattice/dimer.d Lifting of degeneracy.e Forbidden band becoming active.f Several components possible and/or combination tones with a degenerated component. | ||||
| — | 3075 | b | 3090 | ν2 + ν13 + ν18E1u |
| — | 3054 | b | 3070 | ν13 + ν16A2u + E1u |
| 3027 sh | 3024 | b | 3035 | ν12E1u |
| — | 1954 | b | 1960 | ν7 + ν19E1u |
| 1477 | 1479 | b,c,d | 1478 | ν13E1u |
| 1471 sh | ||||
| 1387 sh | 1386 | c,d,f | 1385 | ν2 + ν20E2u |
| 1378 sh | ||||
| — | 1285 sh | 1308 | ν9B2u | |
| 1244 sh | 1243 | b,c,d,f | 1245 | ν11 + ν20E1u |
| 1244 | 1224 sh | |||
| — | 1171 | 1177 | ν17E2g | |
| 1138 sh | 1141 sh | 1147 | ν10B2u | |
| — | 1086 | e | 1097 sh | ν4 + ν20E2g |
| 1044 | 1043 sh | b,ce | 1035 | ν14E1u |
| 1031 | ||||
| 1008 sh | 1007 sh | e | 1008 | ν6B1u or ν7B2g |
| 850 | 857, 849 sh | b,c,d,e | 849 | ν11E1g |
| — | 793 | 793 | ν17 − ν20A2u | |
| 673 | 679 sh, 671 | c,e | 668 | ν4A2u |
| — | 592 | e | 608 w | ν18E2g |
| T = 293 K | T = 80 K | Int.a | C6H12 | Assignment |
|---|---|---|---|---|
| a Interpretation of band splitting.b Superposition of guest and host bands.c Different orientations of the guest molecule in the crystal lattice/dimer.d Lifting of degeneracy.e Forbidden band becoming active.f Several components possible and/or combination tones with a degenerated component. | ||||
| 3131 | 3130 | 3155 | ν3 + ν19 + ν32Eub | |
| 2ν19 + ν32Eu | ||||
| 3071 | 3075 | 3094 | ν5 + ν19 + ν31Eub | |
| 2925 | 2919 | 2952 | ν12A2ub | |
| — | 2898 | ν25Eu/ν1A1g | ||
| — | 2881 | 2898 | ν25Eu/ν1A1gb | |
| — | 2846 | 2847 | ν26Eu/ν2A1gb | |
| 2661 | 2662 | 2660 | ν21 + ν28Eu | |
| 2476 | 2476 | 2477 | ν14 + ν22Eu/2ν21A1g + Eg | |
| — | 2365 | 2366 | ν3 + ν30Eu/ν19 + ν30Eu | |
| — | 2128 | 2136 | ν21 + ν31Eu | |
| 1446 | 1444 | c,d | 1450 | ν14A2u/ν27Eub |
| 1429 | 1429 | |||
| 1386 | 1389 | c,d,f | 1350 | ν4 + ν32Eub |
| 1374 | ||||
| 1243 | 1243 | c,d | 1257 | ν29Eu |
| 1225 | 1226 | |||
| 1047 | 1050 | 1039 | ν5 + ν32Eub | |
| 1012 | 1015 | c,e | 1014 | ν23 + ν32A2u + Eub |
| 933 | 936 | c,e | 941 | ν15A2g |
| 928 | ||||
| 903 | 906 | c,d | 904 | ν30Eu |
| 898 | ||||
| 866 | 867 | c,d | 862 | ν31Eu |
| 845 | 846 | |||
| — | 802 | 821 | ν5A1gb | |
| 781 | 785 | 806 | ν23Eg | |
| 524 | 522 | 524 | ν16A2uν25Eu/ν1A1gb | |
| 1 | 1·C6H6·1 | 1·C6H12·1 | ||||
|---|---|---|---|---|---|---|
| T = 293 K | Irel | T = 80 K | Irel | T = 80 K | Irel | Assignment‡ |
| 3599 | 3599 | 3590 | ν(NH) | |||
| 3479 | 30 | 3482 | 30 | 3482 | 30 | |
| 3339 | 100 | 3408 | 70 | 3408 | 70 | |
| 3309 | 100 | 3354 | 100 | 3350 | 100 | |
| 3221 | 70 | 3290 | 100 | 3283 | 80 | |
| 3138 | 3203 | 70 | 3258 | 70 | ||
| 3134 | 3226 | 65 | ||||
| 3196 | 60 | |||||
| 3134 | ||||||
| 3076 | 3081 | 3081 | ν2; ν20a.(CH) | |||
| 3030 | 100 | 3025 | 100 | 3025 | 100 | ν20a; ν20b (CH) |
| 3002 | 20 | 2998 | 20 | 2998 | 20 | ν7b, ν20b; ν20a (CH) |
| 2952 | 2952 | 2952 | νas (CH3) | |||
| 2926 | 2927 | 2927 | νs (CH3)νas (CH2) | |||
| 2869 | 100 | 2869 | 100 | 2869 | 100 | νs (CH3) |
| 2857 | 95 | 2859 | 95 | 2857 | 95 | |
| 2760 | 50 | 2763 | 50 | 2763 | 50 | ν13 + ν17 |
| 2730 | 100 | 2733 | 100 | 2731 | 100 | |
| 1889 | 1889 | 1893 | ν7 + ν19 | |||
| 1705 sh | 1705 | 40 | 1705 sh | 24 | ν (CO) | |
| 1682 | 65 | |||||
| 1658 | 1666 | 100 | 1665 | 100 | ||
| 1648 | 70 | |||||
| 1602 | 1616 | 80 | sh | 80 | β (NH), ν8a; ν8a | |
| 1604 | 100 | 1608 | 100 | β (NH) | ||
| 1584 | 80 | sh | 80 | β (NH) | ||
| 1553 | 100 | 1554 | 100 | 1558 | 100 | β (NH) |
| 1516 | 80 | 1512 | 80 | 1514 | 80 | ν19a |
Because it is not possible to rule out any hydrogen bonding between the urea units in the calixarene monomer 1 itself, a reference system based on model compounds 2–5 (Fig. 3) can be used to obtain complexation induced shifts (CIS) and estimate the strength of such hydrogen bonds in the monomer as well as in the dimer 1·guest·1 (Table 4). The chosen model substances 2–5 reflect important parts of the calix[4]arene 1 concerning bond strength and substitution pattern. In the latter case, the weight of the substituents must reflect the real situation rather than their actual chemical structure. Further details about the general procedure used for the vibrational analysis have been published earlier.30
| 1 | 1·C6H6·1 | BSa | CISb | |
|---|---|---|---|---|
| (T = 293 K) | (T = 80 K) | |Δ|/cm−1 | |Δ|/cm−1 | Assignment |
| a BS: band splitting.b CIS = complexation induced shift; Δ = ν(1·C6H6·1) − ν(model substance).c 3221, 3309, 3339 are split into two components, e.g. 3221 into 3203 and 3290. | ||||
| 3599 | 3599 | 19–99c | 9–69 | ν (NH) |
| 3479 | 3482 | |||
| 3339 | 3408 | |||
| 3309 | 3354 | |||
| 3221 | 3290 | |||
| 3138 | 3203 | |||
| 3134 | ||||
| 1658 | 1705 | 58 | 8–65 | ν (CO) |
| 1682 | ||||
| 1666 | ||||
| 1648 | ||||
| 1602 | 1616 | 62 | 11–21 2 | β (NH) |
| 1604 | β (NH) | |||
| 1584 | 2 | β (NH) | ||
| 1553 | 1554 | β (NH) | ||
| 1418 | 1431 | 29 | 8–21 | ν (C–N) |
| 1402 | 1418 | |||
| 1402 | ||||
| 640 | 638 | — | 2 | γ (NH) |
| sh | ||||
| 619 | 619 | — | 5 | |
| sh | ||||
| 601 | 601 | 37 | 4–33 | γ (CO) |
| 593 | 594 | |||
| 585 | 586 | |||
| 578 | 576 | 2 | ||
| 564 | ||||
| 553 | 553 | — | ||
According to the NMR data in solution,8 the dimers posses S8-symmetry while the X-ray structure of a comparable urea capsule9 reveals C4 symmetry. In case of deviation from this symmetry, C2 or C1 must be considered as the corresponding point groups. According to the vibrational spectroscopic analysis of cyclohexane,31 this guest belongs to the point group D3d or has lower symmetry when the structure deviates from the chair conformation; benzene belongs to the point group D6h. Hence it follows for included cyclohexane or benzene, respectively, that all vibrations are IR- and Raman active and all degeneracies are lifted.
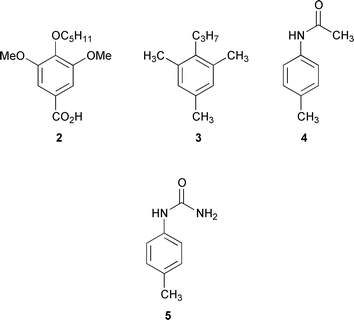 | ||
| Fig. 3 Model compounds 2–5 used for the vibrational analysis of the empty cavitand and molecular capsules. | ||
Vibrational analysis of 1·C6H6·1
The FTIR spectra (KBr disks) obtained for 1·C6H6·1 are shown in Fig. 4 and the results of the analysis of these data are summarised in Tables 1 and 3.Apart from the lifting of degeneracy, the splitting of several absorptions (e.g. 1245 → 1244, 1224 cm−1, cf.Table 1) of included benzene is clearly observed.
Because benzene molecules are found only inside the capsules, there are two possible explanations for this observation. Either there are two independent orientations of the capsule in the crystal lattice or included benzene has two major orientations inside the cavity. Because the line width of the included benzene is similar to that of pure benzene and interactions between encapsulated benzene molecules are unlikely due to size-limitations, the band width is presumably a result of fast rotation of the guest molecules around their longitudinal axes at the two main orientations.
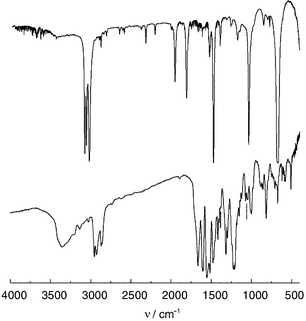 | ||
| Fig. 4 IR spectra of benzene and 1·C6H6·1 (bottom) (T = 293 K, KBr disks). | ||
Most frequencies of benzene are shifted downwards (e.g. 3090 → 3075). Large effects up to 16 cm−1 for the C–H and 23 cm−1 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C valence vibrations are observed, while the average shift for all absorptions is about 11 cm−1. Few frequencies are shifted to higher values with shifts up to 9 cm−1. This can be explained as complexation-induced shift (CIS) by CH–π interactions32 of the benzene molecules in the inner cavity. For the 1 ∶ 1 complex of p-tert-butylcalix[4]arene with benzene a similar averaged CIS is observable (12 cm−1).16
C valence vibrations are observed, while the average shift for all absorptions is about 11 cm−1. Few frequencies are shifted to higher values with shifts up to 9 cm−1. This can be explained as complexation-induced shift (CIS) by CH–π interactions32 of the benzene molecules in the inner cavity. For the 1 ∶ 1 complex of p-tert-butylcalix[4]arene with benzene a similar averaged CIS is observable (12 cm−1).16
However, in this case a maximum shift of 26 cm−1for the C–H and 30 cm−1 for the CC valence vibration, respectively, is observable indicating a somewhat stronger contact of benzene in the cavity of p-tert-butylcalix[4]arene. This difference can be rationalized by additional CH–π interactions of the tert-butyl groups of the p-tert-butylcalix[4]arene skeleton towards the included guest which increase the binding strength. Such an additional binding force is not possible for the urea–calixarene 1, because the urea moieties are involved in hydrogen bonds which hold the capsule together.
Comparison of IR data of the monomer and the dimers (Table 3) clearly reveals some significant shifts of the frequencies. As expected, the shift of the amide bands in the monomer is up to 69 cm−1 and in the dimer up to 99 cm−1, compared to model compounds.
Due to dimerization there is a splitting of some bands of the calixarene 1 into two components, e.g. the methyl group vibrations of the N-tolyl-ureido group and the vibrations of the pentyloxy group (e.g. 2926 → 2927 and 2869 cm−1). The ratio of the split components is 1 ∶ 1. Because the splitting is mainly observed for molecular regions on the outside of the capsule, influences of the guest molecule are not likely. Therefore, the splitting is probably due to a C4 symmetrical arrangement in which both calixarenes are not exactly twisted by 45° as required for a S8 symmetrical arrangement. This is in accordance with the crystal structure of urea calixarene 1 (R1 = CH2CO2Et) in which a twist of 43° was observed.9 This observation is diagnostic irrespective of whether a dimer or monomer is present.
For the calixarene monomer 1 two types of NH groups are present, i.e. a calixaryl-NH and a tolyl-NH. Because one expects symmetrical [νs (NH)] and anti-symmetrical [νas (NH)] vibrations for both types of amide band, involving these NH-groups in identical hydrogen bonds would lead to a signal set consisting of four components. However, each of these bands is split again into two, partly overlapping, components: one is shifted downwards (up to 19 cm−1, 3309 → 3290 cm−1), one upwards (up to 99 cm−1, 3309 → 3408 cm−1). This must be caused by the arrangement of the dimer and gives evidence for a weak and a strong hydrogen bond.
The included benzene has no further influence on the conformation and geometry of the dimer.
In summary, the observed complexation induced shifts and shifts derived from the comparison with model compounds 2–5 give evidence for two types of hydrogen bonds between the urea monomers as well as CH–π interactions between included benzene and the aromatic rings of the host molecule.
Vibrational analysis of 1·C6H12·1
The line width of the included cyclohexane is similar to that of benzene. Therefore, a similar dynamic behaviour (rotation) can be assumed.Most frequencies of cyclohexane are shifted up to 33 cm−1 down, with an average shift of about 14 cm−1; few frequencies are shifted to higher values by up to 39 cm−1 with an average shift of 13 cm−1. This can be explained as complexation induced shift, which is clearly stronger compared to the inclusion of benzene. This may be due to the ca. 20% higher molecular volume of cyclohexane compared to benzene which results in a tighter fit of this guest inside the capsule. The different spatial fit of the two different guests inside the cavity is also reflected in solution. However, in the latter case a higher CIS could be detected for benzene (−3.38ppm) compared to cyclohexane (−2.88 ppm) by 1H NMR spectroscopy.10 This is not a contradiction because the complexation-induced shift observed by NMR spectroscopy is mainly based on the fact that a guest molecule is located in the anisotropic cone of the phenyl rings, whereas CIS obtained by FTIR spectroscopy depends on the anisotropic fields induced by all surrounding functional groups.
Comparison of the monomer and the dimer shows, that there are some significant shifts in the frequencies. The shift of the amide bands is up to 99 cm−1, the other shifts are up to 16 cm−1.
All amide bands of the dimer are split into four components. These components are again split caused by the arrangement of the dimer (cf.1·C6H6·1). Because the CO stretching mode of both capsules (1666 for C6H6 and 1665 cm−1 for C6H12) is very similar but significantly different from the monomer (1658 cm−1), one can assume that the hydrogen bonding towards this oxygen atom is of comparable strength in both dimeric systems. The included cyclohexane has no further influence on the conformation and geometry of the dimer. The splitting of the signals for both benzene and cyclohexane is therefore due to two different arrangements of the guest molecules in the cavity of the capsule or by two types of crystallographically independent capsules. On the basis of the experimental data
available up to now, it is not possible to distinguish between the two explanations.
Again, due to the dimerization some bands are split into two components, and the complexation induced shifts obtained as before give evidence for two types of hydrogen bond between the calixarene monomers and CH–π interactions of the included cyclohexane to the aromatic rings of the calixarenes.
Some low lying vibrations which cannot be assigned to any of the modes of the subunits have to be characterised as specific for the calixarene capsule in toto. They are denoted as “calixarene ring mode”.
Conclusion
By the use of FTIR spectroscopy, the complexes 1·C6H6·1 and 1·C6H12·1 could be characterised. The capsules are held together by two types of hydrogen-bond bridges from two types of different urea NH groups, one showing strong and the other less favourable interactions as indicated by the splitting of the amide bands. The structures of the monomers and corresponding subunits in the dimer are nearly identical; the included guest molecules have little influence on the overall geometry of the surrounding non-covalently assembled container molecule. The mobility of benzene and cyclohexane, respectively, is similar taking the half-width of the corresponding absorption into consideration. Compared with 1·C6H6·1, the IR data of the capsule 1·C6H12·1 exhibit larger shifts, which suggests stronger interactions. Because the overall geometry of the capsule is nearly independent from the guest, which is rotating fast on the IR timescale, the splitting observed for the included solvent molecule is likely to be due to two main orientations of the guests inside the capsule. However, on the basis of the current experimental data, it remains unclear whether the guest molecules are located at two different sites in the capsule or two different types of capsules are existing in the crystal.Three tables containing fully assinged IR data of tetraureido calix [4] arene 1, their clathrates with cyclohexane and benzene and e the corresponding model substances 2–5 as well as 1H NMR spectra of the capsule 1·C6H12·1 in solution.
Experimental
The preparation of the calixarene 1 and the capsules have been described earlier.8,10,34 Infrared spectra were recorded on a FTIR spectrophotometer IFS 113v (Bruker) using KBr windows and a DGTS detector, with a resolution of approximately 0.5 cm−1. Raman spectra were recorded with the Raman-Laser spectrophotometer Dilor XY (multi- and single-channel detector, resolution 1 cm−1) using an Ar-Laser (Coherent, exciting line 514.53 nm). The temperature of the samples was 293 K and 80 K, respectively. Host–guest ratios of the samples used for the IR analysis were determined by NMR spectroscopy10 and thermogravimetric analysis as described earlier.15Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Scha 685/2-2, 2–4 and Bo523/14-1) and the Fonds der Chemischen Industrie. Generous support from Professor Dr G. Maas is gratefully acknowledged.References
- D. C. Gutsche, Calixarenes, in Monographs in Supramolecular Chemistry, ed. F. J. Stoddart, vol. 1, Royal Society of Chemistry, Cambridge, 1989 Search PubMed.
- D. C. Gutsche, Calixarenes Revisited, in Monographs in Supramolecular Chemistry, ed. F. J. Stoddart, Royal Society of Chemistry, Cambridge, 1998 Search PubMed.
- Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed.
- Z. Zhong, A. Ikeda, M. Ayabe, S. Shinkai, S. Sakamoto and K. Yamaguchi, J. Org. Chem., 2001, 66, 1002 CrossRef CAS.
- N. Cuminetti, M. H. K. Ebbing, P. Prados, J. de Mendoza and E. Dalcanale, Tetrahedron Lett., 2001, 42, 527 CrossRef CAS.
- Y. L. Cho, D. M. Rudkevich and J. Rebek, Jr., J. Am. Chem. Soc., 2000, 122, 9868 CrossRef CAS.
- A. Shivanyuk, V. Böhmer and E. F. Paulus, Angew. Chem., 1999, 111, 3091 CrossRef; A. Shivanyuk, V. Böhmer and E. F. Paulus, Angew. Chem., Int. Ed., 1999, 38, 2906 CrossRef CAS.
- M. O. Vysotsky, I. Thondorf and V. Böhmer, Angew. Chem., 2000, 112, 1309 CrossRef; M. O. Vysotsky, I. Thondorf and V. Böhmer, Angew. Chem., Int. Ed., 2000, 39, 1264 CrossRef CAS.
- O. Mogck, E. F. Paulus, V. Böhmer, I. Thondorf and W. Vogt, Chem. Commun., 1996, 2533 RSC.
- M. O. Vysotsky and V. Böhmer, Org. Lett., 2000, 2, 3571 CrossRef CAS.
- M. O. Vysotsky, I. Thondorf and V. Böhmer, Chem. Commun., 2001, 1890 RSC.
- O. Mogck, M. Pons, V. Böhmer and W. Vogt, J. Am. Chem. Soc., 1997, 119, 5706 CrossRef.
- N. Chopra, R. G. Chapman, Y. F. Chuang, J. C. Sherman, E. E. Burnell and J. M. Polson, J. Chem. Soc., Faraday Trans., 1995, 91, 4127 RSC.
- C. A. Schalley, R. K. Castellano, M. S. Brody, D. M. Rudkevich, G. Siuzdak and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 4568 CrossRef CAS.
- J. Schatz, F. Schildbach, A. Lentz and S. Rastätter, J. Chem. Soc., Perkin Trans. 2, 1998, 75 RSC.
- J. Schatz, F. Schildbach, A. Lentz, S. Rastätter, J. Schilling, J. Dormann, A. Ruoff and T. Debaerdemaeker, Z. Naturforsch., Teil B, 2000, 55, 213 Search PubMed.
- J. Schatz, A. C. Backes and H.-U. Siehl, J. Chem. Soc., Perkin Trans. 2, 2000, 609 RSC.
- B. T. G. Lutz, G. Astarloa, J. H. van der Maas, R. G. Janssen, W. Verboom and D. N. Reinhoudt, Vib. Spectrosc., 1995, 10, 29 CrossRef CAS.
- J. W. M. Nissink, H. Boerrigter, W. Verboom, D. N. Reinhoudt and J. H. van der Maas, J. Chem. Soc., Perkin Trans. 2, 1998, 1671 RSC.
- J. W. M. Nissink, H. Boerrigter, W. Verboom, D. N. Reinhoudt and J. H. van der Maas, J. Chem. Soc., Perkin Trans. 2, 1998, 2541 RSC.
- J. W. M. Nissink, H. Boerrigter, W. Verboom, D. N. Reinhoudt and J. H. van der Maas, J. Chem. Soc., Perkin Trans. 2, 1998, 2623 RSC.
- B. Paci, G. Amoretti, G. Arduini, G. Ruani, S. Shinkai and T. Suzuki, Phys. Rev. B: Condens. Matter, 1997, 55, 5566 Search PubMed.
- R. G. Janssen, W. Verboom, B. T. G. Lutz, J. H. van der Maas, M. Maczka and J. P. M. van Duynhoven, J. Chem. Soc., Perkin Trans. 2, 1996, 1869 RSC.
- J. A. Kanters, A. Schouten, E. Steinwender, J. H. van der Maas, L. C. Groenen and D. N. Reinhoudt, J. Mol. Struct., 1992, 269, 49 CrossRef CAS.
- B. Brzezinski, H. Urjasz and G. Zundel, J. Phys. Chem., 1996, 100, 9021 CrossRef CAS.
- L. Frkanec, A. Visnjevac, B. Kojic-Prodic and M. Zinic, Chem. Eur. J., 2000, 6, 442 CrossRef CAS.
- D. M. Rudkevich, Chem. Eur. J., 2000, 6, 2679 CrossRef CAS.
- W. C. Moreira, P. J. Dutton and R. Aroca, Langmuir, 1994, 10, 4148 CrossRef CAS.
- J. W. M. Nissink and J. H. van der Maas, Appl. Spectrosc., 1999, 53, 528 Search PubMed.
- J. Dormann, A. Ruoff, J. Schatz, O. Middel, W. Verboom and D. N. Reinhoudt, J. Phys. Org. Chem., 2001, 14, 707 CrossRef CAS.
- H. Takahashi and T. Shimanouchi, J. Mol. Spectrosc., 1964, 13, 43 CrossRef CAS.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature and Consequences, Wiley-VCH, New York, Chichester, Weinheim, Brisbane, Singapore, Toronto, 1998, ch. 9, pp. 141–144 Search PubMed.
- G. Varsányi, Assignments for vibrational spectra of seven hundred benzene derivatives, London, 1974, vol. 1 Search PubMed.
- O. Mogck, V. Böhmer and W. Vogt, Tetrahedron, 1996, 52, 8489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: three tables containing fully assigned IR data of tetraureido calix[4]arene 1, its complex with cyclohexane and benzene, and the corresponding model substances 2–5 as well as 1H NMR spectra of the capsules 1·C6H6·1 and 1·C6H12·1 in solution. See http://www.rsc.org/suppdata/p2/b1/b108055p/ |
| This journal is © The Royal Society of Chemistry 2002 |