Enantioselective photocyclization of p-toluidides of α,β-unsaturated carboxylic acids in solution. A mechanistic and preparative study
Pilar
Formentín
a,
María J.
Sabater
a,
Michelle N.
Chrétien
b,
Hermenegildo
García
*a and
Juan C.
Scaiano
*b
aInstituto de Tecnología Química CSIC-UPV, Universidad Politécnica de Valencia, Apartado 22012, 46071-Valencia, Spain
bDepartment of Chemistry, Centre for Catalysis Research and Innovation, University of Ottawa, Ottawa, K1N 6N5,, Canada
First published on 27th November 2001
Abstract
Photolysis of p-toluidides of methacrylic (1a) and cyclohex-2-enecarboxylic (1b) acids in nitrogen-saturated cyclopentane solution yields the corresponding 2-quinolones with over 90% chemoselectivity at almost complete conversion. In the presence of substoichiometric amounts (0.1 equivalents) of chiral inductor, low to moderate enantiomeric excesses (ee) are observed in the photo-product. Ephedrine gave the highest ee (37% ee for the photocyclization of 1a) in a series of 11 chiral inductors including alcohols, amines, aminoalcohols, α-amino and α-hydroxy acids. In the case of the irradiation of 1b in the presence of chiral inductors, both diastereo- and enantioselectivity were observed. A weakly absorbing transient species (λmax 400 nm) was detected following 308 nm laser excitation and was assigned to the zwitterionic enolate intermediate resulting immediately after the concerted electrocyclic ring closure. The lifetime of this intermediate is unaffected by oxygen but is quenched by trifluoroacetic acid (kq = 3.76 × 107 M−1 s−1) and ephedrine (kq = 1.19 × 107 M−1 s−1).
Introduction
Asymmetric photochemistry is attracting much current interest as evidenced by several reviews which have appeared related to this topic.1–3 In principle, the main advantage of photo-induced enantioselectivity over thermal ground-state reactions is the lower temperature at which the photochemical reactions can be conducted. The photocyclization of acrylanilides to 3,4-dihydroquinolin-2(1H)-ones has long been studied4–6 and has found some application in the synthesis of alkaloids.7–9 In the present work we report our study on the enantioselective photocyclization of p-toluidides of α,β-unsaturated carboxylic acids 1a,b to the corresponding dihydroquinolinones 2a,b. This reaction has been previously studied in solution at 5–10 °C in the presence of chiral inductors derived from tartaric acid and reported to proceed in low enantiomeric excess (13% ee).10,11 More recently, it has been found that room temperature, solid-state irradiation of 1 ∶ 1 inclusion crystals of acrylanilides in a chiral cyclic acetal of 1,1,4,4-tetraphenylbutanetetrol host results in a 46% yield of the corresponding 2-quinolone with 98% ee.12,13 The key step of the asymmetric induction in crystals was proposed to be the concerted suprafacial sigmatropic 1,5-H migration from the ortho position of the anilide ring to the α-position of the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. Remarkably, photochemical cis–trans isomerization of the CC double bond was not observed in the solid-state irradiation of crotonanilide. This photochemical isomerization was the predominant photoprocess for the photolysis
of crotonanilides in solution.14
O group. Remarkably, photochemical cis–trans isomerization of the CC double bond was not observed in the solid-state irradiation of crotonanilide. This photochemical isomerization was the predominant photoprocess for the photolysis
of crotonanilides in solution.14
In the present work we have combined a product study to assess ee with laser flash photolysis in order to gain insight into the mechanism of the photocyclization. Based on this study, we propose that in solution the key step in chiral induction is asymmetric protonation of the enolate intermediate. This mechanism is different from the concerted sigmatropic H-migration proposed for the solid state irradiation of inclusion crystals.12,13
Results and discussion
Anilides 1a,b were obtained by reacting p-toluidine with the corresponding acyl chloride and were recrystallized from ethanol before irradiation. Photolysis of p-toluidide 1a in benzene does not lead to any significant conversion after 48 h of irradiation.15 This could be explained by a filtering effect of the benzene solvent which has a UV absorption spectrum similar to substrate 1a. In contrast, irradiation of toluidide 1a in cyclopentane under nitrogen for 2–4 h leads to dihydroquinolinone 2a with high selectivity at almost complete conversion (Scheme 1). Given the high chemoselectivity of the photochemical reaction under these conditions, we proceeded to perform the irradiation in the presence of a series of chiral inductors. The results are listed in Table 1. The most relevant parameter in this table is the ee for each irradiation. The ee were determined by integrating the area of the HPLC peaks corresponding to each enantiomer using a UV detector monitoring at 256 nm coupled with an on-line polarimeter. The absolute configuration of the predominant stereoisomer was found to be R, based on a previous assignment in solution.11 Analysis of the ee versus conversion of substrate 1a in the presence of (−)-ephedrine indicates that ee is independent of conversion and constant during the irradiation time. Irradiation of an enantiomerically enriched solution of 2a in cyclopentane showed that no photochemical epimerization of the product occurs under the irradiation conditions. These blank controls indicate no variation of the ee with extent of conversion or irradiation time, allowing for a straightforward comparison of the ee for each chiral inductor. The highest ee was obtained using ephedrine as the chiral inductor at −40 °C. A decrease in the reaction temperature from 24 to −40 °C has a notable beneficial effect on the resulting ee. | ||
| Scheme 1 | ||
| 1a | 1b | ||||
|---|---|---|---|---|---|
| Chiral inductor | Selectivity (%) | Ee (%) | Selectivity (%) | cis ∶ trans ratio | Eea (%) |
| a Enantiomeric excesses are given for the cis isomer with the value for the trans isomer indicated in brackets. | |||||
| No inductor | 96 | — | 94 | 1.1 | — |

|
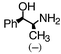
96 | 37 | 93 | 0.9 | 26 (2) |
|
93 | 22 | 74 | 0.5 | 14 (3) |

|
96 | 23 | 74 | 0.3 | 3 (1) |
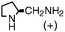
|
94 | 25 | 78 | 0.6 | 3 (7) |
The influence of the chiral inductor concentration at a constant (10−3 M) concentration of 1a was studied for the case of ephedrine at −40 °C. A linear increase of ee with ephedrine concentration was observed at lower concentrations until a limiting ephedrine concentration (∼10−4 M) is reached, at which point the ee levels off. No further improvement of the ee for 2a occurs with increased ephedrine concentration. It is noteworthy that the limiting ephedrine concentration is about one order of magnitude lower than the concentration of substrate 1a.
The fact that the ee for 2a does not increase continuously with chiral inductor concentration does not support the intermediacy of a substrate-ephedrine complex as previously proposed for the mechanism of asymmetric induction in solution. The fact that the highest ee for 2a is obtained for substoichiometric amounts of ephedrine suggests that there is a critical step in the mechanism of chiral transmission where the inductor is acting as a catalyst. The previous observation raises the question of the photocyclization mechanism and the actual intermediates involved. This question was addressed by laser flash photolysis.
Upon 308 nm laser excitation of a ∼10−4 M hexane solution of 1a, a transient absorption spectrum was recorded (Fig. 1). The transient was very long lived and did not significantly decay in 2 ms, the longest monitoring time available to our nanosecond system. Assignment of this transient to the intermediate 3a in Scheme 2 was done based on: (i) the similarity of this spectrum to those for related cyclohexadienyl intermediates and a lifetime in the range expected for this type of long-lived intermediate;16 (ii) the detection of fluorescence from the singlet excited state of compound 1a that decays on a much shorter time scale (ns) than the transient shown in Fig. 2 and; (iii) the chemical reactivity of this species including the lack of reactivity with oxygen (ruling out a triplet excited state) and quenching by trifluoroacetic acid and ephedrine (see Fig. 2). The zwitterionic intermediate 3a would be formed by electrocyclic ring closure of the anilide from the α,β-unsaturated carboxylic acid (Scheme 2).
 | ||
| Fig. 1 Transient spectrum recorded 2.88 μs after 308 nm laser excitation of a solution of p-toluidide 1a in hexane (∼10−4 M) under nitrogen. | ||
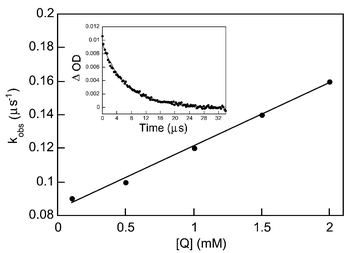 | ||
| Fig. 2 Plot of kobsversus concentration of trifluoroacetic acid (TFAA). Inset: transient decay monitored at 400 nm after 308 nm excitation of a ∼10−4 M solution of 1a containing 1.5 × 10−3 M TFAA. | ||
 | ||
| Scheme 2 | ||
The increased rate of disappearance of the transient in the presence of inductors and acids is particularly relevant in interpreting the results of the asymmetric induction using catalytic amounts of chiral inductor. Since the asymmetric center is created during protonation at the CO α-position of 3a, we propose that the role of the chiral inductor is to act as an acid providing the proton in a stereoselective manner. The general range of pKa values for the species involved in this step is compatible with our proposal.17 The rate constants for quenching of the observed intermediate by trifluoroacetic acid and (−)-ephidrine were determined to be kq
= 3.76 × 107 M−1 s−1 and kq
= 1.19 × 107 M−1 s−1, respectively.
This mechanism for asymmetric induction in the photocyclization of anilides in solution is obviously different from that proposed for the enantioselective irradiation of crotonanilide inclusion crystals.12,13 In the latter case a concerted, stereoselective 1,5-H migration was assumed to be the key step in the photoreaction. The chiral environment provided by the crystal lattice of the host would be responsible for enantioselectivity in the H migration; bimolecular processes are ruled out in the crystalline state. It is obvious that this mechanism for asymmetric induction, based on the immobility of substrate and chiral inductor, cannot operate in solution. Our proposal of asymmetric protonation of a photochemically generated enolate closely resembles the mechanism proposed for the deconjugation of α,β-unsaturated esters18–21 as well as the Norrish type II dealkylation of α-alkyl substituted indanones.22,23 In these cases a photochemically generated prochiral enol undergoes enantioselective protonation by a chiral inductor analogous to the mechanism in Scheme 2.24,25
We also carried out the irradiation of the p-toluidide of cyclohex-2-enecarboxylic acid (1b). The cyclic structure of the α,β-unsaturated carboxylic acid will avoid the occurrence of CC double bond photoisomerization that could disguise the asymmetric induction by concurrent photocyclization of trans as well as cis isomers in solution. In the case of 1b, two asymmetric centers are concurrently formed in the photocyclization. The enantioselective photocyclization of anilides related to 1b has been previously studied in solution11 as well as in inclusion crystals.12 As with 1a, the only product observed was the corresponding dihydroquinolinone 2b as a mixture of cis
and trans stereoisomers (Scheme 3). The results obtained are also collected in Table 1.
 | ||
| Scheme 3 | ||
As can be seen in Table 1, photocyclization of 1b to the cis- and trans-quinolones 2b also occurs with a high chemoselectivity at almost complete substrate conversions. The reaction in the absence of chiral inductor gave a cis ∶ trans diastereomeric ratio close to 1. It is clearly demonstrated in Table 1 that the nature of the chiral inductor distinctively influences both the diastereomeric ratio and the ee. The trans was the preferred diastereomer for irradiations in the presence of chiral inductors and different ee were measured for each cis and trans isomer. This indicates that the chiral inductor favors a certain configuration of the β-carbon at the electrocyclic ring closure.
In conclusion, photocyclization of anilides of α,β-unsaturated carboxylic acids in solution in the presence of chiral inductors affords the corresponding 3,4-dihydro-2(1H)-quinolones with essentially total chemoselectivity and with low to moderate enantiomeric and diastereomeric selectivities. Among the series of chiral inductors, ephedrine in substoichiometric concentrations at low temperature yielded the highest ee (37%) for the irradiation of the p-toluidide of methacrylic acid. We propose that the process by which the chiral induction occurs in solution is different from that reported for the irradiation of inclusion crystals in the solid state. A transient was detected for the first time by laser flash photolysis and assigned to the prochiral, zwitterionic, enolate intermediate arising from the electrocyclic ring closure. The fact that this transient reacts with acids suggests that chiral induction occurs by asymmetric protonation at the α-carbon of the enolate rather than through a concerted 1,5-H migration.
Experimental
Synthesis of anilides 1a,b
Thionyl chloride (1.70 g, 14 mmol) was added to a solution of methacrylic or cyclohex-2-enecarboxylic acid (10 mmol) in CCl4 (20 ml) and the mixture magnetically stirred at room temperature for 2 h. After this time, the solution was concentrated under reduced pressure and a fresh aliquot of CCl4 was added and removed under vacuum to reduce the residual amount of SOCl2 present. A solution of toluidine (10 mmol) dissolved in CH2Cl2 (20 ml) was added to this crude mixture containing the corresponding acyl chloride, and the mixture stirred at room temperature for 2 h. After this time, the solvent was removed under reduced pressure and the resulting solid was recrystallized from EtOH. Toluidides 1a,b were fully characterized by combustion chemical analysis (C, H and N) and by their IR, 1H NMR, 13C NMR, and MS spectroscopic properties. All the data were in agreement with their structure (vide infra).Irradiation procedure
A solution of toluidides 1a,b in cyclopentane (200 ml) was purged with a nitrogen stream for at least 15 min before irradiation. Photolyses were carried out in quartz tubes under magnetic stirring using a 125 W medium pressure mercury lamp. The irradiation vessel was maintained at the required temperature using a cryostat. The course of the reaction was periodically followed by analyzing the photolysis mixture using an isochratic Waters HPLC coupled with a polarimeter and a UV detector monitoring at 256 nm. For the HPLC analyses of the reaction mixtures a chiral column (CHIRALCEL OB) and a 4 ∶ 1 mixture of THF and i-PrOH as eluent were used. At the end of the irradiation, the solvent was removed using a rotary evaporator and the residue analyzed by 1H and 13C NMR (Varian Gemini+, 300 MHz) in CDCl3. The residue was also analyzed by GC-MS (HP 5644 A) using a 25 m capillary column of 5% cross-linked phenylmethylsilicone. The corresponding photo-products were spectroscopically characterized. In the case of the irradiation of substrate 1b, the corresponding 2-quinolone was characterized as a mixture of cis and trans stereoisomers.Laser flash photolysis
Samples were excited with 308 nm pulses (6 ns pulse width, ∼85 mJ per pulse) from a Lumonics EX-530 excimer laser using a Xe–HCl–Ne mixture. Signals from the monochromator/photomultiplier were captured by a Tektronix 2440 digitizer and transferred to a PowerMacintosh computer programmed in the LabVIEW 4.1 environment from National Instruments. Detailed descriptions of similar laser systems have been provided elsewhere.26,27 Solutions of toluides 1a and 1c in spectroscopic grade hexane (∼10−4 M) were fully deaerated in Suprasil 7 × 7 mm quartz cells for 20 minutes prior to photolysis. All measurements were recorded using a flow system to avoid the complications of photo-product build-up. The emission spectrum of compound 1a was measured at room temperature in nitrogen-purged cyclopentane solution with an Edinburgh FS900 spectrofluorimeter using a Xe lamp as the excitation source. The emission decay was shorter than the ns response time of the instrument.Analytical and spectroscopic data of compounds 1a,b
Acknowledgements
Financial support by the Canadian NSERC through an operating grant (J.C.S.) and to the Spanish DGICYT (H.G., project MAT00–1676–CO2) is gratefully acknowledged. M.J.S. also thanks the Generalidad Valenciana for funding (GVDOC99-0203).References
- H. Rau, Chem. Rev., 1983, 83, 535 CrossRef CAS.
- Y. Inoue, Chem. Rev., 1992, 92, 471.
- S. R. L. Everitt and Y. Inoue, in Organic Molecular Photochemistry, eds. V. Ramamurthy and K. S. Schanze, Marcel Dekker, New York, 1999, p. 71 Search PubMed.
- Y. Ogata, K. Takaki and I. Ishino, J. Org. Chem., 1971, 36, 3975 CrossRef CAS.
- J. D. Coyle, Chem. Rev., 1978, 78, 97 CrossRef CAS.
- A. Gilbert and J. Baggot, Essentials of Molecular Photochemistry, Blackwell, Oxford, 1991 Search PubMed.
- J. F. Wolfe and M. A. Ogliaruso, in The Chemistry of Functional Groups, ed. S. Patai, John Wiley and Sons, Chichester, 1979, vol. 2, part B, p. 1063 Search PubMed.
- A. G. Schultz, Acc. Chem. Res., 1983, 16, 210 CrossRef CAS.
- I. Ninomiya and T. Naito, in Advances in Heterocyclic Chemistry, ed. A. Brossi, Academic Press, San Diego, 1983, vol. XXII, p. 189 Search PubMed.
- I. Ninomiya, S. Yamauchi, T. Kiguchi, A. Shinohara and T. Naito, J. Chem. Soc., Perkin Trans. 1, 1974, 2285 Search PubMed.
- T. Naito, Y. Tada and I. Ninomiya, Heterocycles, 1984, 22, 237 Search PubMed.
- K. Tanaka, O. Kakinoki and F. Toda, J. Chem. Soc., Chem. Commun., 1992, 1053 RSC.
- H. Hosomi, S. Ohba, K. Tanaka and F. Toda, J. Am. Chem. Soc., 2000, 122, 1818 CrossRef CAS.
- M. C. Barbera, H. Garcia, M. A. Miranda and J. Primo, An. Quim., 1995, 91, 95 Search PubMed.
- Previous studies of the enantioselective photocyclization of acrylanilides were carried out in benzene–ethyl ether, 3 ∶ 1.
- A. Sanjuan, M. Alvaro, H. Garcia and J. C. Scaiano, J. Am. Chem. Soc., 1998, 120, 7351 CrossRef CAS.
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley and Sons, New York, 1995 Search PubMed.
- O. Piva, R. Mortezaei, F. Henin, J. Muzart and J.-P. Pete, J. Am. Chem. Soc., 1990, 112, 9263 CrossRef CAS.
- R. M. Duhaime, D. A. Lombardo, I. A. Skinner and A. C. Weedon, J. Org. Chem., 1985, 50, 873 CrossRef CAS.
- R. Mortezaei, O. Piva, F. Henin, J. Muzart and J.-P. Pete, Tetrahedron Lett., 1986, 27, 2997 CrossRef CAS.
- F. Henin, J. Muzart, J.-P. Pete and O. Piva, New J. Chem., 1991, 15, 911 Search PubMed.
- O. Piva, J. Org. Chem., 1995, 60, 7879 CrossRef CAS.
- F. Henin, J. Muzart, J.-P. Pete, A. M’oungou-M’assi and H. Rau, Angew. Chem., Int. Ed. Engl., 1991, 30, 416 CrossRef.
- L. Duhamel, P. Duhamel, S. Fouquay, J. J. Eddine, O. Peschard, J. C. Plaquevent, A. Ravard, R. Solliard, J. Y. Valnot and H. Vincens, Tetrahedron, 1988, 44, 5495 CrossRef CAS.
- C. Fehr and J. Galindo, J. Am. Chem. Soc., 1988, 110, 6909 CrossRef CAS.
- J. C. Scaiano, J. Am. Chem. Soc., 1980, 102, 7747 CrossRef CAS.
- J. C. Scaiano, M. Tanner and D. Weir, J. Am. Chem. Soc., 1985, 107, 4396 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |