Condensation of triformylmethane with adenosine: novel cyclic adducts derived from 1,3-dicarbonyl compounds
Kari
Neuvonen
*,
Niangoran
Koissi
and
Harri
Lönnberg
Department of Chemistry, University of Turku, FIN-20014 Turku, Finland
First published on 30th November 2001
Abstract
Condensation of triformylmethane (TFM) with adenosine has been studied in pyridine and aqueous dioxane. One 1 ∶ 1 (2) and two 2 ∶ 1 (6 and 7) TFM–adenosine adducts were isolated. The structural assignment of these products by 1H and 13C NMR and UV spectroscopy and MS spectrometry suggested that the appearance of the 2 ∶ 1 adducts is connected to a competitive self-condensation of TFM, the stable end product of which is benzene-1,3,5-tricarbaldehyde. The intermediates on the reaction pathway can be reacted with adenosine affording a new procedure for nucleic acid base modification. The mechanisms of formation and the role of intramolecular hydrogen bonding in stabilization of cyclic adenosine adducts are discussed.
Introduction
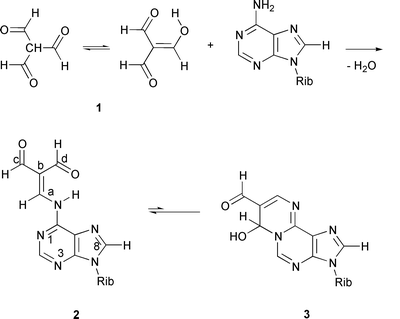
1,3-Dicarbonyl compounds and α,β-unsaturated aldehydes are known to generate cyclic adducts with nucleic acid bases.1 While a number of compounds falling in these categories, such as malonaldehyde (propanedial) and acrolein (pronenal), are produced endogenously as metabolites of lipid peroxides,2 several structurally related compounds, such as halogenated aldehydes3 and halogenated hydroxyfuranones,4 belong to well established xenobiotic mutagens. Accordingly, the reactions of bifunctional carbonyl compounds with nucleic acid bases in general are of interest as a potential source of miscoding of DNA synthesis.Triformylmethane (TFM, 1), first prepared by Arnold and ![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) emli
emli![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) ka,5 is an interesting example of 1,3-dicarbonyl compounds. While it in principle may be regarded as an equilibrium mixture of keto and enol tautomers (Scheme 1), in DMSO-d6 solution, for example, the presence of only the enol form can be observed by NMR spectroscopy. Accordingly, TFM is simultaneously a 1,3-dialdehyde and an α,β-unsaturated aldehyde. In spite of these structurally interesting features that make TFM a potential mutagen, data on its reactions with nucleic acid bases are scarce. It has been shown that TFM forms with cytosine base a rather stable cyclic N3,N4-carbinolamine† adduct,6
and with guanine base an N1,N2-carbinolamine adduct, which undergoes reversible dehydration to the corresponding fully conjugated adduct, 7-formyl-3-(β-D-ribofuranosyl)pyrimido[1,2-a]purin-10(3H)-one.7 The present work describing the reaction of TFM with adenosine, is a systematic continuation of these investigations.
ka,5 is an interesting example of 1,3-dicarbonyl compounds. While it in principle may be regarded as an equilibrium mixture of keto and enol tautomers (Scheme 1), in DMSO-d6 solution, for example, the presence of only the enol form can be observed by NMR spectroscopy. Accordingly, TFM is simultaneously a 1,3-dialdehyde and an α,β-unsaturated aldehyde. In spite of these structurally interesting features that make TFM a potential mutagen, data on its reactions with nucleic acid bases are scarce. It has been shown that TFM forms with cytosine base a rather stable cyclic N3,N4-carbinolamine† adduct,6
and with guanine base an N1,N2-carbinolamine adduct, which undergoes reversible dehydration to the corresponding fully conjugated adduct, 7-formyl-3-(β-D-ribofuranosyl)pyrimido[1,2-a]purin-10(3H)-one.7 The present work describing the reaction of TFM with adenosine, is a systematic continuation of these investigations.
 | ||
| Scheme 1 | ||
Results and discussion
It has been previously shown that condensation of TFM with compounds possessing primary amino groups yields aminomethylene derivatives instead of imines.8 Depending on the molar ratio of TFM and the amine, di- or triimines can also be formed.8 The reaction of TFM with adenosine in pyridine, however, gave a single product, even when a three-fold excess of adenosine was used. This TFM adduct may be assigned as an aminomethylene derivative, viz.N6-(2,2-diformylvinyl)adenosine (2, Scheme 1), not an imine. This assignment receives support from comparison of the 1H and 13C NMR chemical shifts with those observed earlier9,10 for related acyclic adducts of nucleosides. In particular, the observed vicinal coupling between the a-H and H-N6 protons (12.4 Hz) is a clear indication of the suggested enamino structure. This value is in good agreement with the magnitude of the analogous coupling constants recorded for various aminomethylene derivatives of TFM (12–16 Hz)8 and for N6-(2-formylvinyl)adenosine (11.4 Hz).9 The lowfield formyl proton (d-H) is coupled to the a-H proton (4JH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CHO
= 3.3 Hz) due to a favourable planar W coupling pathway, also consistent with the proposed structural assignment.
C–CHO
= 3.3 Hz) due to a favourable planar W coupling pathway, also consistent with the proposed structural assignment.
Although the 1 ∶ 1 TFM adducts of adenosine and cytosine are both aminomethylene tautomers, they exhibit dissimilar ring–chain tautomerism. While the cytosine adduct has been shown to be predominantly in a cyclic carbinolamine form,6 no similar cyclization could be observed with 2 by NMR spectroscopy.
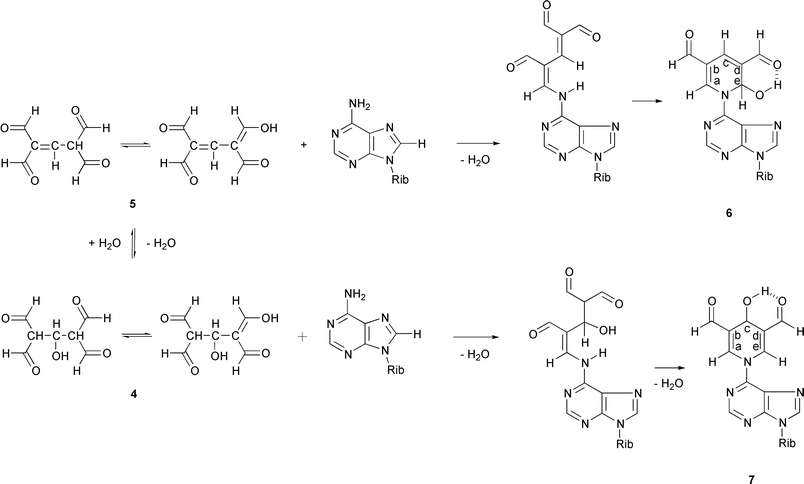
In aqueous dioxane, the condensation between adenosine and TFM resulted in a mixture of 2 and two 2 ∶ 1 TFM–adenosine adducts (6, 7; Scheme 2). Furthermore, the presence of a 3 ∶ 1 TFM–adenosine adduct in the crude reaction mixture could be observed by HPLC/ESI-MS. When the condensation was carried out under aqueous conditions, benzene-1,3,5-tricarbaldehyde could be isolated as a minor reaction product.
 | ||
| Scheme 2 | ||
The exact mass measurements (ESI+-MS) suggest that the [M + H]+ ions of compounds 6 and 7 both exhibit the same elemental composition, viz. C17H18N5O7. The fragmentation pathways, however, are not identical. Scrutiny of the percentage intensities of the [M + H2O + H]+, [M + H]+ and [M − H2O + H]+ ions shows that the intensity ratio [M + H]+
∶ [M − H2O + H]+ in the ESI+ mass spectra is practically the same with both compounds studied. However, the intensity ratio [M + H2O + H]+
∶ [M − H2O + H]+ is 0.87 for compound 6 and
0.35 for compound 7. In other words, compound 6 exhibits a more prominent [M + H2O + H]+ ion. Tentatively, this difference can be accounted for by the divergent ability of 6 and 7 to form covalent hydrates by addition of water to the OCH–CC–N– double bond, which process also occurs in hydrolysis of enamines into ketones. According to this interpretation, the mono-hydrate of compound 6 possessing a 2,6-dihydroxy substituted tetrahydropyridine system is more stable than the corresponding 2,4-dihydroxy substituted system formed by 7. In conditions used for HPLC/ESI-MS experiments, however, 6 and 7
are equilibrated.
The structural assignment of 6 and 7 may be based on the NMR spectroscopic data. Interestingly, compound 6 appears as a mixture of two diastereomers exhibiting roughly the same mole fractions. The diastereomers are probably equilibrated via an open-chain form. The 1H NMR spectrum shows the presence of two partly overlapping AX systems for the diastereomeric CHOH protons. The 13C NMR spectrum is even more sensitive in this respect. The shift differences are, however, too small to allow any reliable assignment of the diastereomers in question. According to the 1H and 13C NMR spectra, compound 7 is more symmetrical than compound 6, and it could hence be assigned to possess a 3,5-diformyl-4-hydroxy-1,4-dihydropyridine
moiety. However, even now two sets of protons assignable to the formally symmetric dihydropyridine system are observed. In other words, the CHO and CHN protons are magnetically non-equivalent. The a-H and e-H protons give an AB spectrum (4JH–C–N–C–H
= 1.28 Hz). It may be tentatively assumed that an intramolecular hydrogen bond, having the CHOH group as the donor and the formyl group as the acceptor, is formed. This hydrogen bond seems to be strong enough to render the 1,4-dihydropyridine ring dissymmetric on the NMR time-scale.
The 1H and 13C NMR chemical shifts of compound 6 are in excellent agreement with those recorded for a closely related derivative bearing a methyl group at C-c. The latter compound has been obtained by a reaction of malonaldehyde with 2′-deoxyadenosine in a buffered aqueous solution, and is supposed to be formed by intermediary formation of 2,4-dihydroxymethylene-3-methylglutaraldehyde from two molecules of malonaldehyde and one molecule of acetaldehyde.11 Significant differences occur only between the shift values of C-b/-d and c-H or C-c due to the change of substitution at C-c.
The structural assignment of 6 and 7 is supported by characteristic differences in their long-range H–H correlations (long-range COSY). Compound 6 shows a correlation between c-H and both b-CHO and d-CHO, on one hand, and between a-H/e-H and b-CHO/d-CHO, on the other hand. Compound 7, in turn, shows a correlation between the following protons: a-H, c-H; a-H, b-CHO; c-H, b-CHO; e-H, c-H and e-H, d-CHO. These correlation patterns support the preceding assignment, suggesting a more symmetric structure for compound 6.
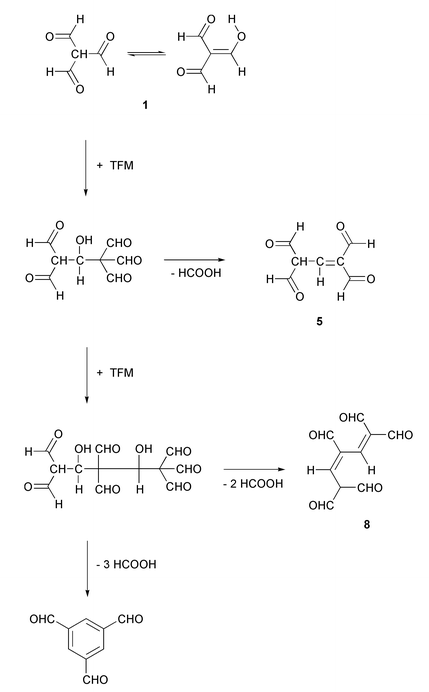
In addition to the adducts described above, HPLC/ESI-MS analysis of the crude reaction mixture obtained in aqueous dioxane clearly showed the appearance of the [M + H]+ ion of a 3 ∶ 1 TFM–adenosine adduct at m/z 458 (see Fig. 1). The elemental composition of the [M + H]+ ion was C20H20N5O8, suggesting that the adduct is formed by condensation (loss of water) of adenosine with the intermediate 8 derived from three TFM molecules, as depicted in Scheme 3.
 | ||
| Scheme 3 | ||
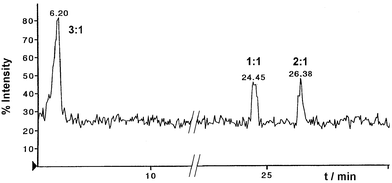 | ||
| Fig. 1 Total ion current plot on the first quadrupole of the instrument between m/z 120–1300 of the mass spectrum for the TFM–adenosine reaction mixture. | ||
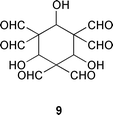
The appearance of the 2 ∶ 1 and 3 ∶ 1 TFM–adenosine adducts in addition to the expected 1 ∶ 1 adduct suggests that TFM undergoes self-condensation before reacting with adenosine. Formation of benzene-1,3,5-tricarbaldehyde as a side product lends further support for this proposal. In fact, two different self-condensation products of TFM have previously been documented, viz. benzene-1,3,5-tricarbaldehyde12 and 2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene-4,8-dicarbaldehyde.13 Formally, benzene-1,3,5-tricarbaldehyde can be formed from three molecules of TFM by loss of three molecules of formic acid (so-called dehydroxyformylation). A cyclic six-membered structure 9 has been proposed as an intermediate.12 Accordingly, the formation of the TFM–adenosine adducts described above can be rationalised with the aid of TFM–benzene-1,3,5-tricarbaldehyde transformation, as depicted in Scheme 3. In all likelihood the intermediates of this transformation are trapped at two different levels, a dimeric (5) and trimeric level (8), before the final cyclization into benzene-1,3,5-tricarbaldehyde. Our results hence suggest that the formation of benzene-1,3,5-tricarbaldehyde proceeds by a step-wise mechanism via open-chain intermediates instead of a cyclic one. The appearance of the two isomeric dihydropyridines 6 and 7 is intriguing. Formation of the 1,2-dihydropyridine derivative 6 can be explained by condensation of adenosine with 1,1,3,3-tetraformylpropene (5). Intramolecular nucleophilic attack of N6 on a formyl group of the initially formed aminomethylene derivative results in formation of 6 (Scheme 2). Formation of the 1,4-dihydropyridine derivative 7 may, in turn, be explained by intermediary formation of the hydrate 4 of the 1,1,3,3-tetraformylpropene, or by equilibration of 6 to 7. On prolonged standing in solutions the 3 ∶ 1 TFM–adenosine adduct is decomposed into 7.
Experimental
CAUTION: TFM is a new reagent for nucleic acid bases. It has not yet been determined whether facile chemical alteration of the structure is accompanied by mutagenicity in living organisms. Nevertheless, caution should be taken in the handling and disposal of the compound.Spectroscopic methods
The NMR spectra were acquired on a JEOL Alpha 500 NMR spectrometer equipped with either a 5 mm normal configuration tuneable probe or a 5 mm inverse z-axis field-gradient probe operating at 500.16 MHz for 1H, and 125.78 MHz for 13C. The deuterium resonance of the solvent was used as a lock signal. The spectra were recorded at 30 °C in DMSO-d6 (if not otherwise stated); 1H and 13C spectra were referenced internally to tetramethylsilane (both 0 ppm).1D proton spectra were acquired with single-pulse excitation, 45° flip angle, pulse recycle time of 9 s and with spectral widths of 8 kHz consisting of 65 k data points (digital resolution 0.11 Hz per pt), zero-filled to 128 k prior to Fourier transformation. Exponential or Lorentz–Gaussian resolution enhancement was usually applied prior to Fourier transformation. Double-quantum filtered (DQF-COSY) spectra were acquired in phase-sensitive mode with spectral widths appropriately optimised from the 1D spectra, and processed with zero-filling (×2, ×4) and exponential weighting (1 Hz) applied in both dimensions prior to Fourier transformation.
1D carbon spectra were acquired with single-pulse excitation, 45° flip angle, pulse recycle time of 3.5 s, and with spectral widths of 34 kHz consisting of 64 k data points (digital resolution 0.52 Hz per pt), zero-filled to 128 k and with 1 Hz exponential weighting applied prior to Fourier transformation. DEPT 135° spectra were acquired with similar spectral windows and with a pulse delay time of 3 s. CHSHF (C,H correlation with partial homonuclear decoupling in f1) experiments were acquired with spectral widths appropriately optimised from the 1D spectra and processed with zero-filling (×2, ×4), a 2π/3-shifted sinebell function, and exponential weighting applied in both dimensions prior to Fourier transformation. The CHSHF spectra utilised a 1JHC coupling of 145 Hz.
The UV spectra of the compounds were recorded on a diode-array detector (Shimadzu SPD-6A) coupled to the HPLC system.
Mass spectrometric methods
The HPLC/ESI+ mass spectra were acquired using a Perkin Elmer Sciex API 365 triple quadrupole LC/MS/MS mass spectrometer connected to two Perkin–Elmer Series 200 micro pumps, a Perkin Elmer 785A UV/VIS detector and Perkin–Elmer Series 200 autosampler (Perkin–Elmer, Norwalk, CT, USA). The separations of compounds were carried out on a Hypersil HyPURITY™ Elite C18 column (4.6 × 150 mm, 5 μm, ThermoHypersil Ltd) using acetonitrile–water (15 ∶ 85 v/v) as an eluent. The accurate masses were measured from ESI+-MS spectra recorded on a Fisons ZabSpec-oaTOF instrument (Manchester, UK). Ionization was carried out using nitrogen as both the nebulizing and bath gas. A potential of 8.0 kV was applied to the ESI needle. The temperature of the pepperpot counter electrode was 90 °C. The samples were introduced by loop injection at a flow rate of 20 μL min−1 using water–acetonitrile–acetic acid (80 ∶ 20 ∶ 1 v/v/v).Chromatographic methods
Analytical HPLC separations were performed on a RP-18 column (SpherisorbODS2, 4 × 125 mm, 5 μm, Hewlett-Packard Ltd). The column was eluated first isocratically for 5 min with a 0.01 M phosphate buffer (pH 7.1), the acetonitrile content of which was then increased from 0 to 30% in 25 min. The flow rate was 1 mL min−1. The preparative isolation of the compounds was carried out on a LiChroprep RP-18 column (37 × 440 mm, 40–63 μm, Merck KGaA). The evaporated fractions, which according to 1H NMR spectroscopy contained the modified nucleosides, were dissolved in 5 mL of water and then further purified on a LiChrospher RP-18 column (10 × 250 mm, 5 μm, Merck KGaA).Materials
The starting materials employed were commercial products of Sigma or Aldrich, and they were used without further purification. Dioxane was of spectroscopic grade, and the solvents otherwise of analytical grade. Pyridine was dried by refluxing on calcium hydride and subsequent distillation. Crude TFM14 was purified by sublimation (Büchi GKR-50 glass tube oven; 45 °C, 1 mm Hg). The white crystalline solid obtained melted at 105 °C (lit.14 104–106 °C).| 2 | 6 | 7 | |
|---|---|---|---|
|
a
1′-H–5″-H and 2′-OH, 3′-OH and 5′-OH are protons in the ribosyl unit; the assignment of signals of the sugar moiety was based on the connectivities in the H–H COSY spectra.
b
3
J
H–CN–H
= 12.4 Hz, 4JH–CC–CHO
= 3.3 Hz.
c
c-CHO syn to a-H, d-CHO anti to a-H.
d
An AB system further split into doublets.
e
,
f
e and f refer to the two different diastereomers exhibiting roughly the same mole fractions and possessing closely related 1H chemical shifts. Due to the relatively small chemical shift differences their mutual assignment was not possible.
g
The OH protons of the two diastereomers overlapping.
h
The assignment is tentative.
i
Due to the intramolecular hydrogen bonding, a-H and e-H protons (an AB system, 4JH–C–N–C–H
= 1.28 Hz) and, on the other hand, b-CHO and d-CHO protons, are magnetically non-equivalent. Because of the small chemical shift differences no mutual assignment of 1H chemical shifts of the protons in question was possible.
j
An AB system (J
= 11.9) further split into multiplets.
|
|||
| 2-H | 8.76 (s) | 8.84 (s) | 8.79 (s) |
| 8-H | 8.88 (s) | 8.950 (s)e | 8.92 (s) |
| 8.949 (s)f | |||
| a-H | 9.17 (dd)b | 9.542 (t, J = 1.4);e 9.540 (t, J = 1.4)f | 9.118i or 9.101i |
| c-H | 7.74 (m) | 4.78 (s, br) | |
| e-H | 7.542 (d, 3J = 6.9);e 7.527 (d, 3J = 7.3)f | 9.118i or 9.101i | |
| e-OH | 6.93 (d, 3J = 7.5)g | ||
| N-H | 12.36 (d)b | ||
| b-CHO | 9.61 (s)h | 9.436 (d, J = 0.64) or 9.434 (d, J = 0.53)i | |
| c-CHO | 9.68 (s)c | ||
| d-CHO | 9.95 (d)b,c | 9.70 (s)h | 9.436 (d, J = 0.64) or 9.434 (d, J = 0.53)i |
| 1′-Ha | 6.05 (d, J = 5.3) | 6.087 (d, J = 5.3);e 6.085 (d, J = 5.3)f | 6.09 (d, J = 5.2) |
| 2′-H | 4.63 (t, J = 5.2) | 4.61 (m) | 4.60 (q, J = 5.3) |
| 3′-H | 4.22 (t, J = 4.4) | 4.21 (q, J = 4.2) | 4.21 (q, J = 4.7) |
| 4′-H | 4.01 (q, J = 3.8) | 4.00 (q, J = 3.9) | 4.00 (q, J = 4.0) |
| 5′-H | 3.72 (dd, J = 12.1; 4.0)d | 3.72 (m)d | 3.72j |
| 5″-H | 3.60 (dd, J = 12.1; 4.0)d | 3.60 (dt, J = 12.1; 4.5)d | 3.60j |
| 2′-OH | 5.55 (s) | 5.57 (d, J = 5.6) | 5.56 (d, J = 5.8) |
| 3′-OH | 5.24 (s) | 5.25 (d, J = 4.9) | 5.23 (d, J = 5.2) |
| 5′-OH | 5.12 (s) | 5.12 (t, J = 4.9) | 5.11 (t, J = 5.4) |
| 2 | 6 d | 7 | |
|---|---|---|---|
| a C-1′–C-5′ carbons in the ribosyl unit. b syn to a-H. c anti to a-H. d Compound 6 appears as a roughly equimolar mixture of two diastereomers possessing closely related 13C chemical shifts. Therefore, no mutual assignment of 13C chemical shifts of the different isomers was possible. e Due to the intramolecular hydrogen bonding, C-a and C-e are magnetically non-equivalent. Because of the small chemical shift difference, however, no mutual assignment of 13C chemical shifts of the carbons in question was possible. | |||
| C-2 | 151.97 | 151.56 | 151.44 |
| C-4 | 152.23 | 152.83 | 152.79 |
| C-5 | 122.05 | 122.72 | 121.41 |
| C-6 | 146.77 | 149.09 | 146.36 |
| C-8 | 144.72 | 143.99 | 143.68 |
| C-a | 151.43 | 146.97 | 141.76e |
| C-b | 114.92 | 116.13; 116.11 | 122.59 |
| C-c | 134.58 | 22.14 | |
| C-d | 129.46 | 122.59 | |
| C-e | 71.56; 71.53 | 141.71e | |
| b-CHO | 187.80 | ||
| c-CHO | 189.93b | 189.97 | |
| d-CHO | 191.81c | 191.22 | 189.97 |
| C-1′a | 88.07 | 88.02; 87.96 | 88.06 |
| C-2′ | 73.93 | 73.92 | 73.95 |
| C-3′ | 70.19 | 70.11 | 70.03 |
| C-4′ | 85.78 | 85.71 | 85.63 |
| C-5′ | 61.15 | 61.05 | 60.97 |
References
- L. J. Marnett, in DNA Adducts: Identification and Biological Significance, eds. K. Hemminki, A. Dipple, D. E. G. Shuker, F. F. Kadlubar, D. Segerbäck and H. Bartsch, IARC Scientific Publications No. 125, IARC, Lyon, 1994, pp.151–163 Search PubMed.
- H. Esterbauer, P. Eckl and A. Ortner, Mutat. Res., 1990, 238, 223 CAS.
- C. Malaveille, H. Bartsch, A. Barbin, A. M. Camus, R. Montesano, A. Croisy and P. Jacquignon, Biochem. Biophys. Res. Commun., 1975, 63, 363 CrossRef CAS; A. Barbin, H. Bresil, A. Croisy, P. Jacquignon, C. Malaveille, R. Montesano and H. Bartsch, Biochem. Biophys. Res. Commun., 1975, 67, 596 CrossRef CAS.
- L. Kronberg, R. Sjöholm and S. Karlsson, Chem. Res. Toxicol., 1992, 5, 852 CrossRef CAS.
- Z. Arnold and J. emlika, Collect. Czech. Chem. Commun., 1960, 25, 1318 CAS.
- K. Neuvonen, C. Zewi and H. Lönnberg, Acta Chem. Scand., 1996, 50, 1137 Search PubMed.
- N. Koissi, K. Neuvonen, T. Munter, L. Kronberg and H. Lönnberg, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1761 Search PubMed.
- Z. Arnold, M. Budĕ
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) ínský and M. Pankova, Collect. Czech. Chem. Commun., 1991, 56, 1019 CAS.
ínský and M. Pankova, Collect. Czech. Chem. Commun., 1991, 56, 1019 CAS. - V. Nair, G. A. Turner and R. J. Offerman, J. Am. Chem. Soc., 1984, 106, 3370 CrossRef CAS.
- F. Le Curieux, T. Munter and L. Kronberg, Chem. Res. Toxicol., 1997, 10, 1180 CrossRef CAS.
- F. Le Curieux, D. Pluskota, T. Munter, R. Sjöholm and L. Kronberg, Chem. Res. Toxicol., 1998, 11, 989 CrossRef CAS.
- J. emlika, J. Krupika and Z. Arnold, Collect. Czech. Chem. Commun., 1962, 27, 2464 CAS.
- Z. Arnold and M. Budĕínský, J. Org. Chem., 1988, 53, 5352 CrossRef CAS.
- M. Budĕínský, P. Fiedler and Z. Arnold, Synthesis, 1989, 858 CAS.
Footnote |
| † The IUPAC name for carbinolamine is aminomethanol. |
| This journal is © The Royal Society of Chemistry 2002 |